
Abstract
Purpose of Review
Primary sclerosing cholangitis (PSC) is a chronic cholestatic disorder characterized by inflammation of intrahepatic and/or extrahepatic bile ducts leading to stricturing, biliary fibrosis, cirrhosis, and liver failure. PSC is highly associated with inflammatory bowel diseases (IBD) and bears significant risk for cholangiocellular and colorectal cancer. To date, no medical treatment has been proven in randomized controlled trials to improve transplant-free and overall patient survival. However, numerous innovative therapeutic concepts are currently tested in phase 2 to phase 3 clinical trials. Based on currently suggested pathogenetic mechanisms of PSC, such drugs target its immunopathogenesis and nuclear and membrane receptors regulating bile acid transport and metabolism, gut microbiota, and liver fibrosis. The purpose of this review is to discuss recent advances in targeted medical treatment options for PSC.
Recent Findings
While a large carefully designed phase 2b trial targeting fibrosis development in PSC failed (simtuzumab), another compound was promoted from phase 2a to phase 3 trial based on significant improvements of alkaline phosphatase (AP) and excellent safety profile (norursodeoxycholic acid, norUDCA).
Summary
Ongoing trials evaluate numerous different targets considered to be involved in PSC pathogenesis, with so far, no clear advantage of either compound. This must be attributed to the still unknown cause of PSC. It may turn out that only combination therapy may reach a breakthrough. The fact that numerous studies isochronally test the same drug or therapeutic concept like in the case of vancomycin or faecal microbiota transplantation (FMT) demands improved and coordinated international strategies for study design to develop more effective drug pipelines, which is one major target of the International PSC Study Group (IPSCSG).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Primary sclerosing cholangitis (PSC) is a rare chronic cholestatic liver disease characterized by inflammation and subsequent multifocal strictures of medium- to large-sized bile ducts which may finally lead to biliary cirrhosis and liver failure [1]. Besides cirrhosis, PSC patients are at high risk for complications such as dominant bile duct strictures, bacterial cholangitis, and cholangiocellular as well as colorectal cancer [2, 3•, 4]. In contrast to primary biliary cholangitis (PBC), the efficacy of ursodeoxycholic acid (UDCA) in PSC is controversially discussed. Although UDCA was shown to improve histology and/or serum biochemistry in PSC, in particular alkaline phosphatase (AP), which, however, so far only is established as outcome-relevant prognostic parameter in PBC [5], there is no convincing evidence for positive effects on the need for liver transplantation or death. Since high-dose UDCA treatment has even been associated with increased mortality, the American Association for the Study of Liver Diseases (AASLD) currently recommends against the use of UDCA in PSC while the American College of Gastroenterology (ACG) and the European Association for the Study of the Liver (EASL) follow a more liberal strategy by discouraging UDCA doses beyond 28 mg/kg/day [6,7,8], since specific subpopulations might definitely profit from UDCA treatment [9,10,11]. Such a differentiated strategy is in some way supported by retrospective studies demonstrating that normalization of AP is associated with a survival benefit, either when using UDCA at intermediate doses of 17–23 mg/kg/day, but also without any medication [9,10,11]. Nevertheless, liver transplantation is currently the only definitive treatment for progressive and complicated PSC or end-stage liver disease. Therefore, we need novel medical treatment strategies [3•]. However, precision treatment with one single drug for PSC is unlikely and the search for effective treatment is hampered by numerous facts: (i) PSC as an umbrella term for an apparently mixed bag probably contains various disease phenotypes, with substantial differences in presentation, clinical course, associated complications, and type of progression; (ii) clinical diagnosis of PSC is imprecisely and therefore unsatisfying, since the gold standard is imaging by ERCP or MRCP that still only allows late diagnosis in many cases and has low specificity; (iii) most importantly, the cause of PSC still is unknown.
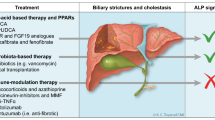
Current pathogenetic models and concepts for PSC include (i) dysregulation of the immune signalling, (ii) increased gut permeability with subsequent delivery of pathogen-associated molecular patterns (PAMPs, including bacterial polysaccharides and endotoxins) to the liver, potentially associated with (iii) the dysbiosis of the gut microbiome, and (iv) the toxic bile hypothesis postulating direct luminal damage of the bile duct wall. Such models were recently concisely reviewed elsewhere [1, 12] and should be considered as a kind of crutch, since there is no unifying and convincing concept to date. Interestingly, there is no hard evidence for a robust genetic backbone in PSC pathogenesis with the exception of some human leukocyte antigens [13]. Nevertheless, recent discoveries in regard to the regulation of bile acid synthesis, transport, and metabolism; the identification of novel target molecules potentially engaged in PSC pathogenesis (e.g. VAP-1 and LOXL2); and the credible role of gut-derived microbes stimulated the development of innovative treatment strategies. Consequently, numerous drugs have been investigated or are currently tested in clinical phase 2 to 3 trials and are therefore focus of this review. The following subdivision of diverse therapeutic principles is arbitrary in its nature, since some drugs may have substantially overlapping modes of action (e.g. farnesoid X receptor (FXR) ligands typically modulating bile acid turnover may also have anti-inflammatory actions) while other treatment strategies may not fit clearly into any currently defined therapeutic concept (e.g. faecal microbiota transplantation, FMT). The dynamic in the field is pleasant and astonishing, but comprises the risk that this article is out of date when just published.
Nuclear and Membrane Receptor Ligand-Based Therapies
Farnesoid X Receptor Agonists
Negative feedback activation of the nuclear bile acid receptor FXR regulates bile acid turnover both within the liver (via SHP-LRH-1/HNF-4α) and from the gut (via FGF19-FGFR4/β-klotho) [14, 15]. Synthetic FXR agonists therefore reduce potentially toxic levels of bile acids in the liver and in the enterohepatic circulation, but may also display anti-inflammatory properties, reduce portal hypertension, and diminish gut permeability [16,17,18,19,20,21].
Bile Acid–Derived FXR Agonists
Obeticholic acid (OCA) is the 6-ethyl derivative of naturally occurring FXR agonist chenodeoxycholic acid (CDCA) with about 100 times higher potency than CDCA itself. In a phase 3 trial in 217 patients with PBC on UDCA or intolerant to it (i.e. the POISE study), OCA was found to reduce serum levels of AP significantly more often than placebo, reaching the composite endpoint of AP levels of less than 1.67 times the upper limit of the normal range, with a reduction of at least 15% from baseline, and a total bilirubin level at or below the upper limit of the normal range in about half of the patients randomized to OCA. Dose-dependent pruritus occurred as one major side effect [22].
Consequently, OCA was tested in a phase 2a, double-blind, placebo-controlled trial in 77 PSC patients for 24 weeks with AP-dependent titrating doses of 1.5–3 mg/day and 5–10 mg/day, respectively, with 1:1:1 randomization versus placebo (the AESOP study; NCT02177136; Table 1). Both doses overall reduced AP levels by 22% reaching statistical significance for the high-dose regiment. In PSC patients with baseline UDCA medication, OCA significantly decreased serum AP levels by about 15% with both doses, whereas in those patients without baseline UDCA, somewhat greater AP reductions were observed, significantly for the higher dose regime at 12 weeks, by 30%. A grave adverse event was as observed in earlier OCA study dose-dependent pruritus, causing discontinuation of the study in 4/25 patients in the high-dose group. A 2-year open-label long-term extension of the study is ongoing [23]. Mechanistically of potential importance for its side effects, OCA is highly preserved within the enterohepatic circulation, which may lead to significant enrichment especially in patients with advanced liver disease. Moreover, both intestinal FXR activation and hepatic FXR activation downregulate bile acid synthesis from cholesterol. Consequently, any FXR activator is supposed to increase low-density lipoprotein (LDL) cholesterol via downregulation of LDL receptor (LDLR), but also to decrease high density-lipoprotein (HDL) cholesterol by inducing scavenger receptor group B type 1 (SR-BI) [24]. The phase 2b FLINT trial with OCA in patients with nonalcoholic steatohepatitis (NASH) that demonstrated significant improvements in histological features of NASH confirmed the anticipated changes in serum cholesterol profiles [25] that also were seen in the POISE study [22]. Decreased bile acid synthesis via FXR agonists in principle also may result in biliary cholesterol supersaturation and increased gallstone susceptibility [26]. However, so far, no clinical data on increased gallstone incidence during OCA treatment have been reported.
Non–bile acid–Derived FXR Agonists and FGF19 Analogues
GS-9674
Non-steroidal FXR agonists have been synthesized in the hope to preserve the attractive therapeutic potential of targeting FXR such as decreased hepatic bile acid levels and potential anti-inflammatory effects, and in parallel to avoid potential adverse effects of bile acid derivatives such as pruritus or drug toxicity. In addition, synthetic non-steroidal FXR agonists are also designed to show higher degree of receptor specificity, less enterohepatic circulation, and improved bioavailability, which might result in favourable extrahepatic effects as well [27].
To the best of our knowledge, there is currently no publication on the effects of GS-9674 (previously called Px-102) in animal models for cholestatic liver diseases available. Rather, in a mouse model of diet-induced obesity, GS-9674 was shown to reduce hepatic steatosis and fibrosis, as well as serum levels of cholesterol, ALT, and AST [28]. Furthermore, dose-dependent antifibrotic effects and lowered portal pressure were observed in portal hypertensive rats [29]. A phase 2 trial recently evaluated the safety, tolerability, and efficacy of 30 mg and 100 mg GS-9674 in 52 PSC patients (NCT02943460). At week 12, cilofexor 100 mg led to significant reductions in serum AP and other liver biochemistry. Grade 2 or 3 pruritus were observed in 14% with 100 mg, 20% with 30 mg, and 40% with placebo (Trauner M et al. Hepatology 2019 https://doi.org/10.1002/hep.30509) (NCT02943460; Table 1).
NGM282
FGF-19 is FXR activation dependently produced in the liver, gallbladder, and the distal small intestine and may despite its short half-life potentially stimulate cell proliferation in the liver and gut [30,31,32,33,34]. Synthetic FGF-19 derivatives may provide a prolonged inhibition of bile acid synthesis via the FGFR4/β-klotho signalling axis, which might be beneficial in cholestatic liver disease, but may also have less cell proliferative activity. NGM282, a synthetic analogue of FGF-19 lacking potential tumorigenic effects, showed dose-dependent reductions in serum AP levels in PBC patients with insufficient response to UDCA [35]. Such a concept was successfully tested in animal models for sclerosing cholangitis [36] and was also evaluated in a randomized controlled trial including 62 patients with PSC (NCT02704364; Table 1). However, according to a press release from NGM, the study did not meet the primary endpoint of a statistically significant change in serum AP levels although significant improvements in biomarkers of hepatic injury and fibrosis and significant reductions in serum bile acids and 7α-hydroxy-cholesten-4-one (C4, marker of bile acid synthesis) were observed (https://www.ngmbio.com/ngm-reports-top-line-results-from-phase-2-study-of-ngm282-in-patients-with-primary-sclerosing-cholangitis-psc/) [3•].
PXR/GR Agonists: Budesonide
For budesonide, a prednisolone analogue and glucocorticoid receptor (GR) agonist with > 90% first-pass hepatic inactivation with additional affinity to the nuclear xenobiotic pregnane X receptor (PXR), no beneficial effects were found in PSC patients so far [37, 38]. However, there may still be a place for alternative PXR ligands, since such a drug would also be anticipated to positively affect autophagy, which may be oppressed in various cholangiopathies including PSC [39, 40]. Candidate drugs should be tested systematically in preclinical models for proof of such an interesting concept.
PPAR Agonists
Anticholestatic properties of peroxisome proliferator-activated receptor (PPAR) agonists are numerous: upregulation of the multidrug resistance 3 receptor (MDR3) thereby enhancing biliary phospholipid secretion and mixed micelle formation; inhibition of bile acid synthesis with upregulation of bile acid detoxification routes; anti-inflammatory effects by suppression of NF-κB transcription; and simultaneous cross-reaction with additional nuclear receptors such as FXR and PXR [3•, 41]. PPAR agonists are now established as useful “second-line therapy” for PBC patients with insufficient UDCA response as supported by the findings of the recently published BEZURSO trial as the largest RCT supporting a beneficial role for bezafibrate, a pan-PPAR agonist [5, 42]. In contrast so far, there is only a limited number of studies in PSC patients [43,44,45] and most importantly no RCT. One promising prospective study with 12-week bezafibrate treatment in 11 PSC patients showed significant improvement of AP and ALT levels and an increase of these parameters after cessation of the study medication [44]. For fenofibrate, a PPAR-α agonist, evidence in PSC is limited to two small open-label studies published so far only in abstract format [46, 47]. For seladelpar, an orally administered potent and selective PPAR-δ agonist that has recently been shown to improve cholestasis in PBC [48], and elafibranor (GFT505), a dual PPAR-α and PPAR-γ agonist that is currently also tested in PBC, no data have been retrieved in PSC so far. These compounds have multiple beneficial anticholestatic effects but it remains to be determined whether these are effective in the treatment of PSC.
The BEZURSO trial convincingly demonstrated that large investigator-initiated multicentre studies with well-defined outcome parameters are feasible.
TGR5 Agonists: INT-767 and INT-777
Secondary bile acids effectively activate the trans-membrane G-protein coupled receptor 5 (TGR5), a cell surface receptor expressed in sinusoidal endothelial cells, Kupffer cells, and cholangiocytes, which represents a potential treatment target for cholestatic liver diseases such as PBC and PSC. Bile acid–derived INT-767 (a dual FXR/TGR5 agonist) and INT-777 (a specific TGR5 agonist) showed promising results with ameliorated liver injury in the Mdr2−/− mouse cholangiopathy model [49]. As a caveat, cholangiocarcinomas were shown to have increased TGR5 expression [50]. It remains uncertain whether pharmacological TGR5 activation may consequently stimulate cholangiocarcinoma formation or progression [50]. The future of TGR5 targeting drugs therefore might be vague, and currently, there are no clinical trials testing TGR5 agonists in PSC patients.
All-trans Retinoic Acid
All-trans retinoic acid (ATRA) activates the nuclear receptor complex FXR/retinoid X receptor (RXR) with consequent repression of CYP71A and bile acid synthesis [51]. Based on promising findings in animal models of cholestasis (Mdr2−/− mice, bile duct–ligated rats) [52, 53], ATRA was evaluated as a potential treatment for PSC. A small pilot study in 15 PSC patients that were administered moderate-dose UDCA (15–23 mg/kg/day) in combination with ATRA (45 mg/m2/day) for 12 weeks did not meet the primary endpoint of a 30% reduction in serum AP, although a significant decrease in serum ALT and C4 levels was observed [54]. Lower dose ATRA (10 mg twice daily) is currently investigated in a phase 2 study (NCT03359174; Table 1).
norUrsodeoxycholic Acid
There is no evidence so far that norursodeoxycholic acid (norUDCA) signals via a known receptor [55]. Results of a recently published phase 2a study in PSC showed that norUDCA significantly reduced serum AP levels in a dose-dependent manner [56••]. A total number of 161 patients were included in this multicentre RCT to either 500, 1000, or 1500 mg of norUDCA per day or placebo. In a 12-week treatment phase, norUDCA reduced AP levels by − 12.3% (p = 0.029) in the 500 mg per day, − 17.3% (p = 0.003) in the 1000 mg per day, and − 26.0% (p < 0.0001) in the 1500 mg per day groups when compared with placebo. Moreover, norUDCA significantly reduced serum transaminases and gamma glutamyl transferase (GGT) in a dose-dependent fashion. The safety profile was excellent. The most frequently observed adverse events in the norUDCA treatment arms were abdominal pain, fatigue, nasopharyngitis, headache, and pruritus but importantly not different to placebo [56••]. Consequently, a phase 3 trial comparing the effects of norUDCA with placebo is currently recruiting PSC patients in Europe (NUC5/PSC, EudraCT Number: 2016-003367-19; Table 1). norUDCA was also successfully tested in a phase 2a trial in NALFD patients [57]. The therapeutic effects of norUDCA may be pleotropic and also not restricted to the liver, with induction of bicarbonate-enriched choleresis, but also anti-inflammatory and antifibrotic properties [58,59,60,61,62].
Cytokine/Chemokine Mediator Targeting Therapies
The failed immunosuppressive treatment strategies tested in PSC are numerous and were summarized carefully in detail elsewhere [63]. Treatment failure of immunosuppression in PSC may have plenty of reasons ranging from patient selection and study design to the mechanisms of the drugs tested so far. Several strategies to selectively act on recently identified immunotargets in PSC that now are about to be tested are based on more or less solid evidence. These treatment strategies include the anti-vascular adhesion protein-1 (VAP1) antibody timolumab (NCT02239211), the chemokine receptor 2 and chemokine receptor 5 (CCR2/CCR5) antagonist cenicriviroc (PERSEUS; NCT02653625), and the α4β7 integrin blocker vedolizumab (NCT03035058).
The most solid theoretical backbone is currently given for timolumab, a fully human, monoclonal, anti-VAP-1 antibody, since VAP-1 expression was shown to be significantly induced in PSC patients and there is also positive preclinical evidence for anti-VAP-1 treatment. VAP-1, a trans-membrane sialoglycoprotein expressed on human hepatic endothelium, drives inflammation in liver disease by supporting lymphocyte adhesion and transendothelial migration. Thereby, VAP-1 also promotes the formation of fibrosis [64, 65]. Treatment with an anti-VAP-1 antibody was shown to prevent fibrosis in murine models of liver injury [66]. Timolumab (BTT1023) is currently tested within the BUTEO trial, a single-arm, two-stage, open-label, multicentre, phase 2 clinical trial in adults with PSC (NCT02239211; Table 1).
The CCR2/CCR5 antagonist cenicriviroc has been shown to have anti-inflammatory and antifibrotic effects on animal models of fibrosis and nonalcoholic steatohepatitis (NASH). A phase 2 trial in PSC evaluating the effect of a 24-week treatment with cenicriviroc 150 mg/day in patients with PSC (NCT02653625) was completed in December 2017, but results are yet to be published (Table 1).
Vedolizumab, an anti-α4β7 integrin monoclonal antibody, is used for IBD treatment. The ligand for α4β7 integrin, MADCAM-1, which is usually expressed in the gut, was also found in the liver sinusoidal endothelium in PSC. Binding of α4β7 integrin to MADCAM-1 causes migration of gut-primed, mucosally activated T cells to the liver [67,68,69]. Vedolizumab, by blocking α4β7 integrin, might therefore play a role in reducing lymphocyte recruitment to the liver and thereby ameliorating hepatic and biliary inflammation in PSC. To date, only small studies evaluated the effect of anti-integrin therapy with vedolizumab in patients with IBD and concomitant PSC. Despite showing efficacy in ameliorating IBD, the impact of vedolizumab on liver biochemistry was rather modest and disappointing [70,71,72]. One retrospective multicentre study of 34 PSC patients receiving vedolizumab for concomitant IBD demonstrated no overall change in serum AP, other liver biochemistry, or the Mayo PSC risk score after 30 weeks [72]. Results from a study of pooled data from patients with PSC-IBD treated with vedolizumab from several European and North-American centres collected by the IPSCSG are about to be published in the near future. Of note, a phase 3 trial with vedolizumab for patients with PSC-IBD (NCT03035058) was withdrawn in early 2018.
Antifibrotic Treatment Strategies
Simtuzumab
Lysyl oxidase like-2 (LOXL2) plays an important role in fibrosis by promoting stabilization of the extracellular matrix, as well as in chemotaxis, cell growth, and cell mobility and may also be engaged in the regulation of bile duct permeability [73,74,75,76]. Induction of LOXL2 activity was demonstrated in fibrotic liver diseases including PSC [76, 77]. Targeting LOXL2 using the monoclonal antibody simtuzumab therefore seemed to represent an attractive treatment option in PSC due to its potential antifibrotic effects, the therapeutic property to seal leaky bile ducts, and, by inhibiting epithelial-mesenchymal transition (EMT), also the potential to prevent cholangiocarcinoma.
Two different doses of simtuzumab administered over 96 weeks were investigated in a placebo-controlled phase 2b study including 234 PSC patients with half of them having bridging fibrosis or cirrhosis at baseline [78••] (Table 1). Simtuzumab was well tolerated; however, this study failed to show any clinical benefit. As such, there were no significant differences in hepatic collagen content, Ishak fibrosis stage, or decrease in AP compared with placebo [78••]. There was not even an effect on liver fibrosis as determined by various tests including measurement of hepatic collagen content [78••]. However, this study provided pivotal information on the clinical course of PSC and useful hints for the design of future PSC treatment trials, and thus should be discussed open-mindedly, for the best of our PSC patients [79]. Of note, despite promising preclinical results with antimouse/rat LOXL2 antibodies in various animal models [80,81,82,83], the anti-LOXL2 strategy with simtuzumab also failed in liver fibrosis due to chronic hepatitis C or NASH, pancreatic cancer, and lung fibrosis [80].
Antibiotics
Due to the strong association between PSC and IBD and the growing evidence for the role of the gut microbiome in PSC development [84], treatment strategies targeting the gut microbiome such as FMT (NCT02424175) or gut-selective antibiotic eradication with vancomycin (NCT01802073, NCT01085760), rifaximin (NCT01695174), or minocycline (NCT00630942), with potential immunomodulatory effects, have been tested [3•] (Table 1).
The glycopeptide antibiotic vancomycin is poorly absorbed when given orally and probably the most promising candidate for PSC treatment [3•] since it was also shown to act as an immune modulator by reducing T cell–mediated cytokine release [85, 86]. Small controlled trials, case series, and case reports in both, adults, and children reported a significant drop in AP [87]. However, the total number of patients treated as well as the treatment periods in the single studies is too small to currently recommend oral vancomycin as a long-term PSC treatment [85]. For that purpose, larger, long-term double-blind controlled trials of vancomycin versus placebo are needed. We identified six different registered clinical trials enrolling PSC patients for vancomycin treatment which urgently calls for better international study coordination.
While a 12-week open-label pilot study evaluating oral rifaximin (550 mg twice daily) in 16 PSC patients showed no significant improvements in serum AP, bilirubin, GGT, or Mayo risk score [88], an open-label study of minocycline (100 mg orally twice daily) in 16 PSC patients over 1 year demonstrated significant improvements in serum AP and Mayo PSC risk score [89]. Besides antimicrobial properties, therapeutic effects of minocycline may be due to its anti-inflammatory and anti-apoptotic activities such as inhibition of inducible nitric oxide synthase, upregulation of interleukin-10, direct suppressive effect on B and T cell function, and inhibition of cell death pathways by reducing proapoptotic and proinflammatory enzyme activation [89, 90].
FMT
FMT has been shown to be effective for recurrent Clostridium difficile infections with colitis, may be of benefit in ulcerative colitis, and was tested in various gastrointestinal disorders including irritable bowel syndrome [91]. The literature on the effects of FMT in general is very hard to interpret, as current protocols differ substantially in regard to route and frequency of application, preparation of the transplanted stool, use or abstinence of upstream antibiotic treatment, randomization, and, probably most importantly, well-defined control groups. Despite numerous uncertainties and potential risks, diagnostic and therapeutic standards for the performance of FMT only recently have been published [92].
An interim analysis of an ongoing open-label trial investigating safety and efficacy of FMT in PSC patients revealed that half of the patients had a decline in AP of ≥ 50% from baseline, without reported adverse events (NCT02424175; Table 1) [3•], which sounds spectacular. However, currently, no single full paper is published on the effects of FMT in PSC.
We have to avoid premature conclusions on this novel approach, in particular, since FMT in its broadest bears the risk of self-application of “stool preparations” that are touted by different distributors. FMT may prematurely reach “clinics,” since this is already discussed intensively in different patient forums.
Other Concepts Currently Tested
The apical sodium–dependent bile acid transporter (ASBT) is pivotal for reabsorption of conjugated bile acids in the ileum [93]. Pharmacological inhibition of ileal ASBT with non-absorbable ASBT inhibitors, so-called IBAT (ileal bile acid transporter) inhibitors, leads to a lower bile acid pool, which is associated with improved liver histology in animal models of cholestatic liver disease and NASH [93]. In cholestatic liver diseases with pruritus, the interruption of the enterohepatic circulation of bile acids with IBAT inhibitors showed promising effects on cholestatic pruritus. However, at least in adults, moderate to severe abdominal side effects substantially limit their application [93]. The ASBT inhibitor maralixibat (CAMEO; NCT02061540) has already been tested in 27 patients with PSC. However, according to the preliminary results given on clinicaltrials.gov, no clinically relevant change in liver biochemistries was observed (Table 1) [3•].
Other drugs that have been or are currently tested in PSC according to clinicaltrials.gov include hymecromone (NCT02780752), mitamycin C (NCT01688024), small molecules (HTD1801, NCT03333928; DUR-928, NCT03394781), curcumin (NCT02978339), and sulfasalazine (NCT035615849). Also, intra-arterial injection of umbilical cord mesenchymal stem cells (UCMSC) is listed as an active but not recruiting trial in PSC (Table 1).
Conclusions
Future medical treatment of PSC might include a combination therapy but we currently lack a convincing concept for that. Theoretically, a potential treatment regimen in PSC may include synergistically acting anticholestatic norUDCA and fibrates. Targeting at an inflammatory gut-liver axis in PSC may contain novel therapeutic antibodies, antibiotics, sulfasalazine, and FMT. Until we will know the cause for PSC, which still remains the main conundrum for successful treatment development, we should discuss and design combination treatment trials in the IPSCSG with approaches tested successfully in phase 2 to 3 clinical trials to avoid performing underpowered or poorly designed studies. We feel that earlier diagnosis of PSC will also be a key for future successful treatment. Therefore, patients with enigmatic AP elevations and abdominal pain or discomfort should liberally be endoscoped to carefully search for often mild to moderate and only right-sided PSC-IBD. Efforts for new diagnostic tests aiming at earlier PSC detection should be increased, since MRCP and ERCP still do only detect the footprints of the disease and are therefore an imperfect gold standard.
The dynamics of the development of novel therapeutic strategies for PSC underscores the importance of the IPSCSG, an organization that was founded in Oslo in 2010. The aim of the IPSCSG is to coordinate basic and clinical research projects and to optimize study design for the best benefit and medical progress of PSC patients.
Abbreviations
- AASLD:
-
American Association for the Study of Liver Diseases
- ACG:
-
American College of Gastroenterology
- AP:
-
Alkaline phosphatase
- ATRA:
-
All-trans retinoic acid
- C4:
-
7α-Hydroxy-cholestene-4-one
- EASL:
-
European Association for the Study of Liver
- FGF-19:
-
Fibroblast growth factor 19
- FGFR:
-
FGF receptor
- FMT:
-
Faecal microbiota transplantation
- FXR:
-
Farnesoid X nuclear receptor
- HNF:
-
Hepatic nuclear factor
- LRH:
-
Liver receptor homologue
- LOXL-2:
-
Lysyl oxidase like-2
- GR:
-
Glucocorticoid receptor
- MADCAM:
-
Mucosal addressin cellular adhesion molecule-1
- MDR-3:
-
Multidrug resistance protein 3
- NF-κB:
-
Nuclear factor kappa B
- norUDCA:
-
24-norUrsodeoxycholic Acid
- OCA:
-
Obeticholic acid
- PBC:
-
Primary biliary cholangitis
- PPAR:
-
Peroxisome proliferator–activated receptor
- PSC:
-
Primary sclerosing cholangitis
- PXR:
-
Pregnane X receptor
- RCT:
-
Randomized controlled trial
- SHP:
-
Small heterodimer partner
- UDCA:
-
Ursodeoxycholic acid
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Karlsen TH, Folseraas T, Thorburn D, Vesterhus M. Primary sclerosing cholangitis - a comprehensive review. J Hepatol. 2017;67(6):1298–323. https://doi.org/10.1016/j.jhep.2017.07.022.
Boonstra K, Weersma RK, van Erpecum KJ, Rauws EA, Spanier BW, Poen AC, et al. Population-based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis. Hepatology. 2013;58(6):2045–55. https://doi.org/10.1002/hep.26565.
• Goldstein J, Levy C. Novel and emerging therapies for cholestatic liver diseases. Liver Int. 2018;38(9):1520–35. https://doi.org/10.1111/liv.13880 Comprehensive review on novel treatment options for PBC and PSC.
Lazaridis KN, LaRusso NF. Primary sclerosing cholangitis. N Engl J Med. 2016;375(12):1161–70. https://doi.org/10.1056/NEJMra1506330.
European Association for the Study of the Liver. EASL Clinical Practice Guidelines: the diagnosis and management of patients with primary biliary cholangitis. J Hepatol. 2017;67(1):145–72. https://doi.org/10.1016/j.jhep.2017.03.022.
EASL. Clinical Practice Guidelines: management of cholestatic liver diseases. J Hepatol. 2009;51(2):237–67. https://doi.org/10.1016/j.jhep.2009.04.009.
Chapman R, Fevery J, Kalloo A, Nagorney DM, Boberg KM, Shneider B, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology. 2010;51(2):660–78. https://doi.org/10.1002/hep.23294.
Lindor KD, Kowdley KV, Harrison ME, American College of Gastroenterology. ACG Clinical Guideline: primary sclerosing cholangitis. Am J Gastroenterol. 2015;110(5):646–59; quiz 60. https://doi.org/10.1038/ajg.2015.112.
Lindström L, Hultcrantz R, Boberg KM, Friis-Liby I, Bergquist A. Association between reduced levels of alkaline phosphatase and survival times of patients with primary sclerosing cholangitis. Clin Gastroenterol Hepatol. 2013;11(7):841–6. https://doi.org/10.1016/j.cgh.2012.12.032.
Al Mamari S, Djordjevic J, Halliday JS, Chapman RW. Improvement of serum alkaline phosphatase to <1.5 upper limit of normal predicts better outcome and reduced risk of cholangiocarcinoma in primary sclerosing cholangitis. J Hepatol. 2013;58(2):329–34. https://doi.org/10.1016/j.jhep.2012.10.013.
Stanich PP, Bjornsson E, Gossard AA, Enders F, Jorgensen R, Lindor KD. Alkaline phosphatase normalization is associated with better prognosis in primary sclerosing cholangitis. Dig Liver Dis. 2011;43(4):309–13. https://doi.org/10.1016/j.dld.2010.12.008.
Hov JR, Karlsen TH. The microbiome in primary sclerosing cholangitis: current evidence and potential concepts. Semin Liver Dis. 2017;37(4):314–31. https://doi.org/10.1055/s-0037-1608801.
Jiang X, Karlsen TH. Genetics of primary sclerosing cholangitis and pathophysiological implications. Nat Rev Gastroenterol Hepatol. 2017;14(5):279–95. https://doi.org/10.1038/nrgastro.2016.154.
Al-Khaifi A, Rudling M, Angelin B. An FXR agonist reduces bile acid synthesis independently of increases in FGF19 in healthy volunteers. Gastroenterology. 2018;155(4):1012–6. https://doi.org/10.1053/j.gastro.2018.06.038.
Zhang JH, Nolan JD, Kennie SL, Johnston IM, Dew T, Dixon PH, et al. Potent stimulation of fibroblast growth factor 19 expression in the human ileum by bile acids. Am J Physiol Gastrointest Liver Physiol. 2013;304(10):G940–8. https://doi.org/10.1152/ajpgi.00398.2012.
Verbeke L, Mannaerts I, Schierwagen R, Govaere O, Klein S, Vander Elst I, et al. FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci Rep. 2016;6:33453. https://doi.org/10.1038/srep33453.
Verbeke L, Farre R, Trebicka J, Komuta M, Roskams T, Klein S, et al. Obeticholic acid, a farnesoid X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats. Hepatology. 2014;59(6):2286–98. https://doi.org/10.1002/hep.26939.
Mookerjee RP, Mehta G, Balasubramaniyan V, Mohamed Fel Z, Davies N, Sharma V, et al. Hepatic dimethylarginine-dimethylaminohydrolase1 is reduced in cirrhosis and is a target for therapy in portal hypertension. J Hepatol. 2015;62(2):325–31. https://doi.org/10.1016/j.jhep.2014.08.024.
Verbeke L, Farre R, Verbinnen B, Covens K, Vanuytsel T, Verhaegen J, et al. The FXR agonist obeticholic acid prevents gut barrier dysfunction and bacterial translocation in cholestatic rats. Am J Pathol. 2015;185(2):409–19. https://doi.org/10.1016/j.ajpath.2014.10.009.
Ubeda M, Lario M, Munoz L, Borrero MJ, Rodriguez-Serrano M, Sanchez-Diaz AM, et al. Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J Hepatol. 2016;64(5):1049–57. https://doi.org/10.1016/j.jhep.2015.12.010.
Verbeke L, Nevens F, Laleman W. Steroidal or non-steroidal FXR agonists - is that the question? J Hepatol. 2017;66(4):680–1. https://doi.org/10.1016/j.jhep.2017.01.013.
Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P, et al. A placebo-controlled trial of obeticholic cid in primary biliary cholangitis. N Engl J Med. 2016;375(7):631–43. https://doi.org/10.1056/NEJMoa1509840.
Kowdley KV, Bowlus CL, Levy C, et al. The AESOP trial: a randomized, double-blind, placebo-controlled, phase 2 study of obeticholic acid in patients with primary sclerosing cholangitis. Hepatology. 2017;66(6):1254A–5A. https://doi.org/10.1002/hep.29634.
Chavez-Talavera O, Tailleux A, Lefebvre P, Staels B. Bile acid control of metabolism and inflammation in obesity, type 2 diabetes, dyslipidemia, and nonalcoholic fatty liver disease. Gastroenterology. 2017;152(7):1679–94 e3. https://doi.org/10.1053/j.gastro.2017.01.055.
Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385(9972):956–65. https://doi.org/10.1016/S0140-6736(14)61933-4.
Al-Dury S, Wahlström A, Panzitt K, Thorell A, Ståhlman M, Trauner M, et al. Obeticholic acid increases cholesterol saturation and FGF19 in human gallbladder bile. Hepatology. 2018;68:159A.
Gege C, Kinzel O, Steeneck C, Schulz A, Kremoser C. Knocking on FXR’s door: the “hammerhead”-structure series of FXR agonists - amphiphilic isoxazoles with potent in vitro and in vivo activities. Curr Top Med Chem. 2014;14(19):2143–58. https://doi.org/10.2174/1568026614666141112094430.
Liles J, Karnik S, Hambruch E, et al. FXR agonism by GS-9674 decreases steatosis and fibrosis in a murine model of NASH. J Hepatol. 2016;64:PS066.
Schwabl P, Hambruch E, Supper P, Burnet M, Peck-Radosavljevic M, Reiberger T, et al. The non-steroidal FXR agonist GS-9674 reduces liver fibrosis and ameliorates portal hypertension in a rat NASH model. J Hepatol. 2016;64:PS058.
Zhao H, Lv F, Liang G, Huang X, Wu G, Zhang W, et al. FGF19 promotes epithelial-mesenchymal transition in hepatocellular carcinoma cells by modulating the GSK3beta/beta- catenin signaling cascade via FGFR4 activation. Oncotarget. 2016;7(12):13575–86. https://doi.org/10.18632/oncotarget.6185.
Tan Q, Li F, Wang G, Xia W, Li Z, Niu X, et al. Identification of FGF19 as a prognostic marker and potential driver gene of lung squamous cell carcinomas in Chinese smoking patients. Oncotarget. 2016;7(14):18394–402. https://doi.org/10.18632/oncotarget.7817.
Yoo C, Kang J, Kim D, Kim KP, Ryoo BY, Hong SM, et al. Multiplexed gene expression profiling identifies the FGFR4 pathway as a novel biomarker in intrahepatic cholangiocarcinoma. Oncotarget. 2017;8(24):38592–601. https://doi.org/10.18632/oncotarget.16951.
Li Y, Zhang W, Doughtie A, Cui G, Li X, Pandit H, et al. Up-regulation of fibroblast growth factor 19 and its receptor associates with progression from fatty liver to hepatocellular carcinoma. Oncotarget. 2016;7(32):52329–39. https://doi.org/10.18632/oncotarget.10750.
Cheng K, Metry M, Felton J, Shang AC, Drachenberg CB, Xu S, et al. Diminished gallbladder filling, increased fecal bile acids, and promotion of colon epithelial cell proliferation and neoplasia in fibroblast growth factor 15-deficient mice. Oncotarget. 2018;9(39):25572–85. https://doi.org/10.18632/oncotarget.25385.
Mayo MJ, Wigg AJ, Leggett BA, Arnold H, Thompson AJ, Weltman M, et al. NGM282 for treatment of patients with primary biliary cholangitis: a multicenter, randomized, double-blind, placebo-controlled trial. Hepatol Commun. 2018;2(9):1037–50. https://doi.org/10.1002/hep4.1209.
Zhou M, Learned RM, Rossi SJ, DePaoli AM, Tian H, Ling L. Engineered fibroblast growth factor 19 reduces liver injury and resolves sclerosing cholangitis in Mdr2-deficient mice. Hepatology. 2016;63(3):914–29. https://doi.org/10.1002/hep.28257.
van Hoogstraten HJ, Vleggaar FP, Boland GJ, van Steenbergen W, Griffioen P, Hop WC, et al. Budesonide or prednisone in combination with ursodeoxycholic acid in primary sclerosing cholangitis: a randomized double-blind pilot study. Belgian-Dutch PSC Study Group. Am J Gastroenterol. 2000;95(8):2015–22. https://doi.org/10.1111/j.1572-0241.2000.02267.x.
Angulo P, Batts KP, Jorgensen RA, LaRusso NA, Lindor KD. Oral budesonide in the treatment of primary sclerosing cholangitis. Am J Gastroenterol. 2000;95(9):2333–7. https://doi.org/10.1111/j.1572-0241.2000.02323.x.
Fickert P, Wagner M. Biliary bile acids in hepatobiliary injury - what is the link? J Hepatol. 2017;67(3):619–31. https://doi.org/10.1016/j.jhep.2017.04.026.
Nakanuma Y, Sasaki M, Harada K. Autophagy and senescence in fibrosing cholangiopathies. J Hepatol. 2015;62(4):934–45. https://doi.org/10.1016/j.jhep.2014.11.027.
Honda A, Ikegami T, Nakamuta M, Miyazaki T, Iwamoto J, Hirayama T, et al. Anticholestatic effects of bezafibrate in patients with primary biliary cirrhosis treated with ursodeoxycholic acid. Hepatology. 2013;57(5):1931–41. https://doi.org/10.1002/hep.26018.
Corpechot C, Chazouilleres O, Rousseau A, Le Gruyer A, Habersetzer F, Mathurin P, et al. A placebo-controlled trial of bezafibrate in primary biliary cholangitis. N Engl J Med. 2018;378(23):2171–81. https://doi.org/10.1056/NEJMoa1714519.
Lemoinne S, Pares A, Reig A, Ben Belkacem K, Kemgang Fankem AD, Gaouar F, et al. Primary sclerosing cholangitis response to the combination of fibrates with ursodeoxycholic acid: French-Spanish experience. Clin Res Hepatol Gastroenterol. 2018;42:521–8. https://doi.org/10.1016/j.clinre.2018.06.009.
Mizuno S, Hirano K, Isayama H, Watanabe T, Yamamoto N, Nakai Y, et al. Prospective study of bezafibrate for the treatment of primary sclerosing cholangitis. J Hepatobiliary Pancreat Sci. 2015;22(10):766–70. https://doi.org/10.1002/jhbp.281.
Mizuno S, Hirano K, Tada M, Yamamoto K, Yashima Y, Yagioka H, et al. Bezafibrate for the treatment of primary sclerosing cholangitis. J Gastroenterol. 2010;45(7):758–62. https://doi.org/10.1007/s00535-010-0204-x.
Dejman A, Clark V, Martin P, Levy C. Fenofibrate improves alkaline phosphatase in primary sclerosing cholangitis. Gastroenterology. 2013;144:S1028–S9.
Chazouilleres O, Corpechot C, Gaouar F, Poupon R. Fenofibrate improves liver tests in primary sclerosing cholangitis with incomplete biochemical response to ursodeoxycholic acid. Hepatology. 2010;52:488A.
Jones D, Boudes PF, Swain MG, Bowlus CL, Galambos MR, Bacon BR, et al. Seladelpar (MBX-8025), a selective PPAR-delta agonist, in patients with primary biliary cholangitis with an inadequate response to ursodeoxycholic acid: a double-blind, randomised, placebo-controlled, phase 2, proof-of-concept study. Lancet Gastroenterol Hepatol. 2017;2(10):716–26. https://doi.org/10.1016/S2468-1253(17)30246-7.
Baghdasaryan A, Claudel T, Gumhold J, Silbert D, Adorini L, Roda A, et al. Dual farnesoid X receptor/TGR5 agonist INT-767 reduces liver injury in the Mdr2-/- (Abcb4-/-) mouse cholangiopathy model by promoting biliary HCO(-)(3) output. Hepatology. 2011;54(4):1303–12. https://doi.org/10.1002/hep.24537.
Reich M, Deutschmann K, Sommerfeld A, Klindt C, Kluge S, Kubitz R, et al. TGR5 is essential for bile acid-dependent cholangiocyte proliferation in vivo and in vitro. Gut. 2016;65(3):487–501. https://doi.org/10.1136/gutjnl-2015-309458.
Cai SY, He H, Nguyen T, Mennone A, Boyer JL. Retinoic acid represses CYP7A1 expression in human hepatocytes and HepG2 cells by FXR/RXR-dependent and independent mechanisms. J Lipid Res. 2010;51(8):2265–74. https://doi.org/10.1194/jlr.M005546.
Cai SY, Mennone A, Soroka CJ, Boyer JL. All-trans-retinoic acid improves cholestasis in alpha-naphthylisothiocyanate-treated rats and Mdr2-/- mice. J Pharmacol Exp Ther. 2014;349(1):94–8. https://doi.org/10.1124/jpet.113.209353.
He H, Mennone A, Boyer JL, Cai SY. Combination of retinoic acid and ursodeoxycholic acid attenuates liver injury in bile duct-ligated rats and human hepatic cells. Hepatology. 2011;53(2):548–57. https://doi.org/10.1002/hep.24047.
Assis DN, Abdelghany O, Cai SY, Gossard AA, Eaton JE, Keach JC, et al. Combination therapy of all-trans retinoic acid with ursodeoxycholic acid in patients with primary sclerosing cholangitis: a human pilot study. J Clin Gastroenterol. 2017;51(2):e11–e6. https://doi.org/10.1097/MCG.0000000000000591.
Trauner M, Halilbasic E, Claudel T, Steinacher D, Fuchs C, Moustafa T, et al. Potential of nor-ursodeoxycholic acid in cholestatic and metabolic disorders. Dig Dis. 2015;33(3):433–9. https://doi.org/10.1159/000371904.
•• Fickert P, Hirschfield GM, Denk G, Marschall HU, Altorjay I, Farkkila M, et al. norUrsodeoxycholic acid improves cholestasis in primary sclerosing cholangitis. J Hepatol. 2017;67(3):549–58. https://doi.org/10.1016/j.jhep.2017.05.009 Important large phase II, double-blind, randomized, placebo-controlled study demonstrating a favorable effect for nor UDCA in the treatment of PSC.
Traussnigg S, Schattenberg JM, Muenevver D, Wiegand J, Geier A, Teuber G, et al. norUrsodeoxycholic acid (norUDCA) improves non-alcoholic fatty liver disease (NAFLD): Results from a randomized placebo-controlled, double-blind phase IIa study. Hepatology. 2017;66:106A–7A. https://doi.org/10.1002/hep.29500.
Fickert P, Pollheimer MJ, Silbert D, Moustafa T, Halilbasic E, Krones E, et al. Differential effects of norUDCA and UDCA in obstructive cholestasis in mice. J Hepatol. 2013;58(6):1201–8. https://doi.org/10.1016/j.jhep.2013.01.026.
Halilbasic E, Fiorotto R, Fickert P, Marschall HU, Moustafa T, Spirli C, et al. Side chain structure determines unique physiologic and therapeutic properties of norursodeoxycholic acid in Mdr2-/- mice. Hepatology. 2009;49(6):1972–81. https://doi.org/10.1002/hep.22891.
Fickert P, Wagner M, Marschall HU, Fuchsbichler A, Zollner G, Tsybrovskyy O, et al. 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2006;130(2):465–81. https://doi.org/10.1053/j.gastro.2005.10.018.
Sombetzki M, Fuchs CD, Fickert P, Osterreicher CH, Mueller M, Claudel T, et al. 24-nor-ursodeoxycholic acid ameliorates inflammatory response and liver fibrosis in a murine model of hepatic schistosomiasis. J Hepatol. 2015;62(4):871–8. https://doi.org/10.1016/j.jhep.2014.11.020.
Krones E, Eller K, Pollheimer MJ, Racedo S, Kirsch AH, Frauscher B, et al. NorUrsodeoxycholic acid ameliorates cholemic nephropathy in bile duct ligated mice. J Hepatol. 2017;67(1):110–9. https://doi.org/10.1016/j.jhep.2017.02.019.
Karlsen TH, Vesterhus M, Boberg KM. Review article: controversies in the management of primary biliary cirrhosis and primary sclerosing cholangitis. Aliment Pharmacol Ther. 2014;39(3):282–301. https://doi.org/10.1111/apt.12581.
Arndtz K, Hirschfield GM. Primary sclerosing cholangitis and the management of uncertainty and complexity. Frontline Gastroenterol. 2017;8(4):260–6. https://doi.org/10.1136/flgastro-2017-100815.
Arndtz K, Corrigan M, Rowe A, Kirkham A, Barton D, Fox RP, et al. Investigating the safety and activity of the use of BTT1023 (Timolumab), in the treatment of patients with primary sclerosing cholangitis (BUTEO): a single-arm, two-stage, open-label, multi-centre, phase II clinical trial protocol. BMJ Open. 2017;7(6):e015081. https://doi.org/10.1136/bmjopen-2016-015081.
Weston CJ, Shepherd EL, Claridge LC, Rantakari P, Curbishley SM, Tomlinson JW, et al. Vascular adhesion protein-1 promotes liver inflammation and drives hepatic fibrosis. J Clin Invest. 2015;125(2):501–20. https://doi.org/10.1172/JCI73722.
Grant AJ, Lalor PF, Hubscher SG, Briskin M, Adams DH. MAdCAM-1 expressed in chronic inflammatory liver disease supports mucosal lymphocyte adhesion to hepatic endothelium (MAdCAM-1 in chronic inflammatory liver disease). Hepatology. 2001;33(5):1065–72. https://doi.org/10.1053/jhep.2001.24231.
Eksteen B, Grant AJ, Miles A, Curbishley SM, Lalor PF, Hubscher SG, et al. Hepatic endothelial CCL25 mediates the recruitment of CCR9+ gut-homing lymphocytes to the liver in primary sclerosing cholangitis. J Exp Med. 2004;200(11):1511–7. https://doi.org/10.1084/jem.20041035.
Ala A, Brown D, Khan K, Standish R, Odin JA, Fiel MI, et al. Mucosal addressin cell adhesion molecule (MAdCAM-1) expression is upregulated in the cirrhotic liver and immunolocalises to the peribiliary plexus and lymphoid aggregates. Dig Dis Sci. 2013;58(9):2528–41. https://doi.org/10.1007/s10620-013-2755-1.
Westerveld D, Grajo J, Beattie L, Glover S. Vedolizumab: a novel medical intervention in the treatment of primary sclerosing cholangitis. BMJ Case Rep. 2017;2017. https://doi.org/10.1136/bcr-2017-220351.
Coletta M, Paroni M, Caprioli F. Successful treatment with vedolizumab in a patient with chronic refractory pouchitis and primary sclerosing cholangitis. J Crohns Colitis. 2017;11(12):1507–8. https://doi.org/10.1093/ecco-jcc/jjx090.
Christensen B, Micic D, Gibson PR, Yarur A, Bellaguarda E, Corsello P, et al. Vedolizumab in patients with concurrent primary sclerosing cholangitis and inflammatory bowel disease does not improve liver biochemistry but is safe and effective for the bowel disease. Aliment Pharmacol Ther. 2018;47(6):753–62. https://doi.org/10.1111/apt.14525.
Molnar J, Fong KS, He QP, Hayashi K, Kim Y, Fong SF, et al. Structural and functional diversity of lysyl oxidase and the LOX-like proteins. Biochim Biophys Acta. 2003;1647(1–2):220–4. https://doi.org/10.1016/S1570-9639(03)00053-0.
Kagan HM, Li W. Lysyl oxidase: properties, specificity, and biological roles inside and outside of the cell. J Cell Biochem. 2003;88(4):660–72. https://doi.org/10.1002/jcb.10413.
Wu L, Zhu Y. The function and mechanisms of action of LOXL2 in cancer (Review). Int J Mol Med. 2015;36(5):1200–4. https://doi.org/10.3892/ijmm.2015.2337.
Pollheimer MJ, Racedo S, Mikels-Vigdal A, Marshall D, Bowlus C, Lackner C, et al. Lysyl oxidase-like protein 2 (LOXL2) modulates barrier function in cholangiocytes in cholestasis. J Hepatol. 2018;69(2):368–77. https://doi.org/10.1016/j.jhep.2018.04.009.
Vadasz Z, Kessler O, Akiri G, Gengrinovitch S, Kagan HM, Baruch Y, et al. Abnormal deposition of collagen around hepatocytes in Wilson’s disease is associated with hepatocyte specific expression of lysyl oxidase and lysyl oxidase like protein-2. J Hepatol. 2005;43(3):499–507. https://doi.org/10.1016/j.jhep.2005.02.052.
•• Muir AJ, Levy C, Janssen HLA, Montano-Loza AJ, Shiffman ML, Caldwell S, et al. Simtuzumab for primary sclerosing cholangitis: phase 2 study results with insights on the natural history of the disease. Hepatology. 2018. https://doi.org/10.1002/hep.30237 Carefully performed placebo-controlled phase 2b trial of simtuzumab that despite initial expectations failed to show any clinical benefit.
Fickert P. Is this the last requiem for simtuzumab? – “Blessed is who expects nothing, for he shall never be disappointed.” - Alexander Pope. Hepatology. 2018. https://doi.org/10.1002/hep.30309.
Harrison SA, Abdelmalek MF, Caldwell S, Shiffman ML, Diehl AM, Ghalib R, et al. Simtuzumab is ineffective for patients with bridging fibrosis or compensated cirrhosis caused by nonalcoholic steatohepatitis. Gastroenterology. 2018;155(4):1140–53. https://doi.org/10.1053/j.gastro.2018.07.006.
Benson AB 3rd, Wainberg ZA, Hecht JR, Vyushkov D, Dong H, Bendell J, et al. A phase II randomized, double-blind, placebo-controlled study of simtuzumab or placebo in combination with gemcitabine for the first-line treatment of pancreatic adenocarcinoma. Oncologist. 2017;22(3):241–e15. https://doi.org/10.1634/theoncologist.2017-0024.
Raghu G, Brown KK, Collard HR, Cottin V, Gibson KF, Kaner RJ, et al. Efficacy of simtuzumab versus placebo in patients with idiopathic pulmonary fibrosis: a randomised, double-blind, controlled, phase 2 trial. Lancet Respir Med. 2017;5(1):22–32. https://doi.org/10.1016/S2213-2600(16)30421-0.
Ikenaga N, Peng ZW, Vaid KA, Liu SB, Yoshida S, Sverdlov DY, et al. Selective targeting of lysyl oxidase-like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut. 2017;66(9):1697–708. https://doi.org/10.1136/gutjnl-2016-312473.
Hov JR, Kummen M. Intestinal microbiota in primary sclerosing cholangitis. Curr Opin Gastroenterol. 2017;33(2):85–92. https://doi.org/10.1097/MOG.0000000000000334.
Chapman RW. Editorial: vancomycin - a promising option for the treatment of primary sclerosing cholangitis? Aliment Pharmacol Ther. 2018;47(9):1321–2. https://doi.org/10.1111/apt.14586.
Cannon K, Byrne B, Happe J, Wu K, Ward L, Chesnel L, et al. Enteric microbiome profiles during a randomized phase 2 clinical trial of surotomycin versus vancomycin for the treatment of Clostridium difficile infection. J Antimicrob Chemother. 2017;72(12):3453–61. https://doi.org/10.1093/jac/dkx318.
Damman JL, Rodriguez EA, Ali AH, Buness CW, Cox KL, Carey EJ, et al. Review article: the evidence that vancomycin is a therapeutic option for primary sclerosing cholangitis. Aliment Pharmacol Ther. 2018;47(7):886–95. https://doi.org/10.1111/apt.14540.
Tabibian JH, Gossard A, El-Youssef M, Eaton JE, Petz J, Jorgensen R, et al. Prospective clinical trial of rifaximin therapy for patients with primary sclerosing cholangitis. Am J Ther. 2017;24(1):e56–63. https://doi.org/10.1097/MJT.0000000000000102.
Silveira MG, Torok NJ, Gossard AA, Keach JC, Jorgensen RA, Petz JL, et al. Minocycline in the treatment of patients with primary sclerosing cholangitis: results of a pilot study. Am J Gastroenterol. 2009;104(1):83–8. https://doi.org/10.1038/ajg.2008.14.
Garrido-Mesa N, Zarzuelo A, Galvez J. Minocycline: far beyond an antibiotic. Br J Pharmacol. 2013;169(2):337–52. https://doi.org/10.1111/bph.12139.
Vaughn BP, Rank KM, Khoruts A. Fecal microbiota transplantation: current status in treatment of GI and liver disease. Clin Gastroenterol Hepatol. 2018;S1542-3565(18):30751. https://doi.org/10.1016/j.cgh.2018.07.026.
Cammarota G, Ianiro G, Tilg H, Rajilic-Stojanovic M, Kump P, Satokari R, et al. European consensus conference on faecal microbiota transplantation in clinical practice. Gut. 2017;66(4):569–80. https://doi.org/10.1136/gutjnl-2016-313017.
Al-Dury S, Marschall HU. Ileal bile acid transporter inhibition for the treatment of chronic constipation, cholestatic pruritus, and NASH. Front Pharmacol. 2018;9:931. https://doi.org/10.3389/fphar.2018.00931.
Funding
Open access funding provided by Medical University of Graz.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Elisabeth Krones: received an unrestricted research grant and norUDCA from Dr. Falk Pharma GmbH, Freiburg, Germany
Hanns-Ulrich Marschall: Consulting for Albireo, Bayer, Intercept; research grant of Intercept, material support by Albireo, Intercept
Peter Fickert: The Medical University of Graz has filed norUDCA patents for the treatment of liver diseases and arteriosclerosis and P.F. is co-inventor (WO2006119803 and WO20099013334); speaker for Falk Foundation, advisory boards for Dr. Falk Pharma GmbH and Intercept; travel grants and unrestricted research grants from Falk Foundation and unrestricted research grant from Gilead Sciences Inc.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Autoimmune, Cholestatic, and Biliary Diseases
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Krones, E., Marschall, HU. & Fickert, P. Future Medical Treatment of PSC. Curr Hepatology Rep 18, 96–106 (2019). https://doi.org/10.1007/s11901-019-00454-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11901-019-00454-4