
Abstract
The incorporation of hydrogen, which is a relatively unexplored reductant used during ferromanganese (FeMn) production, is an attractive approach to lessen atmospheric gaseous carbon release. The influence of hydrogen on the pre-reduction of carbonate-rich United Manganese of Kalahari (UMK) ore from South Africa was investigated. Experiments were performed in 70 pct CO 30 pct CO2 (reference), 70 pct H2 30 pct H2O, and 100 pct H2 gas atmospheres at 700 °C, 800 °C, and 900 °C. Calculated phase stability diagrams and experimental results showed good correlation. The pre-reduction process involved two reactions proceeding in parallel, i.e., the pre-reduction of higher oxides and the decomposition of carbonates present in the ore. A thermogravimetric (TG) furnace was employed for the pre-reduction of the ore in various atmospheres. The calculated weight loss percentage was used to determine the degree and rate of pre-reduction. It was found that the oxidation state of higher Fe- and Mn-oxides was lowered when treated in 70 pct H2 30 pct H2O and 70 pct CO 30 pct CO2, whereas FeO was metalized when using 100 pct H2. As for the intrinsic carbonates, the majority thereof were decomposed in the CO/CO2 atmosphere at 900 °C, and ≥ 700 °C in the H2/H2O and H2 atmospheres. Additionally, the degree and rate of reduction were accelerated by increasing the pre-reduction temperature and by employing a hydrogen-containing gas atmosphere (70 pct H2 30 pct H2O, and 100 pct H2). Scanning electron microscopy and electron microprobe analysis revealed the presence of three phases in the pre-reduced ore: (i) Mn- and Fe-rich, (ii) Mg- and Ca-rich, and (iii) Mg-, Si-, K-, and Na-rich. It was also found that there were no appreciable differences in porosity and decrepitation of the ores treated in the CO/CO2 and hydrogen-containing atmospheres. The use of a hydrogen atmosphere showed potential for the pre-reduction of carbonate-containing manganese ores as it accelerated the decomposition of the carbonates as well as facilitated the metallization of Fe-oxides present in the ore.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Manganese (Mn) is one of the most versatile additives in the steel industry and it is responsible for decreasing the brittleness while enhancing the strength and abrasion resistance of steel. Additionally, the inclusion of an Mn-alloy during steel production is to deoxidize the respective steel. Mn is also relatively low-cost and abundant.[1,2] The industrial production of ferromanganese (FeMn) alloys is usually performed in a submerged arc furnace (SAF), with solid carbon (C) as the primary reductant. During the production process, carbon monoxide (CO) is generated from the reaction between Mn-oxide and C occurring in the smelting zone (hottest area) of the SAF. The CO permeates toward the upper parts of the SAF where it reacts with the raw ore in the furnace charge, initiating the pre-reduction of higher Mn- and iron (Fe)-oxides present in the ore, as given by Reactions [R1] through [R4].[3,4]
The pre-reduction of the higher Mn-oxides to MnO and Fe2O3 to Fe3O4 (Reactions [R1] through [R4]) is exothermic and will increase the overall temperature of the charge when contacted with CO as the charge descends in the SAF.[3] There are also endothermic reactions taking place during the smelting process, such as the reduction of Fe3O4 to FeO (Reaction [R5]), the decomposition of carbonates (Reactions [R6] and [R7]), and the Boudouard reaction (Reaction [R8]). These endothermic reactions have a significant influence on the C and energy consumption of FeMn production.
A separate pre-treatment, i.e., pre-reduction, unit in FeMn production is relatively unexplored when compared to other metal-producing processes, e.g., ferrochromium production. It is generally accepted by the FeMn industry, that due to the exothermic nature of Reactions [R1] through [R4], such reactions are advantageous to occur within the SAF during smelting procedures.
Including a separate pre-treatment step in the FeMn process can also be advantageous. At temperatures above 800 °C, Reaction (R3) (reduction of Mn3O4 to MnO) and Reaction [R8] (Boudouard reaction) proceed concurrently in the coke bed. Hence, the CO2 produced by Reaction [R3] is consumed by Reaction [R8] to form CO. In addition, C is consumed during the direct reduction reaction, i.e., Mn3O4 is directly reduced to MnO in the presence of C following Reaction [R9]. Both Reactions [R8] and [R9] are endothermic, thereby increasing the overall energy consumption, as well as C consumption.[3,5]
Furthermore, not all the ascending CO is utilized during the pre-reduction process and is ultimately expelled from the SAF as part of the off-gas. Typically, the off-gas contains approximately 60 vol pct CO.[3] The excess consumption of C and energy, and loss of unused CO in the off-gas, can be avoided by implementing a pre-treatment unit where the ore is pre-reduced before entering the furnace.
If the hot CO-rich off-gas is redirected to a pre-reduction process, as a heat source and to partake in the pre-reduction reactions, the overall energy demand of the specific FeMn-producing SAF can decrease by up to 25 pct.[6,7] One example is given by the CDK Kashima Works in Japan where pre-treatment of different Mn sources is performed in a rotary kiln in sequence with the SAF. In addition to a C-source being combusted, CO-rich off-gas from the SAF provides additional energy to the pre-reduction kiln. This additional pre-treatment step leads to a decrease in the overall energy consumption.[7] Furthermore, Tangstad et al. showed that supplying hot Mn ore to a SAF reduced the specific SAF’s energy consumption by approximately 20 pct (i.e., a 400 to 500 kWh/ton FeMn reduction).[7]
To avoid the Boudouard reaction, and hence reduce the C and energy consumption, the CO2-producing reactions (Reactions [R1] through [R7]) should occur at a lower temperature than the Boudouard reaction (Reaction [R8]), which is estimated at around 800 °C. The solid-state pre-reduction of the higher oxides is controlled by the kinetics rather than the thermodynamics of the reaction. Studies have shown that parameters such as ore porosity and particle size, the furnace heating rate, and the gas atmosphere affect the rate of pre-reduction.[4,8,9,10] The gas atmosphere used in industrial furnaces consists of CO/CO2, hence many studies performed on the pre-reduction of Mn ore used a CO/CO2 gas mixture. However, several studies have shown that the inclusion of hydrogen as a reductant in the gas atmosphere reduces the oxygen partial pressure (pO2) and increases the degree and rate of pre-reduction.[8,11]
Bhalla et al. studied the pre-reduction of Mamatwan ore originating from South Africa. The experiments were performed in a TG furnace in several CH4-Ar gas mixtures at 1100 °C and 1200 °C. During the pre-reduction process, CH4 underwent cracking which supplied hydrogen gas (H2) and fine solid C to the surface of the ores, allowing intimate contact. The excess fine C caused the regeneration of the reductants CO and H2 from CO2 and H2O provided by the reduction reactions, which resulted in a low pO2 at the reduction sites.[11]
Ngoy et al. performed non-isothermal experiments in a TG furnace on Nchwaning (also referred to as Assmang, the respective South African mine) and Comilog (Gabon) ores to investigate the effect of hydrogen on pre-reduction. Both ores were subjected to hydrogen-containing CO/CO2 gas mixtures, i.e., 41 pct CO 18 pct H2 41 pct CO2 and 41 pct CO 41 pct H2 18 pct CO2, up to 1000 °C. The author found that for the same pO2, the reduction rate increased by 20 to 30 pct when hydrogen was included in the CO/CO2 gas mixture. It was also seen that a higher reduction rate was achieved with a lower pO2 of the gas mixture with or without hydrogen.[8]
The influence of hydrogen on the rate and degree of pre-reduction of Nchwaning ores was investigated by Schanche et al. The ore was reduced at 600 °C to 800 °C in a CO/CO2 and/or hydrogen atmosphere at various pO2. It was found that the degree and rate of reduction improved with an increase in temperature and a decrease in pO2. It was found that the degree and rate of reduction improved with an increase in temperature and a decrease in pO2. The inclusion of hydrogen caused the reduction rate to increase by a factor of 1.3. and 2.6 for low and high pO2, respectively. The authors assumed that the increase was due to the improved diffusion characteristics of hydrogen (i.e., the ability to penetrate ore particles).[4]
Larssen et al. investigated the potential influence of hydrogen (in the form of hydrogen and water vapor) and the occurrence of the water–gas shift reaction (WGSR) on the pre-reduction kinetics of Comilog and Nchwaning ore. The ores were subjected to CO/CO2/H2, CO/CO2/H2O(g), and CO/CO2 gas atmospheres at different pO2 in a TG furnace. The highest reduction rate was achieved in the CO/CO2/H2 gas atmosphere, while the lowest was obtained in the CO/CO2 gas atmosphere. Furthermore, the highest degree of reduction was achieved at the lower pO2 when hydrogen was present in the gas atmosphere. The authors also found that at low temperatures, the WGSR proceeded to some degree without reaching equilibrium; however, equilibrium was reached at higher temperature.[12]
Most of the latter studies were performed on Mn ore with a low carbonate content, such as Comilog and Nchwaning ores. However, during the pre-reduction of Mn ores rich in carbonates (e.g., UMK, Mamatwan, and Gloria ores originating from the Mamatwan environment of the Kalahari basin in South Africa), CO2 in the off-gas mainly originates from carbonate decomposition. More so, due to the endothermic nature of carbonate decomposition, a fraction of the energy demand during pre-reduction is due to the decomposition reactions, i.e., Reactions [R6] and [R7].[3] Nevertheless, it is beneficial that most of the CO2 formation occurs during the pre-treatment step (i.e., pre-reduction) to mitigate the CO2 supply for the Boudouard reaction during smelting procedures.
Calcite (CaCO3), magnesium carbonate (MgCO3), and dolomite (CaMg (CO3)2) are the most abundant carbonates in Mn ores. The decomposition of calcite (CaCO3) initiates at approximately 900 °C; this temperature corresponds with the temperature required for the Boudouard reaction (Reaction [R8]) to initiate. Nevertheless, the magnesium carbonate (MgCO3) and dolomite (CaMg(CO3)2) decompose at < 800 °C.[3] Using hydrogen as a reductant will yield H2O, which will lower the pCO2, compared to when CO is used as a reductant (Reactions [R1] through [R5]). The lower pCO2 atmosphere will accelerate the decomposition of carbonates. It is therefore important to quantify the effect of hydrogen as a reductant on the rate and degree of carbonate decomposition. Carbonate decomposition is also dependent on temperature and is accelerated at higher temperatures.[13,14]
A study on the pre-reduction and decomposition of Mn ores with hydrogen was performed using 100 pct H2 and 70 pct H2 30 pct H2O atmospheres. UMK ore, a carbonate-rich ore from South Africa, was considered as the case study ore. It is noted that South Africa holds the world’s largest reserve of Mn and that the primary source of Mn in South Africa’s smelting industry is based on the available Mn ores sourced in the Kalahari region, typically referred to as UMK, Nchwaning, Mamatwan, and Wessels ores. As a reference, the ore was subjected to pre-reduction in a 70 pct CO 30 pct CO2 gas atmosphere. The 70 pct CO 30 pct CO2 and 70 pct H2 30 pct H2O atmospheres had similar pO2, which suggests that both atmospheres had comparable thermodynamic driving forces for the pre-reduction reactions (Reactions [R1] through [R5]) and decomposition reactions (Reactions [R6] and [R7]). The pre-reduction and carbonate decomposition reactions were investigated by experimentation using a TG furnace at various temperatures.
Materials and Methods
Experimental
UMK ore with a size fraction of 9.5 to 16 mm was selected for the experiments. UMK is a semi-oxidized (Mn2O3) ore from the Mamatwan environment. The ore mainly consists of braunite I (3(Mn,Fe)2O3·MnSiO3), calcite (CaCO3), dolomite (CaMg(CO3)2, and hausmannite (Mn3O4). Kutnohorite ((Ca,Mn,Mg,Fe)(CO3)2), bixbyite ((Mn,Fe)2O3), and hematite (Fe2O3) is also found but to a lesser degree.[15]
The raw ore was dried at 105 °C to remove any surficial moisture. The dried ore was then divided into individual batches of approximately 156 g which were utilized in the experiments. A TG furnace was used to investigate the solid–gas pre-reduction and decomposition reactions of the ore. Experiments were performed in an Entech VTF 80/15 (Ängelholm, Sweden) vertical resistance tube furnace. The ore was placed in a high-temperature gas-tight stainless-steel (253 MA) double-wall crucible (Figure 1) which was suspended from a balance (Mettler Toledo PR2003DR, Greifensee, Switzerland) to record the weight changes during the experiments. Figure 1 presents a schematic illustration of the crucible used in the experiments. While the crucible was freely suspended, the furnace position could be altered vertically so that the crucible could be inside or outside the furnace hot zone during the heating and cooling periods, respectively. The heating of the furnace was controlled by a Eurotherm PID controller and measured with a calibrated S-type thermocouple. A K-type thermocouple placed in an alumina tube and positioned in the middle of the charge was used to measure the charge temperature. All temperature values reported here are based on the measurements made using the K-type thermocouple, as these measurements were the true temperature experienced by the charge. The furnace was preheated to a target temperature (700 °C, 800 °C, or 900 °C) at a rate of 6 °C/min. Once the furnace reached the target temperature, it was moved vertically to accommodate the crucible within the furnace hot zone. The crucible temperature, measured using the K-type thermocouple, took approximately 12 to 13 minutes to match the target temperature. Hereafter, the crucible was kept in the furnace until the charge weight stabilized, i.e., until the reactions between the charge and respective gaseous atmospheres concluded. This period ranged between 99 and 372 minutes, depending on the atmosphere and temperature.

Flexible gas inlet and outlet pipes were connected to the top of the crucible which allowed multiple gasses to mix before entering the crucible. In all the experiments, the total gas flow rate was 4 L/min. The gas was preheated as it moved along the double wall of the crucible down to the sample (Figure 1). Each gas line had its mass flow controller (Bronkhorst F-201C, United Kingdom). In this investigation, Ar (99.999 pct), CO (99.97 pct), CO2 (99.9992 pct), and H2 (99.9 pct) were used.
In addition, the weight of the sample before and after the experiment was recorded on an external Ohaus Pioneer PA4202 scale (New York) to confirm the net weight loss obtained from the TG data. The samples were pre-reduced at temperatures of 700 °C, 800 °C, and 900 °C. However, due to the heating time, pre-reduction of higher oxides (exothermic) and decomposition of carbonates (endothermic) partly occurred during heating to set temperatures, and thus the samples did not experience a true isothermal condition. After the experiment was completed, the sample was allowed to cool to ambient temperature in Ar.
A portion (25 pct) of the pre-reduced samples was crushed and a fraction thereof was used for chemical analysis. The remainder was pulverized in a Retsch Vibratory Disc Mill RS 200 (Haan, Germany) at 900 rpm for 90 seconds. A sample from the unreduced material was prepared similarly. The grinding set consisted of tungsten carbide grinding rings and a plate. XRD analyses were performed on the pulverized samples by using a Bruker D8 A25 DaVinci X-ray Diffractometer (Billerica) with CuKα radiation and LynxEye™ SuperSpeed Detector. The results were obtained in a total of 7200 seconds at a scanning angle of 2θ and a range of 10 to 90 deg with 0.013 deg step sizes. The DIFFRAC.EVA V5.1 software together with the PDF-4+ database was used to index the phases present in the samples.
The chemical composition of the pulverized raw and pre-reduced ores was examined by X-ray fluorescence (Bruker AXS S4 Pioneer X-Ray fluorescence spectrometer) using the fused bead technique. Permanganometric titration (ASTM 465-11:2017) was employed to measure the excess oxygen above MnO which is expressed as MnO2. The C concentration was analyzed with a LECO (Combustion-IR) instrument and the CO2 concentration was determined by assuming that all C in the ore is in the form of CO2. The loss on ignition (LOI) was thermogravimetrically measured to a stable weight at 950 °C in air. All off-gas measurements were conducted using an NDIR CO/CO2 gas analyser.
The microstructure and elemental composition of the raw material and pre-reduced samples were analyzed in secondary and backscattered electron mode by a scanning electron microscope (SEM) equipped with an energy-dispersive x-ray spectrometer (EDS). A Zeiss—Supra 55VP—FEG-SEM including an EDAX OCTANE PRO-A EDS detector (sensor area 10 mm2) operating at 10 kV and a working distance of 10 mm was used. The samples were mounted in epoxy and polished (Struers, Tegramin-20, Ballerup, Denmark) with traditional wet metallographic methods. The samples were C-coated before SEM–EDS analysis. Element mapping of the samples was done by using Electron Probe Micro Analysis (EPMA). FE-EPMA with JEOL WDS system (JEOL JXA 8500, Sydney, Australia) was used for the EPMA analysis.
Gas analyses of the off-gas were conducted using a CO/CO2 gas analyser. Porosity measurements were performed on raw ore and pre-reduced samples. The porosity was determined by the ratio between the absolute and apparent density. The absolute density was measured by Accupyc 1330 helium pycnometer, while the apparent density was determined by a GeoPyc 1360 pycnometer.
The decrepitation of the reduced samples was measured after the pre-reduction experiments by sieving the charge through a sieving stack consisting of 9.5, 6.7, 4.75, 3.15, 1.6, and < 0.5 mm sieves.
Calculations
A time series of five seconds was used to detect the mass loss of the solid charge during the TG performed in different gas atmospheres at various temperatures. The experimental weight loss data from the TG system was used to calculate the weight loss percentage. The corresponding O/Mn ratio as a function of time is calculated from the weight loss percentage through Eq. [1].
where wt pct loss (t) is the weight loss percentage at time t, theoretical wt pct loss is the theoretical weight loss percentage, O/Mn(t) is the O/Mn mol ratio at time t and O/Mn(i) indicates the initial O/Mn mol ratio before reduction. The O/Mn(i) value is calculated from the chemical analysis of the raw ore (Table II), i.e., the total Mn measured in XRF and the MnO2 measured from the titrimetric method. The O/Mn(i) value can be calculated through Eqs. [2] and [3] when assuming that all the Mn absent in MnO2 is present in MnO, and in this case, the O/Mn(i) is calculated to be 1.39. Previous studies have calculated the O/Mn(i) ratio of UMK ore to be between 1.3[18] and 1.45.[19,20] The variation in the O/Mn(i) ratio can be due to the heterogeneity of the ore and/or to some of the Mn being present as manganese carbonate (MnCO3) which will affect the O/Mn ratio.[20]
Furthermore, the theoretical weight loss is determined by the chemical analysis (Table II) of the raw ore by determining the LOI, CO2, O/Mn, and Fe content of the ore. The calculation is based on three assumptions; (i) all Fe is initially in the Fe2O3 form and reduces to FeO, (ii) Mn reduces from O/Mn(i) to MnO, (iii) and all the C present in the raw ore represents carbonates that will decompose to CO2.
Considering the measuring method of LOI, it is assumed that all carbonates decomposed, and all volatiles were evaporated. Furthermore, according to Figure 2, the most stable oxides of Mn and Fe at 950 °C are Mn3O4 and Fe2O3, respectively.[4] Hence the LOI contains water vapor, carbonates, and decomposed Mn (Mn3O4), and the amount of water vapor can be calculated by subtracting the wt pct CO2 shown in the chemical analysis and the decomposed Mn.[20] The calculated theoretical weight loss for UMK ore is shown in Table I.

The Fe–O and Mn–O stability diagrams show oxygen partial pressure as a function of temperature.[4] The pO2 of (black stripe-dotted, blue, and green lines) are superimposed on the stability diagram. Factsage[21] and HSC Chemistry 10[22] were used for the calculation of the stability diagrams and the pO2, respectively. Factsage databases used: FToxide for oxide database for slags, glasses, ceramics, and refractories; FTmisc for miscellaneous database for sulfides, alloys, etc.; FactPS for FACT pure substances database; Factsage modules for phase diagram and figure modules (Color figure online)
Results and Discussion
Characterization
Table II summarizes the chemical composition of the raw and pre-reduced ore and the O/Mn ratios obtained for the three different gas atmospheres, i.e., 70 pct CO 30 pct CO2, 70 pct H2 30 pct H2O, and 100 pct H2 at three different temperatures, i.e., 700, 800, and 900 °C. The MnO2 value (< 0.05 wt pct) obtained through chemical analysis indicates that all the samples are completely reduced to MnO.
Very little CO2 was detected in the samples subjected to the hydrogen-containing gas atmospheres, i.e.,70 pct H2 30 pct H2O, and 100 pct H2 as well as the sample pre-reduced in 70 pct CO 30 pct CO2 at 900 °C, meaning that almost all the carbonates decomposed in these samples. The percentage of decomposed carbonates was calculated from initial values and the results are shown in Figure 3.

A fraction of the total wt pct of carbonates decomposed in UMK ore as a function of temperature
As stated earlier, no CO2 is generated during the pre-reduction reactions (Reactions [R1] through [R5]) when hydrogen is used as the reductant; hence a lower pCO2, and subsequently, a higher decomposition driving force is obtained during pre-reduction in a 70 pct H2 30 pct H2O atmosphere when compared to a 70 pct CO 30 pct CO2 atmosphere.[13,23,24] Figure 3 shows that a lower percentage of carbonates decomposed at 700 °C (29.4 pct) and 800 °C (48.2 pct) for samples reduced in 70 pct CO 30 pct CO2, whereas near 100 pct carbonate decomposition was achieved for the hydrogen-containing atmospheres.
Figure 4 is compiled to show the phase stability diagram for selected carbonates, where the y-axis represents the partial pressure of CO2 (pCO2), and the x-axis represents temperature. It can be seen that in a 70 pct CO 30 pct CO2 atmosphere (log pCO2 = -0.3 that is, pCO2 = 0.501 atm) , the magnesium carbonates (MgCO3), manganese carbonates (MnCO3), dolomite (CaMg(CO3)2) and calcite (CaCO3) all decompose at different temperatures, i.e., 272 °C, 308 °C, 389 °C, and 810 °C, respectively. Similar observations were made by Olsen et al.[3] and Schanche et al.[4] Thermodynamic calculations suggest that calcite (CaCO3) decomposition initiates at temperatures above 810 °C, while the other carbonates decompose at temperatures below 389 °C. Hence, the carbonates still present in samples pre-reduced in a 70 pct CO 30 pct CO2 atmosphere at 700 °C and 800 °C are most likely calcite (CaCO3).

Phase stability diagram for selected carbonates as a function of log pCO2 and temperature. In the 70 pct CO 30 pct CO2 gas atmosphere the log pCO2 = − 0.3 is indicated by the horizontal, red dotted line. The equilibrium temperature at log pCO2 = − 0.3 for each carbonate is shown by the vertical, red dotted lines. The stability diagram and pCO2 were calculated using HSC Chemistry 10.[22] (Color figure online)
Mineralogical Phase Transformation During Pre-reduction
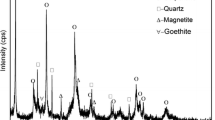
XRD analysis of raw and pre-reduced ores is shown in Figure 5. The chemical formulas of detected phases are shown in the figure caption. The carbonates present in the raw ore were detected as Calcite (CaCO3), Magnesium carbonate (MgCO3) and Kutnohorite ((Ca,Mn,Mg,Fe)(CO3)2). For the samples pre-reduced in CO/CO2, no kutnohorite was detected at 700 °C and 800 °C, while some Ca- and Mg-carbonates were still present. The main Ca- and Mg-carbonate peaks at approximately 29° contained more Ca (0.936) than Mg (0.064) which explains why only calcium oxide (CaO) peaks are visible at 900 °C. As determined using thermodynamical calculations (Figure 4), calcite (CaCO3) is mainly present at 700 °C and 800 °C. Furthermore, all the carbonates decomposed at 900 °C.

XRD patterns of the raw ore and samples pre-reduced in 70 pct CO 30 pct CO2 (a), 70 pct H2 30 pct H2O (b), and 100 pct H2 (c) at 700 °C, 800 °C, and 900 °C. Chemical formula of compounds: calcite (CaCO3); magnesium carbonate (MgCO3) braunite (3(Mn,Fe)2O3·MnSiO3); bixbyite ((Mn,Fe)2O3); hematite (Fe2O3); kutnohorite ((Ca,Mn,Mg,Fe)(CO3)2); manganosite (MnO); calcium oxide (CaO); hausmannite (Mn3O4)
The XRD patterns of the hydrogen-containing gas atmospheres indicate that all the carbonates decomposed in the 700 °C to 900 °C temperature range. These results are in correlation with the chemical analysis shown in Table II and the calculated results in Figure 4.
The XRD of the raw ore showed that braunite (3(Mn,Fe)2O3·MnSiO3) and bixbyite (Mn,Fe)2O3) were the two main Mn-containing phases, hence the original state of Mn before pre-reduction procedures was Mn3+ (Mn2O3).
The XRD patterns of all the pre-reduced samples showed that braunite (3(Mn,Fe)2O3·MnSiO3) and bixbyite ((Mn,Fe)2O3) were pre-reduced to manganosite (MnO) without the formation of hausmannite (Mn3O4). The absence of hausmannite (Mn3O4) in the pre-reduced samples could be due to the initial content in the unreduced ore or that the intermediary hausmannite (Mn3O4) was reduced at a rapid rate.[4]
A study by Zaki et al. showed that a stepwise reduction (more specifically, its decomposition) of synthetic Mn-oxides (MnO2) was only seen in an oxidizing (O2, air) atmosphere. In a reducing atmosphere, the MnO2 reduced directly to MnO without the generation of stable intermediates, i.e., Mn5O8, Mn2O3, and Mn3O4.[25]
Small amounts of braunite (3(Mn,Fe)2O3·MnSiO3)/bixbyite ((Mn,Fe)2O3) were still present in the samples pre-reduced at 700 °C, even though the chemical analysis (Table II) suggests that the Mn-oxides were completely reduced to MnO (O/Mn = 1, MnO2 = < 0.05 wt pct). It has previously been observed that the Fe content of the Mn source lowers the MnO2 measurement from the chemical analysis which explain the discrepancy between the chemical analysis and the XRD analysis.[4] However, the peak intensity was low; hence, the discrepancy between the XRD and chemical analysis is not that significant.
Furthermore, the XRD analysis revealed that metallic Fe was present in samples pre-reduced in 100 pct H2 at all temperatures and low-intensity Fe-oxide peaks were detected in the manganosite (MnO) phase. In Figure 6, the main manganosite (MnO) peak of the sample pre-reduced in 100 pct H2 shifted to the left compared to the sample pre-reduced in 70 pct CO 30 pct CO2, which is indicative of a lower Fe-oxide content. This is in accordance with the observations made in Figure 5(c) regarding metallic Fe, since the formation of Fe warrants its depletion from the oxide phase.[4] In addition, the manganosite (MnO) peak of the sample pre-reduced in 70 pct H2 30 pct H2O is shifted more to the right, indicating a higher Fe-oxide content than the sample pre-reduced in 70 pct CO 30 pct CO2. As was seen in Figures 5(a) and (b), no metallic Fe was detected in samples reduced in a 70 pct CO 30 pct CO2 and 70 pct H2 30 pct H2O atmospheres. The utilization of hydrogen as reductant, hence, improves the reduction of Fe-oxides. This improvement was also seen in previous studies based on the reduction of Fe ores.[26,27,28,29,30]

Main peak from the manganosite (MnO) phase of the sample pre-reduced in 70 pct CO 30 pct CO2, 70 pct H2 30 pct H2O, and 100 pct H2 at 900 °C. The vertical dashed lines indicate the 2θ value (deg) where manganosite (MnO) and Fe- and Mn- oxide (FeO-MnO) peaks form during XRD analysis
The pO2 of the gas atmospheres are included in the Fe–O and Mn–O stability diagram (Figure 2) to show that metallic Fe formation in 70 pct CO 30 pct CO2 and 70 pct H2 30 pct H2O gas atmospheres are not thermodynamically feasible at the given experimental conditions. However, Figure 2 shows that not only kinetically, but also thermodynamically, metallic Fe would form when UMK ore is reduced in 100 pct H2 in the 700 °C to 900 °C temperature range.
SEM Morphology and EPMA on Raw and Pre-reduced UMK Ore
Based on the SEM observations presented in Figure 7, it is evident that two phases were present in the raw ore, i.e., referred to here as light and dark phases. From SEM characterization by Sukhomlinov et al.[14] in a previous non-related study using the same ore type, the lighter phases were rich in braunite (3(Mn,Fe)2 O3·MnSiO3), bixbyite ((Mn,Fe)2O3) and hausmannite (Mn3O4), whereas the darker phases were enriched in Ca and Mg (the main carbonate-associated phases) which were detected at concentrations up to 28 and 4.5 wt pct, respectively. Therefore, the UMK ore consisted of a fine-grained structure (Figure 7(a)), which contained carbonate-rich ovoid and laminae (Figure 7(b)).[14]

Secondary electron SEM micrographs showing the fine-grained structure (a) and carbonate-rich ovoid and laminae (b) of raw UMK ore
EPMA was performed on cross-sectioned and polished raw ore and all samples pre-reduced at 800 °C to determine how the microstructure of the ore changes when hydrogen is used as reductant (Figure 8). EPMA of the raw ore was included as a reference (Figure 8(a)). According to Figures 8(a) through (d), three phases were observed: the Fe- and Mn-rich (Figures 8(a) through (d), white areas), Ca- and Mg-rich (Figures 8(a) through (d), dark gray areas) phases, and an Mg-, Si-, K- and Na-rich (light gray) phase (Figure 8(e)), mainly occurring as a segregated phase. The chemical composition of the detected phases is included in Table AI (Supplementary information).



EPMA performed on cross-sectioned and polished raw ore (a), samples pre-reduced in 70 pct CO 30 pct CO2 (b), 70 pct H2 30 pct H2O (c), and 100 pct H2 (d) atmospheres at 800 °C, and Mg-, Si-, K-, Na-rich phases embedded as a segregated phase in samples pre-reduced in a 70 pct H2 30 pct H2O atmosphere (e)
SEM analysis was performed on cross-sectioned polished samples pre-reduced in the various gas atmospheres at 800 °C to determine the presence and extent of pore formation (i.e., open spaces present in the ore particles), as well as to elucidate the distribution of the phases presented in Figure 8.
The black spots observed in Figure 9 are representative of pores (i.e., open spaces). The average diameter (measured between the two furthest ends) of the pores present in the pre-reduced samples shown in Figures 9(a) through (c) were measured as 2.7, 5.9, and 1.5 µm, respectively. Hence, the largest pores formed during pre-reduction in a 70 pct H2 30 pct H2O atmosphere (Figure 9(b)), whereas smaller pores formed during pre-reduction in the 70 pct CO 30 pct CO2 (Figure 9(a)) and 100 pct H2 (Figure 9(c)) atmospheres. Nevertheless, the pores formed were relatively large when compared to the size of the pores that formed in the Mn-rich area inside reduced Nchwaning ore (average diameter of 0.1 µm).[9] In a non-related study using UMK ore (heated to 400 °C in a synthetic air atmosphere), the pore size diameter ranged from 3.6 to 7.1 µm.[19] Furthermore, Figures 8(e) and 9(d) show how the Mg-, Si-, K-, and Na-rich phase formed in different shapes between the Fe/Mn-rich and Ca/Mg-rich phases. According to the EDS results, the light gray area in Figure 9(d) mainly consisted of Si (12.8 wt pct) and Mg (14.9 wt pct).

Backscattered electron SEM micrographs of cross-sectioned and polished samples pre-reduced in 70 pct CO 30 pct CO2 (a), 70 pct H2 30 pct H2O (b), 100 pct H2 (c) gas atmospheres at 800 °C, and Mg-, Si-, K-, Na—rich phases embedded as a segregated phase in the sample pre-reduced in a 70 pct H2 30 pct H2O gas atmosphere at 800 °C (d)
Considering that Fe was detected by XRD (Figure 5(c)) for the ore pre-reduced in the 100 pct H2 atmosphere, a high magnification (× 10,000) SEM micrograph of the white area (Fe- and Mn-rich phase) seen in Figure 9(c) was prepared and is presented as Figure 10.

Secondary electron micrograph at ×10,000 magnification of the Fe- and Mn-rich phase shown in Fig. 9(c). The sample was pre-reduced in a 100 pct H2 atmosphere at 800 °C
Figure 10 shows the presence of various phases. Of particular importance here was the relatively small and bright spots observed within the Fe- and Mn-rich phase. These spots were analyzed by EDS and contained 7.71 wt pct Fe and 50.7 wt pct Mn. The Fe and Mn contents of the Fe- and Mn-rich phase were determined to be 1.68 and 58.0 wt pct, respectively.
The Mn/Fe ratio of the bright spots and the Fe- and Mg-rich phases were calculated as 6.58 and 34.5, respectively. However, EDS analysis is a semi-quantitative technique, hence the true composition of the bright spots is difficult to determine from the EDS analysis as it is so small and embedded in the large Fe- and Mn-rich phase. More so, considering Castaing’s formula law, the penetration depth of electrons during analysis likely included the underlying sample bulk in the analysis. Thus, based on the Mn/Fe ratio and the XRD patterns (Figure 5(c)) of the samples pre-reduced in 100 pct H2, it is assumed that these bright spots are rich in metallic Fe.
Porosity Analysis of UMK Ore
A higher porosity provides more surface area for gas–solid reactions to proceed which will increase the reduction of higher Fe- and Mn-oxides and improve the reactivity of the ore.[2] However, an increase in porosity leads to a decrease in the ore’s thermal strength.[15,31] A higher porosity makes it more feasible for the ore to absorb moisture, hence when rapidly heated, the ores may crack due to the high pressure caused by the water evaporation (elimination of structural water).[32] In addition, adding fine ore material to the FeMn smelting process will limit the permeability of the gas flow in the furnace and may cause furnace bed sintering. This will have a retarding effect on the reduction rate during the FeMn smelting process,[33] and may lead to potentially catastrophic bed turn-overs.
Porosity analysis was performed on raw ore and samples pre-reduced in all three gas atmospheres at 800 °C. The results can be seen in Table III. The apparent and absolute density of the raw UMK ore particles were the same, hence, the porosity of the raw UMK ore was calculated to be 0 pct. Low porosity of 2.6 pct and 0 to 10 pct was also seen by Biørnstad et al.[19] and Reiersen et al.,[20] respectively. The apparent and absolute density of three raw ore particles was determined to be 3.74, 3.61, and 3.66 g/cm3, respectively.
During pre-reduction procedures, the reduction of higher oxides and carbonate decomposition results in the formation of intra-particle pores. The higher Mn-oxides in all the samples reduced to manganosite (MnO) at 800 °C (Table II). However, the higher Fe-oxides in samples pre-reduced in 70 pct CO 30 pct CO2, and 70 pct H2 30 pct H2O reduced to FeO, while in 100 pct H2 the FeO further reduced to Fe. Furthermore, most of the carbonates decomposed in the hydrogen-containing gas atmosphere at 800 °C while only half of the carbonates (8.8 wt pct, see Table II) decomposed in 70 pct CO 30 pct CO2 at 800 °C. Considering this, one would expect the samples pre-reduced in 100 pct H2 at 800 °C to have the highest porosity, while the samples pre-reduced in 70 pct CO 30 pct CO2 at 800 °C to have the lowest. However, the porosity average in Table III shows that the gas atmosphere used for pre-reduction had an insignificant effect on pore formation. This could be attributed to the heterogeneity (high standard deviation of 7.77) of the ore.
Degree and Rate of Pre-reduction and Carbonate Decomposition
According to the chemical analysis, the majority of weight loss was caused by the decomposition of carbonates. The rest of the weight loss was due to the pre-reduction of Fe- and Mn-oxides (see Table I). Hence the degree of pre-reduction and decomposition of carbonates are presented as the fractional conversion, α, which is defined in Eq. [7].
where wo is the initial weight (wt pct), wh is the highest weight loss (wt pct) obtained from the samples pre-reduced in 100 pct H2 and w(t) (wt pct) is the measured weight as a function of time. When α = 0 no reduction or decomposition has taken place, while when α = 1 the sample is fully reduced (the higher Mn-oxides completely reduced to manganosite MnO) and all the carbonates decomposed. The pre-reduction and decomposition rates are obtained by differentiating α with respect to time. Figure 11 presents the degree and rate of pre-reduction and decomposition from the experiments performed in 70 pct CO 30 pct CO2, 70 pct H2 30 pct H2O, and 100 pct H2 atmospheres at 700 (Figure 11(a)), 800 (Figure 11(b)), and 900 °C (Figure 11(c)).

Experimental data obtained from the TG furnace for samples pre-reduced in 70 pct CO 30 pct CO2, 70 pct H2 30 pct H2O, and 100 pct H2 gas atmospheres at 700 (a), 800 (b), and 900 °C (c) are reported in the form of α and its first derivative, the reaction rate as a function of time. The crucible temperature of the reference experiments is presented on the secondary axis as a function of time. During the reference experiments, quartz was subjected to the same gas atmospheres and temperatures as the ore. The vertical dotted line indicates at what time (between 12 and 13 minutes) the target temperature (700 °C, 800 °C, and 900 °C) was reached
It is evident from Figures 11a through c that within the 70 pct CO 30 pct CO2 gas atmosphere, the degree and rate of pre-reduction and decomposition increased with an increase in temperature. The increase is consistent with other studies conducted at a similar temperature range.[4,14,19] In some cases, the rate of pre-reduction peaked before reaching the target temperature, which is indicative of a high reaction rate. This was indicative that pre-reduction could be performed at lower temperatures or for shorter periods, which would be economically beneficial from an industrial perspective. Prospective research should determine the optimal pre-reduction conditions. In the hydrogen-containing gas atmospheres, only the rate of pre-reduction and decomposition increased with an increase in temperature while the degree stayed more or less the same. The 100 pct H2 and 70 pct H2 30 pct H2O gas atmospheres had relatively the same reaction rate. However, one would expect the 100 pct H2 gas atmosphere to have a higher reduction rate due to its lower pO2[4] and it provides 4 L/min instead of 2.8 L/min of hydrogen during the pre-reduction process. The abnormality can be explained by the following two reasons. Firstly, there is more noise present in the data obtained from the 70 pct H2 30 pct H2O gas atmosphere which makes the results less accurate. Secondly, the weight increase at the beginning (first 12 to 13 minutes) of the pre-reduction in 70 pct H2 30 pct H2O causes the reaction rate to increase.
During the first 5 to 7 minutes of pre-reduction in the 70 pct H2 30 pct H2O gas atmosphere, an increase in weight was observed. The crucible gradually heated up from around 29 °C to the target temperature as it moved into the furnace. The target temperature was reached between 12 and 13 minutes as indicated by the dotted line in Figure 11. The 70 pct H2 30 pct H2O gas enters the crucible at 29 °C causing the water vapor to condense which leads to an increase in weight. As the crucible temperature increases, the condensed water starts to evaporate causing the weight percentage to decrease explaining the negative bump at the beginning of the experiment. The higher the temperature of the furnace the faster the crucible will heat up to reach the target temperature, causing the variation in bump broadness at each target temperature. Reference experiments were run with quartz and a similar bump appeared at the beginning of the pre-reduction process.
The rate of pre-reduction and decomposition was the highest in the hydrogen-containing gas atmospheres. The rate increase was expected when assuming that the diffusion characteristics of the gas atmosphere improve with the presence of hydrogen. Hydrogen has a kinetic diameter of 2.89 Å, which is smaller than that of CO (3.76 Å) and CO2 (3.30 Å) gas.[34] The 100 pct H2 gas had a maximum reduction rate that was 88, 45, and 30 pct higher when compared to the 70 pct CO 30 pct CO2 gas mixture at 700 °C, 800 °C, and 900 °C, respectively. Thus, 100 pct H2 gas had a more significant effect at lower temperatures than at higher temperatures. Similar results were obtained by Ngoy et al.[8] and Schanche et al..[4] In Figures 11a through c, the alpha curves of samples pre-reduced in 70 pct CO 30 pct CO2, 70 pct H2 30 pct H2O, and 100 pct H2 gas atmospheres at 700 (a), 800 (b), and 900 °C (c) flattened after a certain period indicating that all the Mn-oxides in the sample reduced to manganosite; this was also confirmed by the chemical analysis (Table II). In addition, the flattening of the alpha curves shows that the discrepancy between the chemical analysis and the XRD (Figure 5) analysis (as discussed in Section III–B) is negligible.
Furthermore, considering the chemical analysis it is expected that all the 70 pct H2 30 pct H2O alpha curves (Figures 11(a) through (c)) and the 70 pct CO 30 pct CO2 at 900 °C alpha curve in Figure 11(c) end at α = 1 like the 100 pct H2 curves at 700 (a), 800 (b) and 900 °C (c). However, this is not the case and can be explained by the calculated theoretical weight loss (Table I) and XRD analysis (Figure 5). The highest weight loss percentage, i.e., 23 wt pct, was obtained in samples pre-reduced in 100 pct H2. The calculated theoretical weight loss was 21 wt pct, which is 2 wt pct lower than the highest weight loss detected. This weight difference is due to the complete reduction of Fe-oxides (Fe2O3 to Fe) because when assuming that the Fe-oxides reduce to metallic Fe the calculated theoretical weight loss is 23 wt pct (the same as the highest weight loss detected).
Total CO2 Formation from Carbonate Decomposition
The utilization of 100 pct H2 and 70 pct H2 30 pct H2O gas atmospheres as reductants during the pre-reduction process caused the WGSR to take place. The WGSR proceeds according to Reaction [R10].
Hydrogen and water vapor were the input gas, and the CO2 only formed due to the decomposition of carbonates. Hence, CO and water vapor formed endothermically. The sum of the pct CO and pct CO2 detected by the gas analyser was used to determine the relative percentage of CO2 formation from the decomposition of carbonates during the pre-reduction process. These curves are shown in Figure 12.

Total percentage CO2 formation achieved through carbonate decomposition detected by the gas analyser during the pre-reduction of UMK ore in 100 pct H2 and 70 pct H2 30 pct H2O gas atmospheres at 700 (a), 800 (b), and 900 °C (c). The crucible temperature of the gas mixtures as a function of time is presented on the secondary axis. The black lines represent the crucible temperature in the 70 pct CO 30 pct CO2 (dotted), 70 pct H2 30 pct H2O (solid), and 100 pct H2 (dashed) gas atmospheres (Color figure online)
Figure 12 shows that an increase in temperature caused the peaks to intensify and become narrower, meaning that the rate of carbonate decomposition increased with an elevation in temperature while the percentage of carbonate decomposition stayed the same.
The slightly lower total pct CO2 formation peak achieved in 100 pct H2 gas atmosphere could be due to the pct CO and pct CO2 in the off-gas being more diluted when reaching the gas analyser comparing the 70 pct H2 30 pct H2O gas atmosphere. During the pre-reduction in 70 pct H2 30 pct H2O gas atmosphere 30 pct less H2 enters the crucible meaning that 30 pct less H2 will exit the crucible toward the gas analyser. The added water vapor and the water vapor that forms during the pre-reduction process move out of the crucible into a glass bottle where the water vapor condenses, hence it does not affect the gas analyses. Therefore, the percentage of measured CO and CO2 gas in the off-gas per minute will be higher in 70 pct H2 (2.8 L/min) 30 pct H2O gas atmosphere than in 100 pct H2 (4 L/min).
Furthermore, Figure 11 shows that the rate of carbonate decomposition is faster at the beginning of the pre-reduction process when the pCO2 is still low (and the driving force high) but after the maximum total pct CO2 formation is reached the decomposition rate starts to slow down. This could be due to kinetics where the CO2 in the outer part of the particle experiences a lower pCO2 but in the middle of the particle, it sees a pCO2 of 1 atm. Thus, the H2 has to diffuse in and out of the particle; hence, it is natural that the decomposition rate slows down as the decomposition happens more and more to the center of the particle.
As previously mentioned, carbonates decompose faster in the hydrogen-containing gas atmospheres than in the 70 pct CO 30 pct CO2 gas atmosphere (Figure 11); hence, it requires more energy. The higher degree and rate of carbonate decomposition caused the sample temperature in the hydrogen-containing gas atmospheres to be lower in comparison to the sample temperature measured for the 70 pct CO 30 pct CO2 gas atmosphere. The decrease in sample temperature can be seen in Figure 12.
Decrepitation of UMK Ore
In the industry, the presence of undersized Mn-ore material in the furnace will limit the permeability of the pre-reduction zone by stopping the uniform gas flow. The gas flow is responsible for the pre-reduction of the Mn ores higher up in the furnace. Hence, it is desired that the charge fed to the furnace does not decrepitate too much.[19] Decrepitation is caused by several factors. When the ore heats up the outer layer heats up faster than the inside of the ore, causing the areas to expand differently which will increase the internal stress of the ore.[19] Stress can also be caused by phase transformation due to generating an anisotropy that forms cracks that would eventually fracture.[35] Elimination of structural water will cause an increase in vapor pressure inside the pores that can lead to crack formation.[35] Furthermore, the decomposition of carbonates and the loss of oxygen through the pre-reduction of higher oxides of the ore will also contribute to the increase in decrepitation.[32] Figure 13 shows the sample size fraction distribution after pre-reduction of UMK ore in 70 pct CO 30 pct CO2, 70 pct H2 30 pct H2O, and 100 pct H2 at 700 (Figure 13(a)), 800 (Figure 13(b)), and 900 °C (Figure 13(c)).

Size distribution after reduction experiments in 70 pct CO 30 pct CO2, 70 pct H2 30 pct H2O and 100 pct H2 at 700 (a), 800 (b), and 900 °C (c)
According to Figure 13, the size distribution of the UMK ore after pre-reduction shows that 45 to 55 pct of particles are finer than 9.5 mm, indicating that about half of the ore remained in their original particle size (9.5 to 16 mm). Similar results were obtained in Reiersen et al.[20] and Mukono et al.[36] This was observed for all the samples; thus, it can be seen that the employment of 70 pct H2 30 pct H2O and 100 pct H2 as reductants does not change the decrepitation of UMK ore in the 700 °C to 900 °C temperature range compared to 70 pct CO 30 pct CO2.
Furthermore, the slope beyond less than 9.5 mm is flatter at 800 °C than at 700 °C and 900 °C indicating that the samples pre-reduced at 800 °C decrepitated into finer particles. According to the chemical analysis, all the samples were completely reduced and most of the carbonates decomposed in the samples pre-reduced in 70 pct H2 30 pct H2O, 100 pct H2, and 70 pct CO 30 pct CO2 at 900 °C. Even though, the lower temperature CO/CO2 experiments still had some remaining carbonates (Figure 3) it did not decrepitate differently. Hence, it seems like the release of CO2 from the ore during the decomposition of carbonates has an insignificant effect on the degree of decrepitation. These results are also in agreement with the porosity analysis (Table III).
Conclusions
Experiments were performed to investigate the pre-reduction of UMK ore in hydrogen-containing and CO/CO2 gas atmospheres at temperatures 700 °C to 900 °C. According to the chemical analysis, the majority of the weight loss was caused by the decomposition of carbonates (17 wt pct). The rest of the weight loss was due to the pre-reduction of Fe- and Mn- oxides, i.e., 0.89 wt pct (Fe2O3 to FeO) or 2.56 wt pct (Fe2O3 to Fe) and 3.47 wt pct (O/Mn(i) to MnO), respectively. The pre-reduction of higher oxides and the decomposition of carbonates proceeded in parallel.
The higher Mn-oxides in all pre-reduced samples reduced to MnO. The higher Fe-oxides were reduced to FeO in the 70 pct CO 30 pct CO2 and 70 pct H2 30 pct H2O atmospheres, while complete reduction (Fe2O3 to Fe) was achieved in the 100 pct H2 gas atmosphere. Most of the carbonates decomposed in samples pre-reduced in CO/CO2 at 900 °C and in the hydrogen-containing atmospheres at all temperatures. The use of hydrogen as a reductant increased the rate of pre-reduction and carbonate decomposition, which was further accelerated at elevated temperatures.
The XRD results showed that the braunite (3(Mn,Fe)2O3·MnSiO3)/bixbyite ((Mn,Fe)2O3) reduced to manganosite (MnO) without a stable intermediary phase (hausmannite (Mn3O4)). For the CO/CO2 and H2/H2O gas atmospheres, the FeO was contained in the monoxide phase. This was not the case in 100 pct H2 atmosphere where the FeO had been reduced to metallic Fe. Therefore, the use of hydrogen as a reductant allowed the complete reduction, i.e., metallization, of Fe-oxides present in the ore.
Small metallic Fe-rich phases embedded in the Mn- and Fe-rich phase of samples pre-reduced in 100 pct H2 at 800 °C were observed in SEM analysis. The SEM and EPMA showed that the pre-reduced ore consisted of Mg- and Ca-rich, Mn- and Fe-rich, and Mg-, Si-, K-, and Na-rich phases. The employment of hydrogen during the pre-reduction process did not have a significant effect on the porosity or decrepitation of UMK ore. Considering both the process- and environmental-related benefits of using hydrogen as a reductant, it was proposed that a techno-economical study is performed on the use of hydrogen as a reductant during the pre-reduction process. More so, the exploration of the possible incorporation of hydrogen into existing infrastructure is also proposed.
References
W.F. Cannon, Manganese—It turns iron into steel (and does so much more) (U.S. Geological Survey, 2014), https://www.911metallurgist.com/blog/wp-content/uploads/2016/04/Manganese.pdf. Accessed 26 June 2022.
G. Pochart, L. Joncourt, N. Touchard, and C. Perdon: World, 2007, vol. 800, p. 1200.
S.E. Olsen, M. Tangstad, and T. Lindstad: Production of Manganese Ferroalloys, 1st ed. Tapir Academic Press, Trondheim, 2007, p. 247.
T.L. Schanche and M. Tangstad: Minerals, 2021, vol. 11, p. 1097.
K. Swamy, D. Robertson, P. Calvert, and D. Kozak: Infacon, 2001, vol. 9, pp. 293–301.
T. Kazdal, R. Haas-Wittmuess, S. Richter, S. Lang, C. Binder and M. Reuter: International Ferro-Alloys Conference (Infacon XVI) 2021 Proceedings on SSRN, 2021, p. 20.
M. Tangstad, K. Ichihara and E. Ringdalen: Infacon XIV, 2015, pp. 99–106.
D. Ngoy, D. Sukhomlinov, and M. Tangstad: Iron Steel Inst. Jpn. Int., 2020, vol. 60, pp. 2325–31.
K. Berg and S. Olsen: Metall. Mater. Trans. B, 2000, vol. 31B, pp. 477–90.
M. Kumar, S. Ranganathan and S. Sinha: Infacon XI, 2007, pp. 241–46.
A. Bhalla and R.H. Eric: Infacon XIV, 2015, vol. 1, pp. 461–69.
T.A. Larssen and M. Tangstad: Metall. Mater. Trans. B, 2022, vol. 53B, pp. 1–13.
Y. Wang, S. Lin, and Y. Suzuki: Energy Fuels, 2007, vol. 21, pp. 3317–21.
D. Sukhomlinov and M. Tangstad: Norwegian University of Science and Technology, Trondheim, unpublished research, 2021.
M. Visser, H. Smith, E. Ringdalen, and M. Tangstad: Infacon XIII, 2013, vol. 1, pp. 553–66.
T.P. van Kaam: Hvordan Ilmenitt Reduksjon med Hydrogengass Endres ved Introduksjon av Vanndamp, Norwegian University of Science and Technology, Trondheim, 2020.
S.C. Lobo: Experimental Investigations and Modelling of Solid-State Ilmenite Reduction with Hydrogen and Carbon Monoxide, Norwegian University of Science and Technology, Trondheim, 2015.
J.D. Steenkamp, J.E. Olsen and Q.G. Reynolds, Furnace Tapping 101. Springer, 2022.
O. Biørnstad, Decrepitation of Comilog, Assmang and UMK manganese ores during prereduction, Degree of Philosophiae Doctor, Norwegian University of Science and Technology, Trondheim, 2020.
H.S. Reiersen, Behavior of manganese ores during pre-reduction in small scale furnaces, Degree of Philosophiae Doctor, Norwegian University of Science and Technology, Trondheim, 2020.
C.W. Bale, P. Chartrand, S. Degterov, G. Eriksson, K. Hack, R.B. Mahfoud, J. Melançon, A. Pelton, and S. Petersen: Calphad, 2002, vol. 26, pp. 189–228.
A. Roine: Pori, Finland: Outotec, 2018.
J. Criado, F. Gonzalez, and M. Gonzalez: J. Therm. Anal., 1982, vol. 24, pp. 59–65.
J.M. Valverde, A. Perejon, S. Medina, and L.A. Perez-Maqueda: Phys. Chem. Chem. Phys., 2015, vol. 17, pp. 30162–76.
M. Zaki, M. Hasan, L. Pasupulety, and K. Kumari: Thermochim. Acta, 1997, vol. 303, pp. 171–81.
K. Piotrowski, K. Mondal, H. Lorethova, L. Stonawski, T. Szymański, and T. Wiltowski: Int. J. Hydrog. Energy, 2005, vol. 30, pp. 1543–54.
A. Pineau, N. Kanari, and I. Gaballah: Thermochim. Acta, 2006, vol. 447, pp. 89–100.
A. Pineau, N. Kanari, and I. Gaballah: Thermochim. Acta, 2007, vol. 456, pp. 75–88.
D.P.J. Davies, J.J. Ramos-Hernandez, M. Tangstad, E. Ringdalen, J.P. Beukes, D.G. Bessarabov, and S.P. du Preez: Minerals, 2022, vol. 12, p. 534.
J. Davies, M. Tangstad, E. Ringdalen, J.P. Beukes, D. Bessarabov, and S.P. du Preez: Minerals, 2022, vol. 12, p. 911.
M. Tangstad, D. Leroy and E. Ringdalen: The Twelfth International Ferroalloys Congress, Infacon, Helsinki, unpublished research, 2010.
G.L.D. Faria, J.A.S. Tenório, N. Jannotti Jr., and F.D.S. Araújo: Int. J. Miner. Process., 2013, vol. 124, pp. 132–37.
A. Cheraghi, H. Yoozbashizadeh, and J. Safarian: Miner. Process. Extr. Metall. Rev., 2020, vol. 41, pp. 198–215.
N. Mehio, S. Dai, and D. Jiang: J. Phys. Chem. A, 2014, vol. 118, pp. 1150–54.
G.L.D. Faria, N.C.D.S. Viana, N. Jannotti Júnior, C.B. Vieira and F.G.D.S. Araújo: The Twelfth International Ferroalloys Congress Sustainable Future, Infacon, Helsinki, unpublished research, 2010.
H.S.R. T. Mukono, T.L. Schanche, M. Wallin, M. Tangstad: Norwegian University of Science and Technology Trondheim, unpublished research, 2022.
Acknowledgments
This study was funded by the Research Council of Norway (RCN) as the Thanos project (INPART Project 309475) and is gratefully acknowledged.
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Funding
Open access funding provided by NTNU Norwegian University of Science and Technology (incl St. Olavs Hospital - Trondheim University Hospital).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Davies, J., Tangstad, M., Schanche, T.L. et al. Pre-reduction of United Manganese of Kalahari Ore in CO/CO2, H2/H2O, and H2 Atmospheres. Metall Mater Trans B 54, 515–535 (2023). https://doi.org/10.1007/s11663-022-02705-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-022-02705-0