
Abstract
The production of thin-wall ductile iron (TWDI) by high-pressure die-casting (HPDC) is complex because of several metallurgical and microstructural challenges. The present work aims to evaluate the austemperability of components (4 mm thickness) produced by HPDC process. The graphitization kinetics, the pearlite formation during continuous cooling, and the effect of austempering on the evolution of the ausferritic microstructure were investigated using dilatometric tests, microstructural analysis as well as Vickers hardness tests and tensile tests. Results show that components exhibit a brittle behavior because of white structures, small shrinkage cavities, and microporosity in the as-cast condition. Graphitization at 1100 °C allows rapid formation of small graphite particles within a short time (40 s). The critical cooling time (t8/5) to avoid the formation of pearlite upon cooling was found to be 5 s at a martensite start temperature of 193 ± 14 °C. Austempering at 360 °C for 40 min results in an ausferritic microstructure with stable carbon-enriched austenite which provides a high hardness (355 ± 4 HV10) and tensile strength (Rm = 709 ± 65 MPa). The results represent main criteria regarding the producibility of die-casted TWDI, which are helpful for future alloy and heat treatment design.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Austempered ductile iron (ADI) is an attractive material for high-performance components because of its excellent relationship of strength, ductility, and toughness together with its good wear and fatigue resistance. It has therefore been established as an alternative material for high-strength steel forgings or castings in the automotive industry (Ref 1). The high strength-to-weight ratio and stiffness further allow ADI to be a substitute for lightweight materials like aluminum or magnesium at equal mass sections over 3 mm (Ref 2). Concerning sustainability, ADI is superior to all of the materials mentioned above because of its lower embodied energy and good recyclability (Ref 3).
Its exceptional properties can be traced back to its unique “ausferrite” microstructure resulting from a bainitic transformation of austenite during an austempering heat treatment. For this transformation, ductile iron (DI) castings must undergo an austenitization treatment above the A3 temperature first to ensure complete dissolution of pearlite. Rapid quenching to the austempering temperature, which is lower than the pearlitic transition temperature but higher than the martensite start (MS) temperature (Ref 4), provides a metastable austenitic matrix that partially converts into bainitic ferrite during austempering. The cooling rate after austenitization plays a crucial role in the ADI production process. It must be high enough to avoid the formation of pearlite during cooling. Otherwise, only poor mechanical properties of the material can be expected (Ref 5). Within the isothermal austempering stage, individual bainitic ferrite plates, which are separated from each other by thin carbon-enriched layers of austenite, nucleate at the austenite grain boundaries and grow as the reaction proceeds (Ref 4). Consequently, primary metastable austenite (γ0) decomposes into bainitic ferrite (αB) and high-carbon austenite (γHC), while the high-carbon austenite phase ideally possesses thermal stability upon cooling to ambient temperature (stage I reaction) (Ref 6). With increasing the austempering time, high-carbon austenite may decompose into bainitic ferrite and carbides (stage II reaction), a microstructure that is commonly referred to as “bainite” (Ref 6). As coarse carbide precipitates embrittle the microstructure and deteriorate the mechanical properties, the second solid-state reaction is undesired (Ref 7). As described by Darwish and Elliot (Ref 8) and Zahiri at al. (Ref 9), the optimum material properties are attained if the austempering treatment is conducted within a processing window which describes the time interval between the end of the stage I and the beginning of the stage II reaction. However, the retained austenite must be stable against martensitic transformation at ambient temperature. Otherwise, only low toughness and ductility will be achieved. The stability of austenite is primarily determined by its carbon content and varies depending on the applied austempering treatment. Sidjanin et al. (Ref 10) classified the stability of austenite based on its carbon content into the following three types: un-reacted metastable austenite (< 1.2 wt.% C), reacted metastable austenite (1.2-1.7 wt.% C), and reacted stable austenite (1.8-2.2 wt.% C). Although all these types can be termed as “retained austenite” (RA), only the last one is desired in ADI.
As already mentioned, one of many advantages of ADI is its ability to be an lightweight alternative material for thin-wall aluminum castings (Ref 2) that are conventionally produced via high-pressure die-casting (HPDC) (Ref 11), an economic and efficient manufacturing process that is particularly applied if components have high requirements on dimensional accuracy but less on surface roughness (Ref 12). In the HPDC process, the molten metal alloy is injected into a permanent metal mold at high speed (> 30 m/s) and solidifies under high pressure (up to 1300 bar) (Ref 13). The application of a permanent mold thereby allows near-net-shape production of components at high productivity. Therefore, HPDC seems to be a reasonable process to produce thin-wall austempered ductile iron (TWADI) components that offer great potential in terms of resource efficiency compared to conventional thick wall ADI. Labrecque et al. (Ref 14) demonstrated that thin-wall ductile iron (TWDI) is generally producible via gravity casting processes (i.e., sand casting). Khodaverdizadeh and Niroumand (Ref 15) studied the effect of pressure on microstructure and mechanical properties of DI produced by squeeze casting. However, making TWDI components with adequate properties after austempering is ambiguous because of several metallurgical and microstructural challenges. On the one hand, high thermal conductivity of the metal mold causes rapid solidification of the melt. For this reason, DI castings tend to develop undesired white structures, chill zones, and/or microporosity (Ref 16). On the other hand, determining the optimum parameters of the austenitization and austempering treatment that allow fully decomposition of carbides/pearlite, satisfying graphitization, as well as formation of ausferrite with thermally stable austenite is difficult due to the large number of factors that influence the microstructural evolution (e.g., chemical composition of the DI, melt treatment (inoculation and Mg treatment), cooling rate, etc. (Ref 4)). For this reason, many studies on the producibility and properties of TWADI have been conducted in the past years. Jafari et al., for instance, studied the influence of metallurgical impact factors, such as the gating system, sand grain size, and mold coatings on microstructural characteristics and found that TWDI is producible, but graphite spheroids tend to be smaller in size and may be formed at a higher volume fraction and nodularity compared to thick-section castings (Ref 16). Other authors found that preheating the mold to a temperature of 300 °C decreases the fraction of carbides and the shrinkage volume and is therefore appropriate for producing TWDI castings in a permanent mold (Ref 17). Fras´ et al. investigated the effect of nodule count on the austenitization/austempering kinetics and mechanical properties of TWDI. They clarified that neither expensive alloying elements (Ni, Cu or Mo) nor long heat treatment times are required to obtain high-strength TWADI (Ref 18). In this context, the effect of grain refining elements (Ti, Nb, and Zr), and inoculation on the formation of primary structures and the graphite morphology in high-Ni-alloyed TWDI was studied by Gorny et al. (Ref 19). The authors showed that the fading impact of grain refinement and inoculation is linked with changes in the morphology and number of austenite dendrites and eutectic grains. Thus, the refining process is unstable and needs rigid metallurgical control (Ref 19). If no refining treatment is applied, the formation of primary columnar austenite grains is dominated during solidification (Ref 19).
Against this background, it becomes clear that the production of TWADI is still a very complex process and more research needs to be done, especially concerning small section components having a thickness less than 10 mm. It is hence the aim of the present work to contribute to this field by answering the following questions:
-
How does HPDC process influence the evolution of microstructural constituents (white structures, graphite) in TWDI (4 mm thickness)?
-
What is the critical cooling rate to avoid pearlite formation upon cooling to the austempering temperature after austenitization?
-
How does an austempering treatment at 280-400 °C influence the evolution of the ausferritic microstructure (bainitic ferrite morphology, RA carbon content)?
For answering these questions, multiple investigations are carried out in the present study. To evaluate the temperature-time-transformation behavior and the microstructural characteristics of TWDI samples produced via HPDC, heat treatment experiments using dilatometric tests are accompanied by microstructural analysis using optical and scanning electron microscopy (OM, SEM), as well as x-ray diffraction (XRD). These experimental studies are supported by computational thermodynamics based on the CALPHAD method, to predict phase stabilities, and the fraction of graphite in equilibrium condition. Finally, tensile and Vickers hardness tests are conducted on samples in the as-austempered state to examine the effect of austempering on the macro hardness, the tensile strength, and the elongation at fracture at room temperature.
Experimental Methodology
Material and Casting Procedure
An unalloyed commercial spheroidal ductile iron of EN-GJS-500-7C (ASTM A536) type (ACO Eurobar GmbH) has been molten in a commercial laboratory melting furnace. The chemical composition of the received material given in Table 1 was measured via optical spark emission spectrometry (OES, QSG 750, OBLF) and combustion analysis (CA, CS744, Leco).
Subsequently to charging, the furnace was superheated to 1500 °C to transfer the solid material into the liquid phase. Contrary to the conventional procedure, the melt has been neither treated with a Fe-Si-Mg alloy (ferrosilicon-magnesium) for spheroidization nor inoculated using a Fe-Si-based alloy to increase the number of graphite nodules. Both treatments have been excluded for two reasons. First, it was aimed to allow an easy handling of the melt to avoid other treatments being carried out during the HPDC process. Secondly, uncertainties in microstructural characteristics resulting from an inhomogeneous introduction of the additives into the melt within a ladle should be avoided. For the sample production, approximately 0.5 kg of the melt were poured into a ladle from which it was poured into the shot chamber of a vertical cold chamber die-casting machine. The pouring temperature was 1180 ± 15 °C. It was measured manually by K-type thermocouples that were immersed into the melt before pouring it into the shot chamber. When starting the HPDC process, the hydraulically operated plunger injected the molten metal through the shot chamber into the mold cavity. The plunger remained forward and held the pressure for 2 s while the casting solidified. Thereafter, the plunger was retracted, and the lamping unit ejected the casting (cluster).
The mold consisted of two halves with a 100 x 83 x 17 mm dimension (length x width x depth). The cavity in the mold included four identical cylindrical samples in the shape of rods (Ø4.25 x 35.35 mm) and was designed considering an overflow barrier. A 3D-CAD model of the mold cavity and the samples’ geometry is shown in Fig. 1. The mold was made up of a commercial hot-work tool steel (Fe-0.45C-5Co-3Mo-2W-0.4Mn-0.4Si) machined in its conventional hardened and tempered condition. Before each processing cycle, the mold cavity has been preheated to 250 °C to decrease thermal stresses in the mold caused by the cyclic temperature changes. Thus, it was aimed to delay the thermal fatigue of the mold. The preheating was realized by oil that acted as fluid flowing through inserted channels inside the mold. In addition, the mold cavity was coated with a commercial boron nitride-based coating (WP suspension, Ekamold®). This coating was applied onto the tool surface before casting to provide an insulating barrier between the liquid and the mold, on one the hand, and to hinder sticking of the casting to the mold after solidification on the other. After removal of the cast specimens, the clusters were cooled down to room temperature at ambient air before they were further processed with heat treatments and metallographic sample preparation processes.

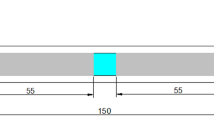
3D model of the mold cavity (a) and the geometry of the rod samples (b); values in mm
After casting, the gating system and the overflow device were cut off the cluster, and the samples were further machined according to Fig. 2(a)-(b). To obtain a proper sample geometry for the dilatometric experiments, samples were deburred and then sectioned in the rod center according to Fig. 2(b). Finally, the surfaces of the samples were ground by SiC grinding paper (80 mesh) to remove the casting skin and to ensure a secure connection of the thermocouple wires to the surface of the samples that were used for the dilatometric tests (Fig. 2c).

Sample preparation stages: (a) removal of samples from the casting (cluster), (b) sample condition after separation, (c) final sample condition (dilatometric sample)
Heat Treatment Experiments
Graphitization
To examine the evolution of graphite particles regarding their fraction and morphology depending on the dwell time, the as-cast rod samples (Fig. 2b) were subjected to a graphitization treatment at a constant temperature of 1100 °C under an inert argon atmosphere in a laboratory muffle furnace (LM-312.27, LINN HIGH TERM). The graphitizing temperature (TG) is significantly higher compared to conventional austenitizing temperatures of about 950 °C (Ref 20). Setting a higher temperature had two reasons. Firstly, higher temperatures provide a higher diffusion rate of carbon, which promotes the decomposition of undesired cementite being present in pearlite and ledeburite that potentially exist in the as-cast condition. Secondly, it was aimed to accelerate the graphitization kinetics by increasing the diffusivity of carbon. Lastly, austenitizing at high temperatures increases the carbon content in austenite (Ref 20), which is beneficial for achieving an ausferritic microstructure with thermally stable retained austenite. For the analysis of the graphite formation kinetics, the samples were graphitized at different graphitizing times (tG) from 40 s to 30 min, directly followed by quenching in water (25 °C) to suppress the formation of graphite and/or pearlite during the cooling process. With this method, reliable quantification of the graphite fraction formed during graphitization and same microstructural properties of the samples prior to the dilatometric tests could be ensured.
Dilatometry
Dilatometric tests were performed on a quenching dilatometer (DIL805, TA Instruments) to analyze the time–temperature transition behavior by recording a continuous cooling transformation (CCT) diagram. For these experiments, graphitized samples (1100 °C/30 min/water quenched) were used. The cylindrical samples (Ø4.00 ± 0.15 x 10.00 ± 0.10 mm) (Fig. 2c) were connected with K-type thermocouples by point welding. In this way, the samples’ temperature could be controlled and verified during the entire testing procedure. At the beginning of the tests, the samples were austenitized at 1100 °C for 1 min and subsequently cooled exponentially to ambient temperature by applying various cooling rates, respectively, t8/5 cooling times. The t8/5 cooling time is defined as the time interval it takes the sample to cool from 800 to 500 °C (Ref 21). It was set to values between 1 and 240 s and realized by the controlled introduction of inert helium gas into the testing chamber (Fig. 3a). The continuous record of the dilatation of the sample during the heat treatment allowed the identification of characteristic phase transition temperatures such as the martensite start (MS) temperature and/or the pearlite start/finish temperature (PS, PF). These critical temperatures were determined according to (Ref 22) using the tangent method technique whereby tangents are applied to the dilatation curve near the beginning and the end of the phase transformation. Based on the characteristic transition temperatures, it was possible to derive a CCT diagram from which essential criteria such as the beginning of the austenite/martensite transition, and the critical cooling time (t8/5,crit) to avoid pearlite formation upon cooling could be drawn.

Schematic time–temperature profiles used for dilatometric experiments: (a) continuous cooling treatments, (b) isothermal austempering treatments
In addition to the continuous cooling experiments, isothermal austempering treatments were conducted at various austempering temperatures (TA) between 280 and 400 °C to determine the impact of the austempering treatment on the bainitic reaction. For this, the samples were rapidly cooled from the austenitizing temperature in (200 K/s, t8/5 = 1 s), and then held at TA for an austempering time (tA) of 40 min. The following cooling to room temperature was carried out at a constant cooling rate of 2 K/s (Fig. 3b).
Computational Thermodynamics
To predict the stability of phases and their fraction in the equilibrium condition, thermodynamic calculations using the CALPHAD method were performed. For these calculations, the commercial Thermo-Calc® software (version 2020b) with the TCFE10 database was applied.
Microstructural Characterization
Metallographic Sample Preparation
The microstructural characterization of the graphitized and austempered samples was carried out in the middle section, as shown in Fig. 2(b). The samples were separated using a wet abrasive cutting machine. The same applies to samples from the dilatometric experiments cut in a transverse direction for the microstructural characterization. For metallographic analysis, all samples were mounted in conductive resin and the surface to be investigated was metallographically prepared by grinding with SiC abrasive paper (80, 220, 500, 1000 mesh), and polishing using diamond suspension with different grain sizes (6, 3, 1 µm). The polished surfaces were contrasted by etching using 4% Nital (96-98 ml ethanol, 2-3 ml nitric acid (HNO3)).
Phase Analysis
As a comparative study to the dilatometric tests, the microstructural constituents that have emerged during continuous cooling and isothermal austempering were quantified by microstructural analysis using optical microcopy (OM, DM2700M, Leica) and scanning electron microscopy (SEM, Tescan Vega3 SBH). Phase fractions of graphite, pearlite, and ferrite were quantified by means of optical image analysis based on ten optical micrographs that were recorded in the as-etched condition at 200-fold magnification. Contrast and threshold adjustments of the images were set within the open-source “ImageJ” software (version 1.48), which was also used to quantify phase area fractions. Contrary to phases like pearlite and ferrite, graphite particles were visually investigated regarding their morphology and nodularity. Therefore, ten optical micrographs were taken at a 500-fold magnification of un-etched samples at random positions. The shape of the graphite particles was classified, and the nodularity (fraction of spheroidal graphite particles) was calculated according to the standards EN ISO 945-1 (Ref 23) and ISO 16112 (Ref 24). The required number of particles of the different roundness classes could be determined within the “ImageJ” software, as well as the particles’ average Feret diameter (\({d}_{\mathrm{Feret}}=2\sqrt{A/\pi }\)) that corresponds to a circle with the same area A as the particle. Regarding the quantification of the graphite fraction, it must be noted that small particles having dFeret ≤ 3 µm were not considered in the analysis according to Khodaverdizadeh and Niroumand (Ref 15). As surface defects appear as graphite in the optical micrograph, it is challenging to distinguish graphite particles from imperfections on the surface. For this reason, the authors suggested to neglect the particle fraction < 3 µm to increase the reliability of the measurement (Ref 15). The morphology of retained austenite and bainitic ferrite was investigated based on high-resolution SEM images that were recorded in the SE (secondary electron) mode at 15.000-fold magnification in the as-austempered condition. The SEM was operated with an acceleration voltage of 10 kV, a working distance of 8 mm, and a beam spot size of 30 nm.
Carbon Content of Austenite and Ferrite Cell Size
The carbon content in the initial austenite (\({C}_{\gamma }^{0}\)) prior to the austempering treatment was calculated based on the measured silicon content (in wt.%), and the applied graphitizing temperature TG (in °C) from the following equation (Ref 7):
The austenite carbon content after the isothermal austempering treatment at different austempering temperatures was calculated by Eq 2 (Ref 25)
where aγ is the lattice parameter of austenite (in nm) and Cγ is the carbon content of austenite (in wt.%). The lattice parameter was determined by XRD analysis based on the (111), (200), and (220) planes of austenite that were measured on polished samples. The used diffractometer (Bragg–Brentano geometry, D2 Phaser, Bruker) was equipped with a Cu radiation source (λKα1 =1.54059 Å, λKα2 = 1.54449 Å) and operated with a current of 10 mA and a voltage of 30 kV. The measurements were taken in a range of 30° to 80° 2θ, with a scanning interval of 0.02 ° 2θ, and a scanning time of 1 s per increment at a temperature of 35 °C. Meanwhile, the sample rotated horizontally at a speed of 10 rpm. Based on the measured diffractograms, the ferrite cell size (d) was derived from the Scherrer equation (Ref 26)
where λ is the wavelength, β is the width of the (100) reflection of ferrite in radians (full width at half maximum, FWHM), and θB is the Bragg angle.
Mechanical Tests
Mechanical tests by means of Vickers hardness tests were performed on a standard Vickers hardness testing machine (Carat 930, ATM) according to DIN EN ISO 6507 (Ref 27). The tests were carried out to evaluate the macro hardness of the samples after continuous cooling and after isothermal austempering. At least five indents were taken in the center zone of the rod samples with a load of 98,1 N (HV10). In addition to the hardness, the tensile strength (Rm), as well as the elongation at fracture (ε), has been measured by tensile tests on a universal tensile testing machine (Sun5, Galdabaini) using tensile test samples having a geometry displayed in Fig. 4. For testing, the samples were loaded with a pre-load of 20 N before a constant strain rate of 0.33 mm/s was applied until fracture. All tests have been performed at room temperature (~25 °C). Average mean values were calculated based on five measurements per sample and heat treatment state.

Schematic sketch of the tensile test sample (values in mm)
Results and Discussion
As-Cast Microstructure
Microstructural analysis was performed on as-cast samples to determine the microstructural constituents and potential casting defects resulting from the HPDC process. Figure 5 shows the optical micrographs of the cross section of a representative sample in various magnifications. At first glance, it becomes apparent that the samples possess microstructural inhomogeneities and typical defects such as pores and cavities in the core region (Fig. 5a). While pores result from gas inclusions caused by the turbulent flow of the melt, cavities in the core region indicate shrinkage of the material during solidification (Ref 28). The shrinkage defects may result from the high undercooling of the melt that is caused by the relatively low temperature and the high thermal conductivity of the used steel mold. The high thermal conductivity accelerates the heat transfer from the solidified outer shell of the sample into the mold and thus increases the solidification rate of the liquid. Since, in contrast to gravity die-casting, no feeders are used in the HPDC process, cavities formed during solidification cannot be closed by subsequent flowing material. Hence, shrinkage cavities may remain in the center of TWDI samples produced via HPDC. Furthermore, inhomogeneities between the microstructures near the surface and in the interior region can be observed, separating the microstructure into two different zones, namely the outer zone, which is 159 ± 22 µm thick, and the center zone representing the core region (Fig. 5b-c). The main difference between these zones is the degree of graphite formation, and the amount of the formed retained austenite (RA). In the outer zone, the microstructure consists of tiny chunky graphite particles that are dispersed in a white ledeburitic microstructure. In contrast, in the center zone, there is a noticeable high number of RA regions. The existence of an outer shell of ledeburite can be traced back to the very high undercooling of the liquid, which suppresses carbon diffusion and thus its ability to form graphite during solidification.

Optical micrographs of a representative sample in the as-cast condition after HPDC process: (a) cross-sectional segment, (b) near-surface region, (c) magnified near-surface region according to (c), (d) center zone microstructure
It is further known that the amount of graphite decreases as the external pressure increases (Ref 21), which is why cast iron produced via HPDC generally has a higher risk for having white structures. Considering the hypoeutectical solidification sequence, which can be derived from its carbon equivalent and the calculated phase diagram (Fig. 6a), it becomes clear why the primary formation of austenite dendrites is favored in this material. The high undercooling of the liquid causes precipitation of the metastable cementite (Fe3C) instead of the stable graphite. In the as-cast condition, the castings correspond to a white casting instead of conventional ductile iron (Ref 20). Nevertheless, the presence of tiny graphite particles near the surface and in the core region of the samples indicate some possibility for graphite particles to form during solidification. The formed microstructure agrees with the findings of Stefanescu et al. (Ref 29), who demonstrated that in low-carbon gray cast iron, graphite nucleates at the austenite/liquid interface without the presence of any foreign inclusions. Apart from ledeburite, it is also noticed that high-carbon-enriched regions can be found as RA predominantly in the center zone. Their presence implies that the formation of graphite spheres was initially intended but then prevented by the rapid cooling and/or the low amount of carbon in the matrix. Either way, the RA regions must possess low amount of carbon because of their low thermal stability, derived from the partial conversion of RA into martensite (Fig. 5d). Therefore, the casting obtained by the applied HPDC process can be described as a RA/martensite containing white iron, which is expected to be brittle. However, it is worth noting that the samples did not fracture during the ejection process out of the mold, which means that possibility to produce TWDI components via the applied HPDC process is given.

Thermodynamic calculations on EN-GJS-500-7: (a) equilibrium phase diagram, (b) equilibrium phase fraction as a function of graphitizing temperature (TG); the dashed line in b) marks the applied TG = 1100 °C
Effect of Graphitization on Graphite Formation
The influence of the graphitization treatment on the graphite formation kinetics was evaluated based on the optical micrographs displayed in Fig. 7.

Optical micrographs of polished samples in the as-quenched condition after graphitization (TG = 1100 °C) at different graphitizing times (tG): (a) 40 s, (b) 2 min, (c) 4 min, (d) 10 min, (e) 20 min, (f) 30 min
It is revealed that subjecting the material to a graphitization treatment at 1100 °C forces graphite particles to form very quickly. As illustrated in Fig. 8(a), short graphitizing times of only 40 s are sufficient to ensure a high graphite fraction (7.0 ± 0.3 vol.%). With increasing graphitizing time, the graphite fraction slightly differs until it remains constant at a time of 25-30 min. The graphite fraction measured in this condition is close to the fraction predicted from thermodynamic calculations (6.8 vol.%, Fig. 6b) which makes it possible to assume that the system has reached its equilibrium condition. The fast graphitization of the material indicates that annealing at 1100 °C allows carbon to have high diffusion rates over wide ranges whereby the microstructure moves toward equilibrium quickly. With increasing dwell times, the graphite particles grow continuously from 5.7 ± 0.7 to 7.0 ± 0.4 µm. Nevertheless, compared to conventionally produced thin and thick wall DI castings (3-60 mm thickness) where graphite nodules may reach diameters of 20-60 µm (Ref 30) or even up to 120 µm (Ref 31), the graphite particles observed in this investigated TWDI material are very small.

Graphite analysis: (a) area fraction of graphite in the as-quenched condition and nodularity and size (dFeret) of graphite (b) as a function of the graphitizing time (tG at 1100 °C); the dashed line in (a) indicates the theoretical volume graphite fraction calculated via Thermo-Calc® (see Figure 6)
Figure 8 shows that the maximum nodularity value is only about 12.9 ± 0.7% after graphitization for 30 min. This value is much lower compared to those in standard DI where round particles (shape VI according to EN ISO 945-1 (Ref 23)) and nodularities of at least 80-85% are desired not to impair the mechanical properties (Ref 32, 33). The present graphite particles have somewhat different shapes. They can be classified into the group of vermicular, chunky, exploded, and partly spheroidal graphite (shapes III-VI), which typically appear in malleable cast iron where graphite particles grow isolated within the matrix (Ref 23). The low number of acceptable spheroids is the reason for the low nodularity. It can be explained by the missing melt treatment with elements such as Mg and/or Ce that are conventionally used to promote the crystallization and spheroidization of graphite in the liquid phase (Ref 34). Impurity elements have a strong influence on the nucleation of graphite, as they form sulfides, oxides, or nitrides, which act as nuclei (Ref 34). Riposan et al. (Ref 35) defined a three-stage model for nucleation of graphite in gray cast iron and described the nucleation of graphite particles on complex sulfides with diameters < 5 µm that have been nucleated on small oxides with diameters < 2 µm. Since the melt has not been treated with inoculants within the present study, it is not surprising that graphite's nodularity is much lower than that in inoculated spheroidal cast irons.
TTT Behavior and Critical Cooling Time
Different microstructures were obtained based on dilatometric experiments depending on the applied cooling time (t8/5) after graphitization at 1100 °C for 30 min. As depicted in Fig. 9(a), cooling to ambient temperature at a high cooling rate (t8/5 = 1 s) results in the formation of hard platelike martensite (α’) with some blocky and film-like RA between the plates. The measured MS temperature is 193 ± 14 °C, which means that the material should be austempered above this temperature to avoid martensite transformation of the metastable austenite. However, the MS temperature is about 30 K below that of a comparable DI material having 3.7 wt.% C, 2.3 wt.% Si, and 0.25 wt.% Mn reported in the literature (Ref 21). The slight decrease may be explained by the unconventionally high austenitizing temperature of 1100 °C that allows higher amount of carbon to be dissolved in the austenitic matrix. As carbon is a strong austenite stabilizer, it gains higher stability against martensitic transformation with increasing carbon content. Thus, the MS temperature drops to lower values. Apart from the effect of the austenitizing temperature on the carbon dissolution in austenite, the measured MS temperatures indicate less carbon being dissolved in austenite after austenitization as expected from empirical calculations. Based on Eq 1, the initial carbon content of austenite (\({C}_{\gamma }^{0}\)) is calculated to be about 1.28 wt.% which seems to be too high with respect to the measured MS temperature of 193 ± 14 °C. This enables the assumption that the empirical equation used (Eq 1) is not suitable for an adequate derivation of \({C}_{\gamma }^{0}\) if very high austenitizing temperatures are involved. This would explain why other authors such as Uhrenius supposed the austenitizing temperature to be within a range of 850-1100 °C to obtain reasonable \({C}_{\gamma }^{0}\) values (Ref 36). Nevertheless, there are other possibilities to estimate \({C}_{\gamma }^{0}\) regardless of the applied austenitizing temperature. Bayati et al. (Ref 37) presented an empirical relationship for the MS temperature (Eq 4) based on the study of Rounds and Rundman (Ref 38), who linked martensite-start and martensite finish temperatures (in °C) by an isopleth through the phase diagram of the Fe-C-Si system (2.5 wt.% Si). According to Eq 4, \({C}_{\gamma }^{0}\) is expected to reach values between 0.74-0.85 wt.%, which are significantly lower than the value calculated by Eq 1.
However, verification of these values is difficult as measurements concerning \({C}_{\gamma }^{0}\) have not been within the scope of the present study. Kawulok et al. (Ref 39), though, recently investigated the effect of strain on the transformation diagram of a hypereutectoid high-carbon low-alloyed steel with 0.99 wt.% C, 0.38 wt. Mn, 0.32 wt.%, 1.45 wt.% Cr (100Cr6). The authors determined critical transformation temperatures via quenching dilatometry. They showed that the MS temperature of the steels’ matrix is about 211 °C considering a full dissolution of carbides during austenitization and quenching at very high cooling rate (150 K/s). As the reported MS value is in good agreement with the average MS temperature measured in this study, the actual initial carbon content of austenite can be expected to be within the range of the calculated carbon contents of 0.85 wt.% and 1.28 wt.% C.

Optical micrographs of the microstructure after continuous cooling at different cooling times (t8/5): (a) 1 s, (b) 5 s, (c) 8 s, (d) 30 s, (e) 60 s, (f) 240 s; as-cast (HPDC) samples were graphitized at 1100 °C for 30 min prior to each cooling step
Apart from the initial amount of carbon in austenite, it is essential to note that an almost complete martensitic microstructure is achievable by applying cooling times of up to t8/5 = 4 s while slightly slower cooling causes microstructural changes. As shown in Fig. 9b, cooling at t8/5 = 5 s is insufficient to reach an almost complete martensitic matrix. Instead, the formation of Widmanstätten ferrite (αW) and pearlite becomes visible in the optical micrograph. Observing the microstructure at higher magnification reveals that “pearlite islands” remain together with proeutectoid ferrite (allotriomorphic ferrite (Ref 20)) along the former austenite grain boundary near the small graphite particles (Fig. 10).

SEM micrograph of the microstructure in the as-quenched condition after cooling at the critical cooling time (t8/5,crit = 5 s)
The transition of austenite into Widmanstätten ferrite can be interpreted as a pre-stage of the bainitic reaction as both have a common nucleation mechanism (Ref 20). According to Bhadeshia, Widmanstätten ferrite forms if the driving force is insufficient to sustain diffusionless growth at a specific temperature (the temperature at below which the nucleation of displacive transformations first becomes detectable) (Ref 20). If the driving force is sufficient, then the austenite converts into bainitic ferrite, and Widmanstätten ferrite does not form at all (Ref 20). Nevertheless, as both phases, Widmanstätten ferrite and pearlite must be avoided, the cooling time of t8/5 = 5 s can be stated as the critical cooling time (t8/5,crit) that should not be exceeded within technical quenching processes. This becomes obvious when looking at the microstructures obtained after slower cooling (t8/5 = 8 s). Here, the transition of austenite into Widmanstätten and/or bainitic ferrite, and pearlite continues while the fraction of martensite decreases (Fig. 9c). Ferrite can be seen to have formed around graphite nodules, leading to the well-known “bull’s-eye” appearance of graphite (Ref 21). By lowering the cooling rate further (t8/5 > 8 s), the undercooling of the austenite decreases continuously, resulting in neither Widmanstätten/bainitic ferrite nor martensite being found in the microstructure. Instead, austenite decomposes into pearlite and ferrite (α) while graphite particles become bigger with increasing t8/5 values and decreasing cooling rates (Fig. 9d-f). This behavior can be explained by the longer diffusion time with decreasing undercooling of austenite which allows carbon to diffuse over wide ranges toward graphite particles to reach the equilibrium condition. However, a higher diffusion rate of carbon goes along with a depletion of carbon in these regions resulting in a higher ferrite fraction around graphite nodules (Fig. 9f).
So, based on the microstructural analysis and the investigation of the dilatation curves, a continuous cooling transformation (CCT) diagram can be constructed according to Fig. 11 where all phase regions, the critical cooling time (t8/5,crit) to avoid formation of pearlite, as well as the average MS temperature are being highlighted.

Continuous cooling transformation (CCT) diagram of EN-GJS-500-7 produced via HPDC process
Effect of Isothermal Austempering on Microstructural Characteristics and Mechanical Properties
Changes in the carbon content of austenite (Cγ), the ferritic cell size (d), and the global hardness were measured after isothermal austempering at temperatures between 280 and 400 °C for 40 min (Table 2). The corresponding changes in the microstructure are shown in Fig. 12. Figure 12(a-b) shows the OM and SEM micrographs of the microstructure after austempering at 280 °C for 40 min. Austempering at low temperatures results in an ausferritic microstructure with very fine sheaves of bainitic ferrite (αB) that are known to form in the lower-bainite region (Ref 20). The reason for the appearance of the fine bainitic ferrite sheaves is the high undercooling of the austenite that generates a high driving force for the bainitic reaction and a large number of bainitic ferrite nuclei (Ref 20).

Optical micrographs (a,c,e,g) and SEM micrographs (b,d,f,h) after austempering at different temperatures (TA at 40 min): (a-b) 280 °C, (c-d) 320 °C, (e-f) 360 °C, (g-h) 400 °C
As the growth of a bainitic ferrite sheave is considered diffusionless, the metastable austenite converts into carbon supersaturated bainitic ferrite platelets without diffusion of carbon between austenite and ferrite (Ref 20). However, subsequently to this transition, the excess of carbon in the bainitic ferrite is partitioned into the surrounding austenitic matrix, whereby the latter will be chemically stabilized against martensitic transformation (Ref 20). The thermal stability of RA is the main criterion when high ductility is desired, but it differs depending on the RA morphology. As shown in Fig. 12(b), the present RA shows two morphologies, a film-like, and a blocky shape. While the film-like RA is trapped between the parallel platelets of bainitic ferrite, the blocky RA is randomly distributed in the matrix. As indicated in the micrograph in Fig. 12(a)-(b), the RA blocks are not thermally stable, as they have partially transformed into untempered high-carbon martensite (α’) upon cooling to ambient temperature. Martensite plates tend to be very thin and can be therefore readily distinguished from bainite (Ref 20). The presence of martensite can be explained by differences in the carbon concentration of film-like and blocky RA. It is known that films of austenite will be enriched by carbon at a higher concentration than untransformed blocky RA, because the diffusion distance from the bainitic ferrite sheave to the surrounding austenite films is much shorter (Ref 40). RA films located within bainitic sheaves refer to high-carbon austenite, while RA blocks correspond to low-carbon austenite (Ref 40).
On the other hand, it is indicated in the dilatation curves (Fig. 13a) that the stage I reaction is still ongoing as the length continuously increases and has not reached a plateau level yet, which corresponds to the processing window. Taken the measured low RA carbon content of 1.44 ± 0.01 wt.% into account (Table 2), it becomes clear that the stage I reaction has not been completed within 40 min. Thus, reacted metastable austenite must be present in the microstructure. As a result, the ausferritic matrix exhibits martensite traces that provide a high hardness (479 ± 17 HV10) but is expected to exhibit low ductility during mechanical loading.

Effect of the austempering temperature (TA) on the bainitic ferrite formation kinetics and the reflection peaks in the x-ray diffractogram: (a) dilatation curves, (b) x-ray diffraction patterns; the dots in a) mark the end of the stage I reaction
Increasing the austempering temperature to 320 °C increases the relative amount of bainitic ferrite aggregates and reduces blocky RA. Although the sheaves of bainitic ferrite show only a slight difference in their shape compared to the ausferritic microstructure obtained at 280 °C (Fig. 12d), their size increased from 27 ± 3 to 36 ± 3 µm (Table 2). Likewise, the RA carbon content increased to 1.56 ± 0.05 wt.% because of the better redistribution of carbon at a higher temperature. Based on the measured carbon content, martensite formation upon cooling could be expected, especially since the stage I reaction has not been completed yet (Fig. 13a). However, martensite plates cannot be distinguished in the micrographs (Fig. 12c-d) but are indicated to be present because of the high hardness of 434 ± 5 HV10 (Table 2).
Isothermal austempering at 360 °C causes further structural changes of the bainitic ferrite morphology and of the RA stability. Compared to the sheaves of bainitic ferrite formed at 320 °C, the sheaves formed at 360 °C appear coarser and show a feathery shape that can be linked to the bainitic sheaves that form in the upper bainite region (Ref 20). As a result, the ferritic cell size increases to 45 ± 2 µm. Like the different morphologies of bainitic ferrite, RA can also be found in different shapes, such as films and blocks, between the single ferrite platelets. In contrast, no evidence of martensite can be found within blocky RA, which correlates with the higher RA carbon content of 1.60 ± 0.02 wt.% measured via XRD analysis and the lower hardness of 355 ± 4 HV10 (Table 2).
Regarding the transformation kinetics, the dilatation curves show that increasing the austempering temperature accelerates the stage I reaction as the time needed to reach the processing window significantly decreases. The desired plateau is reached at an austempering time of only 2000 s (33.3 min) and is decreased to 1000 s (16.6 min) at an austempering temperature of 400 °C (Fig. 13a). However, as Fig. 12(g)-(h) reveals, when increasing the austempering temperature to 400 °C, the relative amount of bainitic ferrite decreases, and conversely to the previously described condition, only blocky RA may be found in the ausferritic microstructure. As all the bainitic ferrite sheaves exhibit a coarse feathery shape, it can be concluded that austempering at 400 °C results in the formation of purely upper bainite structures with a ferritic cell, which is about twice as large as the those that form during austempering at 280 °C (Table 2). Although the amount of bainitic ferrite and RA can be significantly increased by raising the austempering temperature from 360 to 400 °C, the average carbon content in RA remains unaffected. As given in Table 2, the RA carbon content only slightly increases (1.61 ± 0.01 wt.% C). This content is consistent with the carbon content values reported by the study of Bayati and Elliot (Ref 37) and Mallia et al. (Ref 41), who showed that the amount of carbon in RA increases from 0.9-1.1 wt.% up to 1.5-1.8 wt.% C within 10-100 min during austempering at 400 °C . It is also noticed that no carbide precipitates can be found in the microstructure and/or in the XRD pattern given in Fig. 13(b), which means that the processing window is not closed at an austempering time of 40 min. In other words, the stage II reaction has not yet been induced. It is hence possible to assume that the RA owes its highest carbon content to this condition which would explain the absence of martensite. So, based on the presented results, it is concluded that austempering at 360-400 °C provides the maximum carbon content in RA and is suitable to achieve an ausferritic microstructure with thermally stable RA within a short austempering time of 40 min. However, in terms of hardness, it is preferable to set the austempering temperature to 360 °C instead of 400 °C, as higher temperatures lead to higher fraction of blocky RA that, in turn, decreases the global hardness from 355 ± 4 HV10 to 324 ± 5 HV10.
To examine the impact of austempering on the mechanical strength and the ductility of the TWADI material, the tensile strength (Rm) and the elongation at fracture (ε) were determined in the as-cast and the as-austempered (360 °C/40 min) condition at room temperature. Based on the material's microstructure in the austempered condition (see Fig. 12(e-f)) it can be assumed that having dispersed graphite particles with smaller size within the matrix may be beneficial in terms of the mechanical strength of the material. Because graphite in cast iron is treated as a void with almost no strength, graphite particles decrease the load carrying area thereby causing stress concentration at the graphite/matrix interface that favors microcracking at these areas (Ref 42). Fewer and smaller graphite particles increase the free path of the matrix so that it should be able to compensate higher mechanical stresses during tensile loading. Consequently, the negative impact of graphite on microcrack initiation is expected to be lower in HPDC-produced TWADI samples compared to those that are conventionally produced via gravity die-casting (GDC). However, the measured tensile strength and elongation at fracture reveal that the opposite is the case, actually (see Table 3). Although austempering of the HPDC-produced TWDI samples at 360 °C for 40 min allows an increase in strength (Rm = 709 ± 65 MPa) and ductility (ε = 3 ± 1%) compared to the as-cast condition (Rm = 183 ± 21 MPa; ε = 1 ± 0%), the measured values are below those of TWADI material that has been produced via GDC process (Rm = 878 ± 46 MPa; ε = 8 ± 1%). It seems reasonable to assume that this may be explained by the number of casting defects (see Fig. 5) expected to be significantly lower in the GDC processed TWADI samples. Since the GDC process is characterized by a slow casting speed and the presence of feeder systems, it is assumed that GDC processed TWADI samples exhibit much lower density of cavities and/or gas porosity whereby higher tensile strength and ductility may be achieved. Therefore, further studies should focus on reducing microporosity in HPDC-produced samples to obtain TWADI parts with comparable strength and ductility.
Conclusion
The present study aimed to evaluate the austemperability of thin-wall ductile iron (TWDI) components (4 mm thickness) made up by a commercial ductile iron (EN-GJS500-7) that were produced by high-pressure die-casting (HPDC). Microstructural characteristics such as the graphitization kinetics, the formation of pearlite on continuous cooling, as well as the effect of the austempering temperature on the evolution of ausferrite were investigated. Based on the presented results, the following conclusions can be drawn:
-
TWDI components can be produced by HPDC process. In the as-cast condition, the microstructure consists of thermally unstable retained austenite (RA) and a predominantly white ledeburitic/martensitic matrix. Microporosity and small shrinkage cavities can be found near the surface and in the core region.
-
Graphitization of the HPDC as-cast condition at 1100 °C allows rapid formation of graphite within a short period of time (40 s). If no melt treatment has been proceeded, graphite particles remain very small (d < 10 µm) and possess low nodularity (< 10%) after graphitizing for 30 min.
-
The critical cooling time (t8/5) to avoid pearlite formation upon cooling after austenitization was found to be 5 s at a martensite start (MS) temperature of 193 ± 14 °C.
-
Austempering at 360-400 °C for 40 min results in an ausferritic microstructure while an optimum combination of high hardness (355 ± 4 HV10) and thermally stable carbon-enriched RA (Cγ = 1.60 ± 0.02 wt.%) is achieved at 360 °C. However, the tensile strength (Rm = 709 ± 65 MPa) and elongation at fracture (ε = 3 ± 1%) achieved are lower compared to TWADI produced via gravity die-casting (Rm = 878 ± 46 MPa, ε = 8 ± 1%).
References
M. Bahmani, R. Elliott, and N. Varahram, Austempered Ductile Iron: a Competitive Alternative for Forged Induction-hardened Seel Crankshafts, Int. J. Cast Metals Res., 1997, 9(5), p 249–257. https://doi.org/10.1080/13640461.1997.11819666
D.M. Stefanescu, Lightweight Iron Castings: Can They Replace Aluminum Castings?, Foundryman, 2003, 96, p 221–224.
J.R. Keough, Austempered Ductile Iron (ADI)–A Green Alternative, America Foundry Society Applied Process Inc. Technologies Division, Livonia, 2011.
A. Trudel and M. Gagné, Effect of Composition and Heat Treatment Parameters on the Characteristics of Austempered Ductile Irons, Can. Metall. Q., 1997, 36(5), p 289–298. https://doi.org/10.1179/cmq.1997.36.5.289
E. Dorazil, B. Barta, E. Munsterova, L. Stransky, and A. Huvar, High Strength Bainitic Ductile Cast Iron, AFS Int. Cast Metals J., 1982, 22, p 52–62.
R.B. Gundlach and J.F. Janowak, Development of a Ductile Iron for Commercial Austempering, AFS. Trans, 1983, 94, p 377–388.
R.C. Voigt and C.R. Loper, Austempered Ductile Iron-process Control and Quality Assurance, J. Heat Treat., 1984, 3, p 291–309. https://doi.org/10.1007/BF02833124
N. Darwish and R. Elliott, Austempering of Low Manganese Ductile Irons, Mater. Sci. Tech., 1993, 9(7), p 572–585. https://doi.org/10.1179/mst.1993.9.7.572
S.H. Zahiri, E.V. Pereloma, and C.H.J. Davies, Application of Bainite Transformation Model to Estimation of Processing Window Boundaries for Mn-Mo-Cu Austempered Ductile Iron, Mater. Sci. Tech., 2001, 17(12), p 1563–1568. https://doi.org/10.1179/026708301101509610
L. Sidjanin, M. Novovic, and R.E. Smallman, Metallographic Investigations of ADI Materials Metallographische Charakterisierung von ADI-Werkstoffen, Pract. Metall., 1996, 33(1), p 2–16.
M.S. Dargusch, G. Dour, N. Schauer, C.M. Dinnis, and G. Savage, The Influence of Pressure During Solidification of High Pressure Die Cast Aluminium Telecommunications Components, J. Mater. Proc. Techn., 2006, 180(1–3), p 37–43. https://doi.org/10.1016/j.jmatprotec.2006.05.001
L.X. Kong, F.H. She, W.M. Gao, S. Nahavandi, and P.D. Hodgson, Integrated Optimization System for High Pressure Die Casting Processes, J. Mater. Proc. Techn., 2008, 201(1–3), p 629–634. https://doi.org/10.1016/j.jmatprotec.2007.11.250
G. Kaufman and E.L. Rooy, Aluminum Alloy Castings: Properties Processes and Applications, ASM International, Materials Park, 2014.
C. Labrecque, M. Gagné, A. Javaid, and M. Sahoo, Production and Properties of Thin-wall Ductile Iron Castings, Int. J. Cast Metals Res., 2003, 16(1–3), p 313–317. https://doi.org/10.1080/13640461.2003.11819601
H. Khodaverdizadeh and B. Niroumand, Effects of Applied Pressure on Microstructure and Mechanical Properties of Squeeze Cast Ductile Iron, Mater. Des., 2011, 32, p 4747–4755. https://doi.org/10.1016/j.matdes.2011.06.040
H. Jafari, M.H. Idris, A. Ourdjini, M. Karimian, and G. Payganeh, Influence of Gating System, Sand Grain Size, and Mould Coating on Microstructure and Mechanical Properties of Thin-Wall Ductile Iron, J. Iron. Steel Res. Int., 2010, 17(12), p 38–45. https://doi.org/10.1016/S1006-706X(10)60195-1
K.A. Jafar and A.-A. Behnam, Influence of Mold Preheating and Silicon Content on Microstructure and Casting Properties of Ductile Iron in Permanent Mold, J. Iron. Steel Res. Int., 2011, 18(3), p 34–39. https://doi.org/10.1016/S1006-706X(11)60034-4
E. Fras, M. Gorny, E. Tyrala, and H. Lopez, Effect of Nodule Count on Austenitising and Austempering Kinetics of Ductile Iron Castings and Mechanical Properties of Thin Walled Iron Castings, Mater. Sci. Technol., 2012, 28(2), p 1391–1396. https://doi.org/10.1179/1743284712Y.0000000088
M. Górny, M. Kawalec, G. Sikora, E. Olejnik, and H. Lopez, Primary Structure and Graphite Nodules in Thin-Walled High-Nickel Ductile Iron Castings, Metals, 2018, 8(8), p 649. https://doi.org/10.3390/met8080649
H.K.D.H. Bhadeshia, Bainite in Steels: Theory and Practice, 3rd ed. CRC Press, Baco Raton, 2015.
H. Berns and W. Theisen, Ferrous Materials–Steel and Cast Iron, 4th ed. Springer, Berlin, 2010, p 38. https://doi.org/10.1007/978-3-540-71848-2
Standard practice for quantitative measurement and reporting of hypoeutectoid carbon and low-alloy steel phase transformations. A1033-10, ASTM International, 2015
DIN EN ISO 945-1:2019-19, Mikrostruktur von Gusseisen – Teil 1: Graphitklassifizierung durch visuelle Auswertung (ISO 945-1:2019), Beuth-Verlag, 2019, in German, https://dx.doi.org/https://doi.org/10.31030/3091837
ISO 16112:2017-02, Gusseisen mit Vermiculargraphit – Klassifikation, Beuth-Verlag, 2017, in German
C.S. Roberts, Effect of Carbon on the Volume Fractions and Lattice Parameters of Retained Austenite and Martensite, Trans. AIME, 1953, 197, p 203.
B.D. Cullity, Elements of X-Ray Diffraction, 2nd ed. Addison- Wesley, Reading, 1978, p 102
DIN EN ISO 6507-1:2018-07, Metallische Werkstoffe – Härteprüfung nach Vickers – Teil 1: Prüfverfahren (ISO 6507-1:2018), Beuth-Verlag, 2018, in German, https://dx.doi.org/https://doi.org/10.31030/2778746
T. Kanno, Effect of Pouring Temperature, Composition, Mould Strength and Metal Flow Resistance on Shrinkage Cavities in Spheroidal Graphite Cast Iron, Int. J. Cast Metals Res., 2008 https://doi.org/10.1179/136404608X361567
D.M. Stefanescu, G. Alonso, P. Larranaga, and R. Suárez, On the Stable Eutectic Solidification of Iron-Carbon-Silicon Alloys, Acta Mater., 2016, 103, p 103–114. https://doi.org/10.1016/j.actamat.2015.09.043
S. Bockus and G. Zaldarys, Influence of the Section Size and Holding Time on the Graphite Parameters of Ductile Iron Production, Metalurgija, 2009, 48(1), p 19–22.
B. Domeij, J.C. Hernando, and A. Diószegi, Size Distribution of Graphite Nodules in Hypereutectic Cast Irons of Varying Nodularity, Metall. Mater. Trans. B, 2018, 49, p 2487–2504. https://doi.org/10.1007/s11663-018-1274-z
J. Friess, U. Sonntag, and I. Steller, From Individual Graphite Assignment to an Improved Digital Image Analysis of Ductile Iron, Inter. J. Metalcasting, 2020, 14(4), p 1090–1104. https://doi.org/10.1007/s40962-020-00416-3
DIN EN 1563:2019-04, Gießereiwesen – Gusseisen mit Kugelgraphit, Beuth-Verlag, 2019, in German, https://dx.doi.org/https://doi.org/10.31030/2792445
D.M. Stefanescu, G. Alonso, and R. Suarez, Recent Developments in Understanding Nucleation and Crystallization of Spheroidal Graphite in Iron-Carbon-Silicon Alloys, Metals, 2020, 10(2), p 221. https://doi.org/10.3390/met10020221
I. Riposan, M. Chisamera, S. Stan, P. Toboc, C. Ecob, and D. White, Al, Zr–FeSi Preconditioning of Grey Cast Irons, Mater. Sci. Techn., 2008, 24(5), p 579–584. https://doi.org/10.1179/174328408X298842
B. Uhrenius, Hardenability Concepts with Applications to Steel, TMS-AIME, Warrendale, 1978, p 28–81
H. Bayati and R. Elliott, Austempering Process in High Manganese Alloyed Ductile Cast Iron, Mater. Sci. Techn., 1995, 11(2), p 118–129. https://doi.org/10.1179/mst.1995.11.2.118
T.N. Rouns and K.B. Rundman, Constitution of Austempered Ductile Iron and Kinetics of Austempering, Trans. AFS, 1987, 95, p 851–874.
R. Kawulok, I. Schindler, J. Sojka, P. Kawulok, P. Opěla, L. Pindor, E. Grycz, S. Rusz, and V. Ševčák, Effect of Strain on Transformation Diagrams of 100Cr6 Steel, Curr. Comput.-Aided Drug Des., 2020, 10(4), p 326. https://doi.org/10.3390/cryst10040326
Y.N. Taran, K.I. Uzlov, and A.Y. Kutsov, The Bainite Reaction Kinetics in Austempered Ductile Iron, J. Phys. IV France, 1997, 07(C5), p 429–434. https://doi.org/10.1051/jp4:1997568
J. Mallia, M. Grech, and R.E. Smallman, Effect of Silicon Content on Transformation Kinetics of Austempered Ductile Iron, Mater. Sci. Techn., 1998, 14(5), p 452–460. https://doi.org/10.1179/mst.1998.14.5.452
P.Q. Dai, Z.R. He, C.M. Zheng, and Z.Y. Mao, In-situ SEM Observation on the Fracture of Austempered Ductile Iron, Mater. Sci. Eng. A, 2001, 319–321, p 531–534. https://doi.org/10.1016/S0921-5093(01)01088-7
Acknowledgments
The authors gratefully acknowledge financial support from Bundesministerium für Bildung und Forschung (BMBF) to the project “Druckguss-ADI: Entwicklung einer druckgussfähigen ADI-Legierung und eines zugehörigen Druckgussverfahrens mit integraler Wärmeführung” (03XP0203B).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
van gen Hassend, F., Ninnemann, L., Töberich, F. et al. Study on the Austemperability of Thin-wall Ductile Cast Iron Produced by High-Pressure Die-casting. J. of Materi Eng and Perform 31, 1405–1418 (2022). https://doi.org/10.1007/s11665-021-06252-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11665-021-06252-8