
Abstract
Introduction
Synovitis–acne–pustulosis–hyperostosis–osteitis (SAPHO) is an acronym for various osteoarticular and dermatological manifestations that can appear in the same patient. It is a rare syndrome, but since its awareness has increased, there have been more and more such reports in the literature.
Aims
The objectives of this review are to summarize the current state of knowledge on pediatric and adult-onset SAPHO syndrome, and to discuss treatment strategies that should be considered.
Results
The SAPHO syndrome can affect patients of any age, and its etiology is still not known. The syndrome has its cognizable radiological characteristics that are most important in making the diagnosis. There are several diagnostic criteria as well, but they need further validation. No standard treatment protocols are available and current treatment options are not evidenced-based due to the rarity of the syndrome. Therapy is empirical and aimed at easing pain and modifying the inflammatory process. It includes nonsteroidal anti-inflammatory drugs (NSAIDs) as the first-line agents. Antibiotics, corticosteroids, disease-modifying anti-rheumatic drugs, biologicals targeting tumor necrosis factor alpha or interleukin-1, and bisphosphonates have all been used with variable success. Surgery is reserved to treat complications. Even though it is a disease with good long-term prognosis, its treatment remains a challenge and the results are known to be disappointing, especially with the skin component of the disease.
Conclusion
It is expected that these patients present at the time of diagnosis and the treatment should be as early, effective, and safe as possible in order to prevent osteoarticular progression and to limit the adverse events associated with pharmacological drugs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The SAPHO (synovitis–acne–pustulosis–hyperostosis–osteitis) syndrome was first introduced by the rheumatologist Chamot in 1987, and it is characterized by a combination of skin and osteoarticular manifestations. The term attempts to comprise numerous names that have been used in the literature for the last 50 years, describing the above-mentioned characteristics. Some of those names are bilateral clavicular osteomyelitis with palmar and plantar pustulosis, inter-sterno-costo-clavicular ossification, subacute and chronic symmetric osteomyelitis, arthro-osteitis associated with a follicular occlusive triad, sternoclavicular hyperostosis, nonbacterial osteitis, pustulotic arthro-osteitis, chronic recurrent multifocal osteomyelitis (CRMO), Koehler’s disease, pyogenic sterile arthritis, acquired hyperostosis syndrome, or spondyloarthritis hyperostotica pustulo-psoriatica [1–4]. CRMO will be discussed more in detail in this review as well, due to the great confusion that these two terms generate in the literature. Numerous authors have suggested that CRMO and SAPHO lie along the same clinical spectrum. Some believe that CRMO is the pediatric presentation of SAPHO, even though there are some rare descriptions of SAPHO seen in children and seldom in adolescents, as well as descriptions of CRMO in adults. It seems that the differentiating clinical feature is mainly in the localization of inflammation: in pediatric CRMO, the extremities are more often affected, whereas in SAPHO, the axial skeleton with costosternoclavicular region is the focus [5–9].
SAPHO is considered a rare disease and sufficient data on its prevalence are unavailable. It is predominantly found in patients with average ages of 30 and 50 years [10]. Hayem et al. [11] reviewed 120 cases of SAPHO and revealed that there is a female predominance among patients younger than 30 years old at the beginning of the disease. Despite all of this, there is a considerable number of reports on children who suffer from SAPHO, and, today, it is considered that it can evolve at any age [1]. The youngest described patient was only 15 months old [12]. According to some authors, the annual prevalence is estimated at 1/10,000 in Caucasians [6, 13] or 0.00144/100,000 in Japanese [14–16].
The clinical presentation is heterogeneous and patients may, therefore, present to different specialists. SAPHO is well known to dermatologists and rheumatologists, but there are only a few reports in the orthopedic literature. Since the disease can evolve at any age, it is important to present such literature to clinicians dealing with children, especially since various manifestations (pustulosis and osteitis) do not necessarily coincide. Recognizing the disease in time will prevent osteoarticular progression. Otherwise, patients can suffer deformity, loss of function, and increasing pain, which might require wide resections.
A MEDLINE search using SAPHO, SAPHO syndrome, and chronic recurrent multifocal osteomyelitis as keywords was performed and, further, relevant articles from retrieved references were extracted.
The objectives of this review article are to review the etiology, presentation, diagnosis, treatment, pathogenesis, and genetics of the syndrome and to raise awareness of this entity.
Etiology
Whether the SAPHO syndrome represents a clinical entity by itself, should be considered a subset within the family of spondyloarthropathies (due to the frequent affliction of the axial skeleton, enthesitis, and inflammatory bowel diseases), or be considered a variant of another rheumatic disease (i.e., psoriatic arthritis) is still unknown [2].
The pathogenesis of SAPHO is probably multifactorial and it involves a combination of genetic, infectious, and immunological components.
The published data show that HLA-B27 is more frequent in SAPHO, but spondyloarthropathies overlap with SAPHO, and statistical analyses performed on those cohorts resulted in a higher proportion of HLA-B27 SAPHO patients. Therefore, it is no wonder that other studies refute these data and showed that there are no relations between SAPHO and HLA-B27. According to some authors, there is a positive connection with HLA 39 and HLA 61 [4, 17, 18]. Due to some similarities between SAPHO and other autoinflammatory syndromes with a genetic basis and due to familial clustering, several other genes are being studied. Researchers have discovered that genes which seem to play a role in the SAPHO syndrome are located in the chromosome 18: LPIN2 and NOD2. LPIN2 encodes lipin 2, which is involved in modulating apoptosis of polymorphonuclear cells, and mutations of the NOD2 gene may lead to an abnormal immune response to bacterial peptidoglycans via activation of the proinflammatory transcription factor nuclear factor kappa B [19].
There are also hypotheses of infectious disease, suggesting that bone lesions are caused by a low-virulence pathogen [2, 13]. Different types of pathogens were isolated from different bone sites and pustules in the skin, including Staphylococcus aureus [20], Haemophilus parainfluenzae, and Actinomyces, as well as Treponema pallidum, Veillonella, and Eikenella [21]. The most important is Propionibacterium acnes, which is identified more often, but positive cultures can only be seen in a small number of total bone biopsy specimens. The largest number of P. acnes-positive biopsy specimens was proved by Assmann and Simon [2] in their study of 21 SAPHO patients, where 67 % of them were positive. This infectious hypothesis is supported by increased levels of circulating IgA in these patients and there is also evidence that intra-articular injection of inactivated P. acnes in rats can cause erosive joint lesions. On the other hand, according to some of the latest considerations, since P. acnes is found in only two-thirds of biopsies at most and the treatment with antibiotics is effective only for as long as it is taken, it is considered that SAPHO cannot be classified among infections, even due to latent organisms [22–24].
There are various reports on immune system dysfunction in SAPHO [25–29]. According to some of them, humoral immune response is hyperactive and in others, it is hypoactive. This is similar to the cell-mediated immune response that has been reported as normal or hyperactive; total immune system impairment has been reported as well [28]. Hurtado-Nedelec et al. showed that SAPHO is characterized by elevated IL-8 and IL-18 levels. They had not detect any autoantibodies among their SAPHO patients, including rheumatoid factor, anti-CCP2, or antinuclear antibodies. IL-8 and TNFα production by purified polymorphonuclear leukocytes (PMN) were elevated in these patients compared to the controls, but the oxidative burst and IL-18 production were normal. They also showed that, after 28 days of etanercept therapy, PMN, IL-8, and TNFα production was downregulated and TNFα plasma levels were increased [30]. Assman and Simon [2] have shown that the proinflammatory response observed in SAPHO is mediated by the ability of P. acnes to trigger interleukin IL-1, IL-8, and IL-18 and TNFα release by monocytes, keratinocytes, sebocytes, and dendritic cells.
After all, the most probable hypothesis about the etiology of SAPHO is that it is caused by autoimmune reactions in genetically predisposed organisms, triggered by some infectious agent [2].
Regarding the pathogenesis of CRMO, infectious and autoimmune theories have been suggested, but none of them have been proven. There is a significant genetic contribution to pathogenesis and besides LPIN2, several other genes have to be mentioned. A susceptibility locus on chromosome 18q21.3–18q22 affecting the proline-serine-threonine phosphatase interacting protein 2 (PSTPIP2) has been reported [31] in a small German CRMO cohort. Furthermore, the disorder is also associated with polymorphisms of the IL-10 promoter and mutation of IL1RN causing deficiency of the interleukin-1 receptor antagonist (DIRA), an autosomal recessive disorder that presents with CRMO during the neonatal period [32].
Clinical features
SAPHO syndrome should be suspected in patients who present with osteoarticular and/or certain dermatological clinical manifestations.
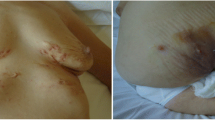
Osteoarticular manifestations involve osteitis, hyperostosis, synovitis, arthropathy, and enthesopathy that present with pain, tenderness, and sometimes swelling over the affected areas and fever. Osteitis is the inflammation of bone, which may involve the cortex and the medullary cavity. Hyperostosis reflects excessive bone growth and may result in enthesopathic new bone formation and joint fusion (Fig. 1). Synovitis mostly manifests as nonerosive oligoarthritis of larger joints. Joint involvement can be primary arthritis or an extension of the osteitis adjacent to the articular structures. Arthritis has been reported in up to 92.5 % of SAPHO cases. The axial skeleton is involved in 91 % and the peripheral joints in 36 % of cases. Besides sternocostal and sternoclavicular joints, which are the most commonly affected, it mainly affects the sacroiliac or hip joints, knees, and ankles. For anterior chest wall disease, three stages have been described (Table 1). The costoclavicular ligament is involved in 48 % of cases, and it is considered a decisive early finding in SAPHO [7, 32, 33].

Bilateral sternoclavicular joint edema in the SAPHO patient
The smallest number of cases in the literature are based on temporomandibular joint involvement [11, 13, 34, 35]. The percentage distribution of arthritis in various parts of the body is demonstrated schematically in Fig. 2.

Percentage distribution of arthritis in the body (SAPHO/CRMO)
Soft tissue surrounding joints and bones can be affected as well. It may be misinterpreted as a neoplastic or lymphatic mass [7, 36], and, although rare, the soft tissue swelling can lead to serious complications, such as thoracic outlet syndrome [11, 36–38].
Enthesopathy can lead to ligament ossification, which can result in the development of bony bridging across joints.
CRMO is an aseptic inflammatory disorder clinically characterized with insidious onset of bone lesions with pain and swelling that is often worse at night, with or without fever. Swelling and warmth can occur over the affected areas. It is most commonly found in the metaphyseal regions of long bones of the lower extremities. Some other sites, such as the clavicules, vertebral bodies, mandible, pelvis, and small bones of the hands and feet, have been shown to be affected as well. Involvement is multifocal, usually unilateral, and it can be accompanied by skin lesions (most often, palmoplantar pustulosis and psoriasis have been described) [32, 39]. As stated earlier, some investigators believe that CRMO is the pediatric presentation of SAPHO, but it seems that the differentiating clinical feature is mainly in the localization of inflammation: in pediatric CRMO patients, the extremities are more often affected and in SAPHO patients, the axial skeleton with costosternoclavicular region is the focus [5].
Typical skin lesions seen in SAPHO patients include palmoplantar pustulosis (PPP) and severe acne [40]. Acne can manifest as acne conglobata, acne fulminans, or hidradenitis suppurativa. Women more often develop PPP and men show severe forms of acne. Pyoderma gangrenosum is the other less frequent manifestation and different forms of psoriasis have also been described [41], as well as Sweet’s syndrome and Sneddon–Wilkinson disease [42]. Skin lesions may vary in severity and may precede (in 50 % of the cases), follow, or occur simultaneously with the onset of arthritis [11]. Usually, the time interval between the onset of skin and osteoarticular manifestations is <2 years [43], but an interval of 38 years has been recorded in the literature [11, 44]. Sonozaki et al. [45] showed that skin lesions precede or follow the onset of osteoarticular lesions within 2 years in about 70 % of patients, while Hayem et al. [11] showed that the skin manifestations anteceded or presented at the same time as the skeletal manifestations in 68 % of their cohort. Dermatological manifestations are known to be resistant to therapy and quite often have a chronic, protracted course.
Radiological features
Radiographs may show expanded bone, sclerosis and osteolysis, periosteal reaction, or enthesopathic new bone formation. Bone scintigraphy delineates increased uptake in affected bone and may reveal asymptomatic disease or abnormalities not apparent on radiographs. The advantage of scintigraphy is the demonstration of multiple sites of involvement, so it is helpful for the elimination of malignancy or infection. Symmetric uptake in the sternoclavicular region with a typical “bull‘s head” appearance shown in bone scintigraphy is characteristic of the SAPHO syndrome (Fig. 3) [46]. It was first described by Freyschmidt and Sternberg [47] but, even though it is considered to be pathognomonic, it is not a very sensitive indicator of SAPHO.

Scintigraphy findings show intensive uptake of the radiopharmaceutical technetium-99m at the sternoclavicular joints and sternum, which represent a “bull’s head“ sign
Magnetic resonance imaging (MRI) will also detect occult lesions, may show findings not seen on plain radiographs, and provide information about soft tissues.
Characteristic radiographic findings are hyperostosis and osteitis. Hyperostosis is radiographically seen as diffuse thickening of the periosteum, cortex, and endosteum, with narrowing of the medullary canal [47]. Both are characterized by increased bone sclerosis [35, 39].
In the early stages, the disease usually manifests as an osteolytic process. As healing progresses, the lytic/sclerotic picture is produced. Characteristic features of osteitis and hyperostosis become more apparent with time [35].
Joint involvement is characterized by arthritis, with joint space narrowing and, sometimes, erosions. There might be periarticular osteopenia. Ligamentous ossifications can be observed as well [32, 37].
Several spine lesions have been described regarding this syndrome, and they include vertebral body corner lesions, nonspecific spondylodiscitis and osteodestructive lesions seen in adults and children, and osteosclerotic vertebral lesions, paravertebral ossification, and sacroiliitis seen in adults.
The term “corner lesion” describes focal cortical erosion at one of the vertebral body corners, which is usually seen in adults. Nonspecific spondylodiscitis is seen as focal erosive changes with sclerosing remodeling of the vertebral end plates, usually anteriorly located at the discovertebral junction. This can be seen in up to 32 % of cases, and single and multiple levels may be found [35]. Takigawa et al. [14] observed nonconsecutive and consecutive multilevel lesions, both at a proportion of 38 %. It may be painful for many weeks but, usually, with time, it becomes asymptomatic. Rarely it is a cause of neurological complications or deformity [35].
Osteodestructive lesions include osteolytic vertebral lesions, usually limited to one vertebrae, with a variable degree of collapse. Collapse may induce kyphosis, spinal canal stenosis, and spinal cord injury. If it is quite marked, it can present as a vertebra plana in children, which is not characteristic of an adult population [14]. Sacroiliitis can be seen and it is usually unilateral. Ankylosis may be present as well, and it is usually connected with the relief of pain [7, 38, 48].
Affection of the long bones is commonly seen among children. Predominantly, the metadiaphyses are affected, especially the distal femur, and proximal and distal tibia. Radiographically, it may manifest as lytic lesions, sclerotic or mixed lesions, and periosteal reaction may eventually develop. MRI is the technique of choice in young patients suspected of SAPHO/CRMO, particularly due to the lack of radiation requirements and its sensitivity in detecting early subclinical lesions. It is seen as bone marrow edema, which shows up as hypointense on T1 and hyperintense on T2 signals in the affected metaphysis. As the disease progresses, hypointense T1 and T2 signals in the medullary space and cortex represent medullary sclerosis and cortical thickening [17]. Lesions are usually multiple and often symmetrical. Involvement of the adjacent epiphysis and altered bone growth are rare [17, 35].
Many of the radiological manifestations of the disease can be seen on plain radiographs. It is important to emphasize that radiographs made during the first 3 months of the disease course are normal in 80 % of cases and all patients had abnormal radiographs at the end of follow-up [38]. Similar findings were shown by Fritz et al. [49]. They found that the sensitivity of conventional radiography in the early stages of the disease is 13 % and, compared to MRI, it shows only 16 % of the lesions seen on MRI. For identifying subclinical foci, whole-body scintigraphy or whole-body MRI is very useful. Actually, if initial radiographs are negative and disease is suspected, bone scintigraphy is used as the next step to detect occult inflammatory lesions and clinically suspected localizations. Because of increased cost, the use of whole-body MRI is recommended for indeterminate cases, monitoring of disease activity, and for better delineation of soft tissue changes. Intravenous contrast will highlight abscesses and other soft tissue changes that may be associated with more aggressive conditions [17]. It should be kept in mind that imaging procedures cannot accurately distinguish among SAPHO/CRMO, malignancy, and osteomyelitis, and such findings should always be interpreted within other clinical and laboratory parameters.
Laboratory tests
There are no laboratory tests that are diagnostic of SAPHO. They can be normal or may show elevated inflammatory markers, such as erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), and elevated levels of components of complements C3 and C4. Mild leukocytosis and mild anemia were observed as well. Compared to healthy controls, these patients have elevated levels of immunoglobulin A [2, 50]. A study searching for some specific antibody profiles for those patients has been conducted recently, but, unfortunately, without any success. Hurtado-Nedelec et al. [30] showed significantly increased levels of IgA in their cohort of 29 SAPHO patients, while the levels of IgM and IgG were normal. This information can possibly be used as an additional tool in making the diagnosis, but further investigations need to be done. Also, some studies exhibit correlation with B39 and B61 [18].
Histopathological findings
Osteitis refers to bone inflammation and appears histopathologically as sterile inflammatory infiltrate [3]. Early during the disease course, the predominant finding is PMN infiltrate. In the intermediate stage, the infiltrate is composed primarily of mononuclear cells and in the late stage, bone trabeculae are enlarged and sclerotic, with an increased number of osteocytes and marrow fibrosis. Skin biopsy of the affected skin shows neutrophilic pseudo-abscesses [30]. SAPHO and CRMO lesions are histologically identical [40].
Diagnostic criteria
There are several published diagnostic criteria for SAPHO and the presence of only one of the inclusion criteria is sufficient for making the diagnosis. The criteria suggested by Kahn and the other by Benhamou are the most frequently mentioned. All of them are preliminary and need further validation (please see Tables 2, 3, and 4). With regard to all of them, it can be said that the criteria made by Kahn and modified in 2003 seems to be the most precise. Even though the existence of such criteria is very helpful in making the diagnosis, it is very doubtful as to whether bone and joint involvement associated with chronic bowel diseases (which is one of the inclusion criteria) can be classified as SAPHO syndrome, since arthropathies associated with inflammatory bowel disease are included in the EULAR/ILAR criteria for juvenile idiopathic arthritis (JIA) [51, 52].
Furthermore, it should be discussed whether CRMO is the pediatric presentation of SAPHO or an entity by itself. For those reasons, these criteria need further modifications.
Diagnosis and differential diagnosis
The diagnosis is usually made by a rheumatologist, who will consult with a dermatologist to treat the skin component of the disease. Making a diagnosis is challenging because not all symptoms are always apparent or present at the same time, or some may be subtle. In making the diagnosis, the above-mentioned clinical, radiological, and laboratory characteristics, as well as diagnostic criteria, are used. Regarding differential diagnosis, early during the disease course, infectious (osteomyelitis) and neoplastic etiology must be excluded. Tumors with local extension should be considered—thyroid cancer, lymphoma or osteosarcoma, metastatic breast, prostate cancer, and neuroblastoma [17]. Psoriatic arthritis with axial skeleton manifestation and pustular psoriasis, a special subgroup of psoriatic disease, can be the cause of diagnostic dilemma. Radiographic signs of osteitis with hyperostosis are not often seen in psoriatic arthritis [53]. Furthermore, differential diagnosis includes Paget’s disease (genetic disease with increased bone turnover, repeated fractures and deformities, markedly elevated level of alkaline phosphatase, and radiographs revealing characteristic mosaic pattern, both osteolysis and osteosclerosis) and Sweet’s syndrome (neutrophilic dermatosis with elevated inflammatory markers that can be accompanied with aching joints). When the clavicle is affected, Tietze’s syndrome (swelling of the costal cartilages, mostly in adults, rare in children) and avascular necrosis of the clavicular epiphysis are considered as well. Regarding differential diagnosis in the pediatric age group, it also includes Ewing’s sarcoma, histiocytosis, Majeed syndrome, or DIRA. Majeed syndrome is an autosomal recessive (AR) disorder that presents with early-onset CRMO and dyserythropoietic anemia. DIRA is an AR disorder that manifests with CRMO in the neonatal period with generalized pustulosis, osteitis, periostitis, and systemic inflammation [17, 32, 54]. Differential diagnosis of SAPHO and CRMO is shown in Table 5.
Finally, it should always be kept in mind that SAPHO and CRMO are diagnosed by exclusion. When only one site is involved in the absence of skin lesions, making the diagnosis can be difficult and biopsy may be needed. Sterile osteitis (little or no medullary change) is one of the major characteristics of this syndrome, but the diagnosis can never be done by histological results alone, and the advantage of biopsy is just to exclude other diagnoses [24, 46].
Treatment
Because to the variety of clinical presentations, the treatment of SAPHO syndrome remains a challenge and outcomes are known to be disappointing, especially with the skin component of the disease. There have been no randomized controlled trials on the effectiveness of various therapies, but nonsteroidal anti-inflammatory drugs (NSAIDs) are generally considered as the first-line treatment option [4]. Antimicrobial therapy is useful in patients with positive biopsy cultures, but it has little or no effect in others. Successful treatment has been reported for doxycycline, azithromycin, sulfamethoxazole/trimethoprim, and clindamycin [20, 55]. Azithromycin acts not only as an antimicrobial, but also as an anti-inflammatory and immunomodulatory drug, and Schilling and Wagner suggest the simultaneous usage of azithromycin together with calcitonin (osteotropic drug) [56]. Other treatment options include colchicine, corticosteroids, bisphosphonates, and disease-modifying agents, such as methotrexate, sulfasalazine, and anti-TNFα therapy. Bisphosphonates act by inhibiting bone resorption and turnover, and by possible anti-inflammatory activity that suppresses the production of IL-1, IL-6, and TNFα [57]. They have no effect on skin lesions. Local corticosteroid injections have also been tried, but this treatment modality has a significant effect only on osteitis lesions [53]. Some authors used corticosteroids orally and, in that case, they will act on both skeletal and skin manifestations. Dermatologists use topical corticosteroids, psoralen plus ultraviolet A (PUVA) photochemotherapy, and retinoids [58]. Disease-modifying agents are only indicated when symptoms persist for at least 4 weeks, despite adequate NSAID therapy. There is increasing evidence of anti-TNFα usage in the treatment of such patients. Case reports and case series on TNFα blockade often demonstrate a marked improvement in the clinical picture, regardless of whether or not this treatment is permanently effective. The most often published cases in the literature are about the use of infliximab in these patients. Usually, 5 mg/kg at weeks 0, 2, and 6 followed by a 6–8-week interval has been used, just like that used in spondyloarthropathies. Lower doses of infliximab and reduction in the duration of intervals have been tested, but it has been noted that decreased infusion intervals like in spondyloarthropathies and lower dosages cannot maintain the remission of disease [58]. Both skeletal and cutaneous lesions responded well in most of the described cases, with exception of PPP, which sometimes failed to respond. In some cases, infliximab induced exacerbation of skin manifestation. Arias-Santiago et al. [59] suggested adalimumab as a possible alternative therapy in such cases, and there are also reports on the successful treatment of SAPHO with etanercept and the IL-1 receptor antagonist anakinra. Anakinra appeared to be helpful in five out of six SAPHO patients, two of which previously failed to respond to TNF blockers [60]. Autologous bone transplantation using microvascular flaps is applied as an experimental treatment procedure [15].
Physiotherapy can always be used as an additional treatment for osteoarticular manifestations. Surgery is considered for patients whose condition has failed to respond to all other therapeutic interventions [61]. Wide resections are reserved to treat complications when patients develop deformity or loss of function with pain [15]. There are several reports in the literature about the surgical treatment of such patients; for example, resection of the medial clavicle or the sternoclavicular joint, which seemed to provide variable improvement in pain, although some authors report no improvement with this intervention [54]. Furthermore, mandibular involvement has been treated with minor surgical procedures, such as decortications and curettage, but extensive extirpation of the cortical jaw was done as well [62].
Clinical course and conclusion
Except for a minority of patients who have a self-limited course, most of them have either relapsing–remitting course or chronic indolent pattern. Over the long term, rheumatic manifestations in most patients show little progression [11]. Maugars et al. [38] revealed that, after an average follow-up of around 12 years, 53 % of patients develop disease at new sites.
Colina et al. [48] identified that female sex, anterior chest wall involvement, peripheral arthritis, skin lesions, and high inflammatory parameters at first presentation are related to the chronic course of the disease.
SAPHO is rare, but as awareness increases, it is being reported more often. It should be suspected when evaluating patients with lytic, sclerotic, or hyperostotic bone lesions and pain.
This paper is an attempt to increase awareness about SAPHO syndrome among orthopedic pediatric surgeons and prompt recognition will help avoid unnecessary examinations, biopsies, surgical treatments, antibiotic therapy, or possible physical and psychological impairments associated with the disease, especially among children. It is important to remember that the skin manifestations and bony involvement may not be present at the same time, and it would be best to refer suspected SAPHO patients to the rheumatologist/dermatologist. Further examination and randomized controlled trials need to be done in order to better understand the disease, as well as to aid the development and establishment of adequate therapies.
References
Kerrison C, Davidson JE, Cleary AG, Beresford MW (2004) Pamidronate in the treatment of childhood SAPHO syndrome. Rheumatology (Oxford) 43:1246–1251
Assmann G, Simon P (2011) The SAPHO syndrome—are microbes involved? Best Pract Res Clin Rheumatol 25:423–434
Matzaroglou Ch, Velissaris D, Karageorgos A, Marangos M, Panagiotopoulos E, Karanikolas M (2009) SAPHO syndrome diagnosis and treatment: report of five cases and review of the literature. Open Orthop J 3:100–106
Khanna L, El-Khoury GY (2012) SAPHO syndrome—a pictorial assay. Iowa Orthop J 32:189–195
Rohekar G, Inman RD (2006) Conundrums in nosology: synovitis, acne, pustulosis, hyperostosis, and osteitis syndrome and spondylarthritis. Arthritis Rheum 55:665–669
Hayem G (2004) SAPHO syndrome. Rev Prat 54:1635–1636
Earwaker JWS, Cotten A (2003) SAPHO: syndrome or concept? Imaging findings. Skeletal Radiol 32:311–327
Tyrrell PN, Cassar-Pullicino VN, Eisenstein SM, Monach JF, Darby AJ, McCall IW (1996) Back pain in childhood. Ann Rheum Dis 55:789–793
Wallach D (2000) Neutrophilic dermatoses: an overview. Clin Dermatol 18:229–231
Van Doornum S, Barraclough D, McColl G, Wicks I (2000) SAPHO: rare or just not recognized? Semin Arthritis Rheum 30:70–77
Hayem G, Bouchaud-Chabot A, Benali K et al (1999) SAPHO syndrome: a long-term follow-up study of 120 cases. Semin Arthritis Rheum 29:159–171
Eleftheriou D, Gerschman T, Sebire N, Woo P, Pilkington CA, Brogan PA (2010) Biologic therapy in refractory chronic non-bacterial osteomyelitis of childhood. Rheumatology (Oxford) 49:1505–1512
McPhillips A, Wolford LM, Rodrigues DB (2010) SAPHO syndrome with TMJ involvement: review of the literature and case presentation. Int J Oral Maxillofac Surg 39:1160–1167
Takigawa T, Tanaka M, Nakanishi K et al (2008) SAPHO syndrome associated spondylitis. Eur Spine J 17:1391–1397
Mochizuki Y, Omura K, Hirai H, Kugimoto T, Osako T, Taguchi T (2012) Chronic mandibular osteomyelitis with suspected underlying synovitis, acne, pustulosis, hyperostosis, and osteitis (SAPHO) syndrome: a case report. J Inflamm Res 5:29–35
Hukuda S, Minami M, Saito T et al (2001) Spondyloarthropathies in Japan: nationwide questionnaire survey performed by the Japan Ankylosing Spondylitis Society. J Rheumatol 28:554–559
Thakur U, Blacksin M, Beebe K, Neilson JC, Dashefsky B, Tagoylo G (2012) Synovitis, acne, pustulosis, hyperostosis and osteitis (SAPHO) and chronic recurrent multifocal osteomyelitis (CRMO): role of imaging in diagnosis. Radiography 18:221–224
De Souza A, Solomon GE, Strober BE (2011) SAPHO syndrome associated with hidradenitis suppurativa successfully treated with infliximab and methotrexate. Bull NYU Hosp Jt Dis 69:185–187
Burgemeister LT, Baeten DL, Tas SW (2012) Biologics for rare inflammatory diseases: TNF blockade in the SAPHO syndrome. Neth J Med 70:444–449
Rozin AP, Nahir AM (2007) Is SAPHO syndrome a target for antibiotic therapy? Clin Rheumatol 26:817–820
Arnson Y, Rubinow A, Amital H (2008) Secondary syphilis presenting as SAPHO syndrome features. Clin Exp Rheumatol 26:1119–1121
Trimble BS, Evers CJ, Ballaron SA, Young JM (1987) Intraarticular injection of Propionibacterium acnes causes an erosive arthritis in rats. Agents Actions 21:281–283
Assmann G, Kueck O, Kirchhoff T et al (2009) Efficacy of antibiotic therapy for SAPHO syndrome is lost after its discontinuation: an interventional study. Arthritis Res Ther 11:R140
Berthelot JM, de la Cochetière MF, Potel G, Le Goff B, Maugars Y (2013) Evidence supporting a role for dormant bacteria in the pathogenesis of spondylarthritis. Joint Bone Spine 80:135–140
Ferguson PJ, Lokuta MA, El-Shanti HI, Muhle L, Bing X, Huttenlocher A (2008) Neutrophil dysfunction in a family with a SAPHO syndrome-like phenotype. Arthritis Rheum 58:3264–3269
Eyrich GK, Langenegger T, Bruder E, Sailer HF, Michel BA (2000) Diffuse chronic sclerosing osteomyelitis and the synovitis, acne, pustolosis, hyperostosis, osteitis (SAPHO) syndrome in two sisters. Int J Oral Maxillofac Surg 29:49–53
Grosjean C, Hurtado-Nedelec M, Nicaise-Roland P et al (2010) Prevalence of autoantibodies in SAPHO syndrome: a single-center study of 90 patients. J Rheumatol 37:639–643
Malmström M, Fyhrquist F, Kosunen TU, Tasanen A (1983) Immunological features of patients with chronic sclerosing osteomyelitis of the mandible. Int J Oral Surg 12:6–13
Montonen M, Lindqvist C (2003) Diagnosis and treatment of diffuse sclerosing osteomyelitis of the jaws. Oral Maxillofac Surg Clin North Am 15:69–78
Hurtado-Nedelec M, Chollet-Martin S, Nicaise-Roland P et al (2008) Characterization of the immune response in the synovitis, acne, pustulosis, hyperostosis, osteitis (SAPHO) syndrome. Rheumatology (Oxford) 47:1160–1167
Golla A, Jansson A, Ramser J et al (2002) Chronic recurrent multifocal osteomyelitis (CRMO): evidence for a susceptibility gene located on chromosome 18q21.3–18q22. Eur J Hum Genet 10:217–221
Ferguson PJ, Sandu M (2012) Current understanding of the pathogenesis and management of chronic recurrent multifocal osteomyelitis. Curr Rheumatol Rep 14:130–141
Dihlmann W, Dihlmann SW (1991) Acquired hyperostosis syndrome: spectrum of manifestations at the sternocostoclavicular region. Radiologic evaluation of 34 cases. Clin Rheumatol 10:250–263
Girschick HJ, Krauspe R, Tschammler A, Huppertz HI (1998) Chronic recurrent osteomyelitis with clavicular involvement in children: diagnostic value of different imaging techniques and therapy with non-steroidal anti-inflammatory drugs. Eur J Pediatr 157:28–33
Depasquale R, Kumar N, Lalam RK et al (2012) SAPHO: what radiologists should know. Clin Radiol 67:195–206
Kahn MF, Hayem F, Hayem G, Grossin M (1994) Is diffuse Sclerosing osteomyelitis of the mandible part of the synovitis, acne, pustulosis, hyperostosis, osteitis (SAPHO) syndrome? Analysis of seven cases. Oral Surg Oral Med Oral Pathol 78:594–598
van Holsbeeck M, Martel W, Dequeker J et al (1989) Soft tissue involvement, mediastinal pseudotumor, and venous thrombosis in pustulotic arthro-osteitis. A study of eight new cases. Skeletal Radiol 18:1–8
Maugars Y, Berthelot JM, Ducloux JM, Prost A (1995) SAPHO syndrome: a followup study of 19 cases with special emphasis on enthesis involvement. J Rheumatol 22:2135–2141
Cotten A, Flipo RM, Mentre A, Delaporte E, Duquesnoy B, Chastanet P (1995) SAPHO syndrome. Radiographics 15:1147–1154
Monsour PAJ, Dalton JB (2010) Chronic recurrent multifocal osteomyelitis involving the mandible: case reports and review of the literature. Dentomaxillofac Radiol 39:184–190
Kahn MF, Khan MA (1994) The SAPHO syndrome. Baillieres Clin Rheumatol 8:333–362
Govoni M, Colina M, Massara A, Trotta F (2009) SAPHO syndrome and infections. Autoimmun Rev 8:256–259
Fruehauf J, Cierny-Modrè B, Caelen Lel-S, Schwarz T, Weinke R, Aberer E (2009) Response to infliximab in SAPHO syndrome. BMJ Case Rep. doi:10.1136/bcr.10.2008.1145
Sugimoto H, Tamura K, Fujii T (1998) The SAPHO syndrome: defining the radiologic spectrum of diseases comprising the syndrome. Eur Radiol 8:800–806
Sonozaki H, Mitsui H, Miyanaga Y et al (1981) Clinical features of 53 cases with pustulotic arthro-osteitis. Ann Rheum Dis 40:547–553
Quirico Rodríguez M, Casáns Tormo I, Redal Peña MC, López Castillo V (2010) The importance of bone scintigraphy in the diagnosis of sapho syndrome. Rev Esp Med Nucl 29:127–130
Freyschmidt J, Sternberg A (1998) The bullhead sign: scintigraphic pattern of sternocostoclavicular hyperostosis and pustulotic arthroosteitis. Eur Radiol 8:807–812
Colina M, Govoni M, Orzincolo C, Trotta F (2009) Clinical and radiologic evolution of synovitis, acne, pustulosis, hyperostosis, and osteitis syndrome: a single center study of a cohort of 71 subjects. Arthritis Rheum 61:813–821
Fritz J, Tzaribatchev N, Claussen CD, Carrino JA, Horger MS (2009) Chronic recurrent multifocal osteomyelitis: comparison of whole-body MR imaging with radiography and correlation with clinical and laboratory data. Radiology 252:842–851
Toussirot E, Dupond JL, Wendling D (1997) Spondylodiscitis in SAPHO syndrome. A series of eight cases. Ann Rheum Dis 56:52–58
Cassidy JT, Levinson JE, Bass JC et al (1986) A study of classification criteria for a diagnosis of juvenile rheumatoid arthritis. Arthritis Rheum 29:274–281
Petty RE, Southwood TR, Baum J et al (1998) Revision of the proposed classification criteria for juvenile idiopathic arthritis: Durban, 1997. J Rheumatol 25:1991–1994
Jung J, Molinger M, Kohn D, Schreiber M, Pfreundschuh M, Assmann G (2012) Intra-articular glucocorticosteroid injection into sternocostoclavicular joints in patients with SAPHO syndrome. Semin Arthritis Rheum 42:266–270
Carroll MB (2011) Sternocostoclavicular hyperostosis: a review. Ther Adv Musculoskelet Dis 3:101–110
Wagner AD, Andresen J, Huelsemann J, Zeidler H (1997) Long-term antibiotic therapy successful in patients with SAPHO syndrome. Arthritis Rheum 40:S62
Schilling F, Wagner AD (2000) Azithromycin: an anti-inflammatory effect in chronic recurrent multifocal osteomyelitis? A preliminary report. Z Rheumatol 59:352–353
Amital H, Applbaum YH, Aamar S, Daniel N, Rubinow A (2004) SAPHO syndrome treated with pamidronate: an open-label study of 10 patients. Rheumatology (Oxford) 43:658–661
Greenspan A, Gerscovich E, Szabo RM, Matthews JG 2nd (1991) Condensing osteitis of the clavicle: a rare but frequently misdiagnosed condition. AJR Am J Roentgenol 156:1011–1015
Arias-Santiago S, Sanchez-Cano D, Callejas-Rubio JL, Fernández-Pugnaire MA, Ortego-Centeno N (2010) Adalimumab treatment for SAPHO syndrome. Acta Derm Venereol 90:301–302
Wendling D, Prati C, Aubin F (2012) Anakinra treatment of SAPHO syndrome: short-term results of an open study. Ann Rheum Dis 71:1098–1100
Taylor HG, Dawes PT (1992) Sterno-costo-clavicular hyperostosis. Br J Clin Pract 46(4):276–278
Wannfors K (2013) SAPHO (synovitis–acne–pustulosis–hyperostosis–oste itis): a multidisciplinary approach. Oral Surg Oral Med Oral Pathol Oral Radiol 116:692–697
Conflict of interest
The author declares that there were no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Rukavina, I. SAPHO syndrome: a review. J Child Orthop 9, 19–27 (2015). https://doi.org/10.1007/s11832-014-0627-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11832-014-0627-7