
Abstract
Purpose of Review
The aim of this study was to examine the synaptic biomarker neurogranin in cerebrospinal fluid (CSF) in different stages of HIV infection and in relation to what is known about CSF neurogranin in other neurodegenerative diseases.
Recent Findings
CSF concentrations of neurogranin are increased in Alzheimer’s disease, but not in other neurodegenerative disorder such as Parkinson’s disease, frontotemporal dementia, and Lewy body dementia. Adults with HIV-associated dementia have been found to have decreased levels of neurogranin in the frontal cortex, which at least to some extent, may be mediated by the proinflammatory cytokines IL-1β and IL-8.
Summary
CSF neurogranin concentrations were in the same range for all groups of HIV-infected individuals and uninfected controls. This either indicates that synaptic injury is not an important part of HIV neuropathogenesis or that CSF neurogranin is not sensitive to the type of synaptic impairment present in HIV-associated neurocognitive disorders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neurogranin is a neuron-specific post-synaptic protein, abundant in excitatory neurons in the cortex, hippocampus, and amygdala [1]. It plays an important part in synaptic plasticity, enhancing synaptic strength by regulating the availability of calmodulin [2, 3]. This process is an essential part in long-term potentiation (LTP), a process believed to be essential to generate memories. Neurogranin knock-out mice display loss of spatial and emotional learning as well as a decrease in LTP induction resulting in disorientation [4]. In humans, synaptic dysfunction (assessed via CSF neurogranin) has been associated with memory performance [5].
Neurogranin can be quantified in CSF with enzyme-linked immunosorbent assay (ELISA), and it has been studied in various neurocognitive disorders. Alzheimer’s disease affects the brain regions where neurogranin is mainly expressed, i.e., the hippocampus, amygdala, and the neocortex. Synapse loss has been shown to be an early event, occurring prior to neuronal death and cognitive decline [6,7,8]. Neurogranin levels are markedly reduced in the frontal cortex and hippocampus in Alzheimer’s disease, indicating loss of post-synaptic elements [9]. The development of novel anti-neurogranin monoclonal antibodies has made it possible to quantify low levels of neurogranin in CSF [10•]. Several studies have shown that CSF neurogranin concentrations are elevated in patients with mild cognitive impairment due to Alzheimer’s disease as well as in patients with Alzheimer’s disease [10•, 11]. There is a correlation between levels of CSF neurogranin and the severity of cognitive decline and brain atrophy in early stages of the disease [12, 13].
Since synapse degeneration occurs in all neurodegenerative processes, it is an unexpected finding that CSF neurogranin concentrations are not increased in other neurodegenerative disorders such as Parkinson’s disease, frontotemporal dementia, Lewy body dementia, progressive supranuclear palsy, or multiple system atrophy, indicating that high CSF neurogranin might be specific for Alzheimer’s disease [14, 15•]. This could be explained by the fact that the main brain regions affected by Alzheimer’s disease are also the regions with the highest expression of neurogranin.
In regard to multiple sclerosis and neuroinflammatory conditions, there is very little published on CSF neurogranin. Although axonal damage may be pronounced in relapsing-remitting multiple sclerosis, in one published study, there were no signs of dendritic spine involvement and CSF neurogranin levels were normal [16].
Soon after transmission, HIV can be detected in the cerebrospinal fluid (CSF) in most individuals [17, 18]. If left untreated, it will lead to a neurodegenerative process with inflammation and neuronal loss that eventually can manifest with HIV encephalitis presenting as subacute HIV-associated dementia (HAD) [19, 20]. Since neurons are not infected by HIV, other mechanisms must be involved in the neuropathological damage. In addition to neuronal injury and death, synaptic disruption also contributes to neurocognitive impairment in HIV-infected individuals. The pathogenesis of neurodegeneration, including synaptic injury, is still only partially understood, but chronic immune activation and a combination of secondary effects due to both viral and host factors have been suggested [21,22,23].
Several CSF biomarkers reflect the intrathecal immune activation and neuronal damage in HIV infection. Neopterin is an important marker of cell-mediated immune activation. It is mainly produced by activated macrophages/monocytes, and its CSF levels increase with increasing immunosuppression in untreated HIV-infected individuals and are highest in patients with HAD and CNS opportunistic infections [24, 25]. CSF neurofilament light protein (NFL) is a sensitive marker of HIV-induced axonal injury [25, 26, 27•]. CSF NFL concentrations are highest in individuals with HAD where neuronal damage and loss are prominent, but some untreated individuals without neurocognitive symptoms with low CD4+ T cell counts may also have high levels.
HIV and Alzheimer’s disease share some similar features in neuropathology. One example is the increased deposition of hyperphosphorylated tau in the hippocampus [28], but there are also differences. Green et al. found ß-amyloid plaques in brains from HIV-infected individuals [29], whereas others [28, 30] have not detected any difference when compared to HIV-negative age-matched controls.
There are also similarities as well as differences in CSF biomarkers between HAD and Alzheimer’s disease. Typical changes in CSF biomarkers in Alzheimer’s disease include increases in total tau (t-tau) and phosphorylated tau (p-tau) and a decrease in Aβ1–42 [31]. In HIV-infected individuals with neuronal injury and cognitive impairment, t-tau can be elevated but p-tau is generally not [32,33,34].
There is a potential for synaptic injury in HIV infection by exposure to viral proteins such as Tat and gp120 and proinflammatory substances released by activated cells in the CNS [22]. There is, however, only scarce information about the role or perturbation of neurogranin in HIV infection, and to our knowledge, nothing about CSF neurogranin levels. In a recently published study, it was found that patients with HIV-associated neurocognitive disorders had significantly reduced expression of neurogranin in frontal cortex tissues compared to uninfected controls [35].
HIV-infected individuals now have long lifespans and therefore are at risk of being affected by diseases that come with age, such as various neurocognitive disorders. It is important to be able to correctly diagnose these conditions. CSF biomarkers add valuable information in situations like this, since they can aid in discriminating between different conditions.
To test the hypothesis that synapses are involved in the neuropathogenic pathway in HIV-1 infection, we have analyzed CSF neurogranin in a cohort of HIV-infected individuals thoroughly classified by systemic progression, CNS symptomatic presentation, and antiretroviral therapy (ART).
Methods
In this cross-sectional study, we used archived CSF samples from two academic centers: Sahlgrenska University Hospital, Gothenburg, Sweden, and San Francisco General Hospital, University of California San Francisco (UCSF), USA. All research protocols and informed consents were reviewed and approved by an ethics committee.
The study consisted of 138 HIV-infected individuals divided into six groups and 13 HIV-negative controls. HIV-infected participants were divided into four groups with untreated subjects without neurological impairment (neuroasymptomatic; NA) stratified by CD4+ T cell count into CD4 < 50 cells/μL (n = 25), CD4 50–199 cells/μL (n = 25), CD4 200–349 cells/μL (n = 25), and CD4 > 350 cells/μL (n = 30), subjects with HAD (n = 11), and subjects on suppressive ART without signs of neurological impairment (n = 33).
CSF Samples and Analytical Methods
Cell counts, HIV RNA, and proteins were analyzed immediately after sampling and remaining aliquots were centrifuged and frozen at − 70 °C until further analysis.
CSF neurogranin was measured by an in-house ELISA using Ng7 as capturing antibody and polyclonal Ng anti-rabbit antibody (ab23570; Upstate Biotechnology, Lake Placid, NY, USA) as detecting antibody, as previously described in detail [11]. All samples were analyzed in one round of experiments using one batch of reagents by board-certified laboratory technicians who were blinded to clinical data. The lower limit of quantification was 125 pg/mL, and intra-assay coefficients of variation were below 10%. CSF NFL was measured using a commercially available sandwich ELISA (NF-light® ELISA kit, UmanDiagnostics AB, Umeå, Sweden). Upper reference values are age-dependent [27•]. CSF neopterin was analyzed using a commercially available immunoassay (BRAHMS, Berlin, Germany) with an upper normal reference value of 5.8 nmol/L [24].
HIV RNA in CSF and plasma was measured using the Roche Amplicor Monitor version 1.5, Roche Taqman assay version 1 or 2 (Hoffman La-Roche, Basel, Switzerland), or Abbott RealTime HIV-1 assay (Abbott Laboratories; Abbott Park, Illinois, USA). Lower limit of detection was 20–40 copies/mL.
Statistical Methods
Statistical analysis was made with SPSS version 21 (IBM for Mac) and graphs made by GraphPad, Prism 7.0. Correlations were analyzed by Pearson correlation analysis. One-way ANOVA with Tukey’s multiple comparisons tests were used for adjusted p values for differences between groups. To reduce skewness, the following parameters were log transformed: plasma HIV RNA, CSF HIV RNA, CSF NFL, CSF neopterin, CSF neurogranin, and CD4+ T cell count.
Results
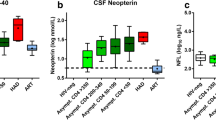
Patient characteristics are presented in Table 1. In contrast to CSF NFL, there were no significant differences in CSF neurogranin levels between any of the studied groups; although, there was a relatively wide range within each group (Fig. 1a). CSF neurogranin concentrations were in the same range for HIV-negative controls as all groups of HIV-infected individuals. CSF NFL concentrations were highest in the HAD group and significantly higher CSF NFL was also found in neuroasymptomatic individuals with low CD4+ T cell counts as compared to those with higher CD4+ T cell counts and HIV-negative controls. Participants with HAD and neuroasymtpomatic individuals with low CD4+ counts also had higher CSF neopterin and NFL levels compared with the other groups (Fig. 1b, c). A weak but statistically significant correlation was found between CSF neurogranin and CSF NFL concentrations (r = 0.38, p < 0.0001) (Fig. 2). The correlation between CSF neurogranin and CSF neopterin was even weaker, but still significant (r = 0.18, p < 0.05), but stronger between CSF NFL and CSF neopterin (r = 0.47, p < 0.0001).

Log concentrations of (a) cerebrospinal fluid (CSF) neurogranin, (b) CSF neopterin, and (c) CSF NFL for the seven groups of participants: HIV-negative; NA neuroasymptomatic untreated individuals, subgrouped by level of blood CD4+ T cell count to CD4 < 50, 50–199, 200–349, and > 349 T-cells/μL; individuals with diagnosed HAD (HIV-associated dementia); and individuals on suppressive antiretroviral therapy without signs of neurological impairment

Correlation between cerebrospinal fluid (CSF) NFL and CSF neurogranin with the same color-coding for the seven different groups of patients as in Fig. 1
Discussion
The pathogenesis leading to neuronal injury and death in HIV-infection is complex and far from completely understood. Our aim was to investigate if early synaptic dysfunction could precede axonal injury in HIV-infected individuals, but CSF neurogranin concentrations were in the same range for all participants, uninfected controls, individuals with CD4+ T cell counts ranging from less than 50 to more than 349, individuals on suppressive ART, and even individuals with HAD. This either indicates that synaptic injury is not an important part of HIV pathogenesis in the CNS or, which probably is more likely, that CSF neurogranin is not a sensitive marker for the type of synaptic impairment that may be ongoing in HIV-associated neurocognitive disorders, in analogy with several other neurodegenerative disorders [15•].
A study published earlier this year found that individuals with HAD had significantly lower expression of neurogranin in tissue from frontal cortex [35]. CSF neurogranin concentrations were not evaluated. The number of participants with HAD was, however, very low (n = 3), and even though they were on ART, they had high plasma viral loads, so the results need to be interpreted with caution.
In Alzheimer’s disease, there is also a marked reduction of neurogranin levels in frontal cortex and hippocampus, the same areas of the brain where neurogranin is mostly expressed. CSF neurogranin levels are increased in Alzheimer’s disease, but not in other studied neurodegenerative disorders [15•]. When quantified with the same method as in this study, individuals with Alzheimer’s disease had significantly higher CSF neurogranin levels than the other groups (median and interquartile range), 463 (275–669) pg/mL in Alzheimer’s disease, compared with 196 (120–297) pg/mL in healthy controls, 120 (120–304) pg/mL in Lewy body dementia, 156 (120–283) pg/mL in Parkinson’s disease, 188 (120–302) pg/mL) in progressive supranuclear palsy, and 191 (120–265) in multiple system atrophy. This could perhaps be due to the fact that different brain regions are affected by various disorders or that the degree of synaptic injury, and hence, neurogranin levels in CSF vary. It has therefore been suggested that CSF neurogranin could be used as a specific biomarker for Alzheimer’s disease.
With an aging population of HIV-infected individuals on ART, it is important to be able to discriminate neurological impairment caused by HIV itself from other neurocognitive disorders. CSF biomarkers are of great value in situations like this, since they can reflect different aspects of the pathogenesis of HIV CNS infection. CSF neurogranin could, perhaps, in combination with other CSF biomarkers, contributes to differentiating between HAD and other causes for neurocognitive decline, for example Alzheimer’s disease [36].
Since HIV does not infect neurons, other mechanisms are responsible for the synaptodendritic injury, neuronal dysfunction, and apoptosis that occurs. Although the exact pathogenesis remains unclear, some of the key steps in this process involve viral invasion of the CNS, which triggers local immune activation mediated by infected perivascular macrophages and microglia interacting with astrocytes. CSF neopterin is mainly produced by macrophages and related cells and is one of the most used markers of cell-mediated intrathecal immune activation [37, 38]. We found the same pattern for CSF neopterin levels as in several other studies [24, 39], with the highest values in those with HAD and thereafter in those with low CD4+ T cell counts. Increased CSF NFL levels are common in advanced stages of HIV-infection, particularly in individuals with HAD [24, 40], something we also found in this study.
This study has several limitations. It has a retrospective cross-sectional design and even though we did not find any significant differences in CSF neurogranin levels in the various groups of HIV-infected participants, it could be of interest to follow CSF neurogranin levels in a longitudinal study, especially after initiation of ART. The total number of participants was relatively large, but some of the individual groups were limited in size.
Conclusions
CSF concentrations of neurogranin are increased in Alzheimer’s disease, but not in other neurodegenerative disorder such as Parkinson’s disease, frontotemporal dementia, Lewy body dementia, progressive supranuclear palsy, or multiple system atrophy. Our results show that CSF neurogranin levels are not increased during any stage of HIV-infection compared to uninfected controls. Since neurogranin is a biomarker of synaptic injury, these findings either indicate that axonal injury occurs in HIV-infected individuals without preceding synaptic damage or that CSF neurogranin is not sensitive to the type of synaptic impairment that may be present in HIV-associated neurocognitive disorders.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Represa A, Deloulme JC, Sensenbrenner M, Ben-Ari Y, Baudier J. Neurogranin: immunocytochemical localization of a brain-specific protein kinase C substrate. J Neurosci. 1990;10(12):3782–92.
Zhong L, Cherry T, Bies CE, Florence MA, Gerges NZ. Neurogranin enhances synaptic strength through its interaction with calmodulin. EMBO J. 2009;28(19):3027–39.
Zhong L, Gerges NZ. Neurogranin and synaptic plasticity balance. Commun Integr Biol. 2010;3(4):340–2.
Pak JH, Huang FL, Li J, Balschun D, Reymann KG, Chiang C, et al. Involvement of neurogranin in the modulation of calcium/calmodulin-dependent protein kinase II, synaptic plasticity, and spatial learning: a study with knockout mice. Proc Natl Acad Sci U S A. 2000;97(21):11232–7.
Casaletto KB, Elahi FM, Bettcher BM, Neuhaus J, Bendlin BB, Asthana S, et al. Neurogranin, a synaptic protein, is associated with memory independent of Alzheimer biomarkers. Neurology. 2017;89(17):1782–8.
DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27(5):457–64.
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30(4):572–80.
Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298(5594):789–91.
Reddy PH, Mani G, Park BS, Jacques J, Murdoch G, Whetsell W Jr, et al. Differential loss of synaptic proteins in Alzheimer’s disease: implications for synaptic dysfunction. J Alzheimers Dis. 2005;7(2):103–17 discussion 173-180.
• Kvartsberg H, Duits FH, Ingelsson M, Andreasen N, Ohrfelt A, Andersson K, et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer’s disease. Alzheimers Dement. 2015;11(10):1180–90. One of the first papers to investigate neurogranin in Alzheimer's disease and that high CSF neurogranin levels can predict progression to dementia.
Kvartsberg H, Portelius E, Andreasson U, Brinkmalm G, Hellwig K, Lelental N, et al. Characterization of the postsynaptic protein neurogranin in paired cerebrospinal fluid and plasma samples from Alzheimer’s disease patients and healthy controls. Alzheimers Res Ther. 2015;7(1):40.
Portelius E, Zetterberg H, Skillback T, Tornqvist U, Andreasson U, Trojanowski JQ, et al. Cerebrospinal fluid neurogranin: relation to cognition and neurodegeneration in Alzheimer’s disease. Brain. 2015;138(Pt 11):3373–85.
Tarawneh R, D'Angelo G, Crimmins D, Herries E, Griest T, Fagan AM, et al. Diagnostic and prognostic utility of the synaptic marker Neurogranin in Alzheimer disease. JAMA Neurol. 2016;73(5):561–71.
Portelius E, Olsson B, Hoglund K, Cullen NC, Kvartsberg H, Andreasson U, et al. Cerebrospinal fluid neurogranin concentration in neurodegeneration: relation to clinical phenotypes and neuropathology. Acta Neuropathol. 2018;136:363–76.
• Wellington H, Paterson RW, Portelius E, Tornqvist U, Magdalinou N, Fox NC, et al. Increased CSF neurogranin concentration is specific to Alzheimer disease. Neurology. 2016;86(9):829–35. This paper reports that CSF concentrations are increased in Alzheimer's disease, but not in other neurodegenerative disorders or in healthy controls.
Novakova L, Axelsson M, Khademi M, Zetterberg H, Blennow K, Malmestrom C, et al. Cerebrospinal fluid biomarkers as a measure of disease activity and treatment efficacy in relapsing-remitting multiple sclerosis. J Neurochem. 2017;141(2):296–304.
Chiodi F, Keys B, Albert J, Hagberg L, Lundeberg J, Uhlen M, et al. Human immunodeficiency virus type 1 is present in the cerebrospinal fluid of a majority of infected individuals. J Clin Microbiol. 1992;30(7):1768–71.
Valcour V, Chalermchai T, Sailasuta N, Marovich M, Lerdlum S, Suttichom D, et al. Central nervous system viral invasion and inflammation during acute HIV infection. J Infect Dis. 2012;206(2):275–82.
Navia BA, Jordan BD, Price RW. The AIDS dementia complex: I. Clinical features. Ann Neurol. 1986;19(6):517–24.
Navia BA, Cho ES, Petito CK, Price RW. The AIDS dementia complex: II. Neuropathology. Ann Neurol. 1986;19(6):525–35.
Ellis R, Langford D, Masliah E. HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev Neurosci. 2007;8(1):33–44.
Everall IP, Heaton RK, Marcotte TD, Ellis RJ, McCutchan JA, Atkinson JH, et al. Cortical synaptic density is reduced in mild to moderate human immunodeficiency virus neurocognitive disorder. HNRC Group. HIV Neurobehavioral Research Center. Brain Pathol. 1999;9(2):209–17.
Festa L, Gutoskey CJ, Graziano A, Waterhouse BD, Meucci O. Induction of interleukin-1beta by human immunodeficiency virus-1 viral proteins leads to increased levels of neuronal ferritin heavy chain, synaptic injury, and deficits in flexible attention. J Neurosci. 2015;35(29):10550–61.
Hagberg L, Cinque P, Gisslen M, Brew BJ, Spudich S, Bestetti A, et al. Cerebrospinal fluid neopterin: an informative biomarker of central nervous system immune activation in HIV-1 infection. AIDS Res Ther. 2010;7:15.
Eden A, Marcotte TD, Heaton RK, Nilsson S, Zetterberg H, Fuchs D, et al. Increased Intrathecal immune activation in virally suppressed HIV-1 infected patients with neurocognitive impairment. PLoS One. 2016;11(6):e0157160.
Jessen Krut J, Mellberg T, Price RW, Hagberg L, Fuchs D, Rosengren L, et al. Biomarker evidence of axonal injury in neuroasymptomatic HIV-1 patients. PLoS One. 2014;9(2):e88591.
• Yilmaz A, Blennow K, Hagberg L, Nilsson S, Price RW, Schouten J, et al. Neurofilament light chain protein as a marker of neuronal injury: review of its use in HIV-1 infection and reference values for HIV-negative controls. Expert Rev Mol Diagn. 2017;17(8):761–70. This review describes CSF NFL concentrations across spectrum of HIV-infection and it also provides more precise estimates of age-related upper limits of CSF NFL concentration.
Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE. Accelerated Tau deposition in the brains of individuals infected with human immunodeficiency virus-1 before and after the advent of highly active anti-retroviral therapy. Acta Neuropathol. 2006;111(6):529–38.
Green DA, Masliah E, Vinters HV, Beizai P, Moore DJ, Achim CL. Brain deposition of beta-amyloid is a common pathologic feature in HIV positive patients. AIDS. 2005;19(4):407–11.
Gelman BB, Schuenke K. Brain aging in acquired immunodeficiency syndrome: increased ubiquitin-protein conjugate is correlated with decreased synaptic protein but not amyloid plaque accumulation. J Neuro-Oncol. 2004;10(2):98–108.
Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368(9533):387–403.
Gisslen M, Krut J, Andreasson U, Blennow K, Cinque P, Brew BJ, et al. Amyloid and tau cerebrospinal fluid biomarkers in HIV infection. BMC Neurol. 2009;9:63.
Krut JJ, Price RW, Zetterberg H, Fuchs D, Hagberg L, Yilmaz A, et al. No support for premature central nervous system aging in HIV-1 when measured by cerebrospinal fluid phosphorylated tau (p-tau). Virulence. 2017;8(5):599–604.
Clifford DB, Fagan AM, Holtzman DM, Morris JC, Teshome M, Shah AR, et al. CSF biomarkers of Alzheimer disease in HIV-associated neurologic disease. Neurology. 2009;73(23):1982–7.
Guha D, Wagner MCE, Ayyavoo V. Human immunodeficiency virus type 1 (HIV-1)-mediated neuroinflammation dysregulates neurogranin and induces synaptodendritic injury. J Neuroinflammation. 2018;15(1):126.
Makitalo S, Mellgren A, Borgh E, Kilander L, Skillback T, Zetterberg H, et al. The cerebrospinal fluid biomarker profile in an HIV-infected subject with Alzheimer’s disease. AIDS Res Ther. 2015;12:23.
Fuchs D, Hausen A, Reibnegger G, Werner ER, Dierich MP, Wachter H. Neopterin as a marker in HIV infection. Clin Chem. 1988;34(2):466–7.
Cano OD, Neurauter G, Fuchs D, Shearer GM, Boasso A. Differential effect of type I and type II interferons on neopterin production and amino acid metabolism in human astrocyte-derived cells. Neurosci Lett. 2008;438(1):22–5.
Griffin DE, McArthur JC, Cornblath DR. Neopterin and interferon-gamma in serum and cerebrospinal fluid of patients with HIV-associated neurologic disease. Neurology. 1991;41(1):69–74.
Brew BJ, Pemberton L, Cunningham P, Law MG. Levels of human immunodeficiency virus type 1 RNA in cerebrospinal fluid correlate with AIDS dementia stage. J Infect Dis. 1997;175(4):963–6.
Funding
The study was supported by grants from the Swedish and European Research Councils, Swedish State Support for Clinical Research (ALFGBG), the Knut and Alice Wallenberg Foundation, the Swedish Brain Foundation, the Torsten Söderberg Foundation, and NIH grants: R01 NS094067 (RWP), R01MH081772 (SS), P01 DA026134 (RWP, project PI).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Aylin Yilmaz, Dietmar Fuchs, Serena Spudich, and Richard W. Price declare that they have no conflict of interest. Henrik Zetterberg has served at scientific advisory boards for Eli Lilly, Roche Diagnostics and Wave, has received travel support from Teva, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB, a GU Ventures-based platform company at the University of Gothenburg. Kaj Blennow has served as a consultant or at advisory boards for Alzheon, BioArctic, Biogen, Eli Lilly, IBL International, Merck, Novartis, Pfizer, and Roche Diagnostics, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB, a GU Venture-based platform company at the University of Gothenburg.
Human and Animal Rights and Informed Consent
The study was approved by the regional ethics committees and informed consent was obtained from the participants. No animal experiments were performed.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Central Nervous System and Cognition
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Yilmaz, A., Fuchs, D., Price, R.W. et al. Cerebrospinal Fluid Concentrations of the Synaptic Marker Neurogranin in Neuro-HIV and Other Neurological Disorders. Curr HIV/AIDS Rep 16, 76–81 (2019). https://doi.org/10.1007/s11904-019-00420-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11904-019-00420-1