
Abstract
A low-alloy medium-carbon bainitic steel was isothermally tempered at 300 °C for up to 24 hours which led to a significant hardness decrease. In order to explain the decreasing hardness, extensive microstructural characterization using scanning and transmission electron microscopy, X-ray diffraction, and atom probe tomography was conducted. The experimental work was further supplemented by thermodynamic and kinetic simulations. It is found that the main underlying reason for the hardness reduction during tempering is related to dislocation annihilation, possibly also with corresponding changes in Cottrell atmospheres. On the other hand, cementite precipitate size, effective grain size of the bainite, and retained austenite fraction appear unchanged over the whole tempering cycle.
Similar content being viewed by others


1 Introduction
Bainitic steels are used in applications such as crankshafts, seat belt buckles, rails or bearings, all of which require high performance in terms of hardness and wear resistance combined with a good toughness. When choosing the optimal material for a certain component, geometrical constraints together with desired properties are determining factors. Alloying must be used to obtain the desired hardenability in order to avoid the formation of unwanted phases leading to poor mechanical properties or thermal cracking, but at the same time the cost should be minimized. The alloying content is therefore kept to a minimum and the processing time is kept short to reduce cost. The bulk mechanical properties of a steel product can often be achieved by a single heat treatment; however, additional processing steps such as coating in order to enhance wear, corrosion, and friction properties of the surface may result in further processing steps at elevated temperature resulting in tempering of the bulk steel.
Bainite was first observed in 1920 by Hultgren,[1] but was at that time identified as “secondary ferrite.” Still, a century after the discovery, the full extent of the formation of bainite[2,3,4] and how the structure correlates to the mechanical properties is not completely understood. There are several studies on the contribution of different bainitic microstructural features to the mechanical properties.[5,6,7,8,9,10] Much focus has been on the effect of the bainitic ferrite sub-units and the retained austenite, but the carbon content and the dislocation density in the ferrite have also been discussed. It should be emphasized that many of the previously investigated steels contained high amounts of Si which are of interest thanks to their excellent mechanical properties, yet their main drawback is long processing times.[11] When processing time is an important factor, low Si bainitic steels are more suitable.
It has been reported that bainitic ferrite in high Si grades is rather stable upon tempering and only small changes in the width of packets have been observed for tempering up to 30 minutes at 550 °C.[9] The high Si content destabilizes the cementite to such an extent that it may not precipitate; instead the remaining austenite in such a case is enriched with carbon and is thus stabilized and remains as films and blocks between the plates and packets of the bainitic ferrite. In the high Si grades, tempering will cause the retained austenite to undergo an eutectoid transformation when the temperature/time is high/long enough for the solutes to partition.[12] This is often given as one of the main reasons for the change in mechanical properties during tempering.[5,6,7,9,13] Low Si grades, on the other hand, have a much lower amount of retained austenite due to the continuous formation of cementite during the bainitic transformation. Still, a significant loss in hardness is observed when low Si grades are exposed to long isothermal treatments after being fully transformed to bainite which thus cannot be explained by transformation of the retained austenite. The reason for the hardness reduction in low Si bainitic steels during tempering has not been thoroughly investigated previously.
Thus, the aim of this study is to investigate how the changes in microstructure relate to the changes in mechanical properties, mainly the hardness, which occur during tempering. After identifying the reasons for the hardness decrease, a future materials design challenge will be to design a more tempering-resistant bainitic material.
2 Experimental Methods
2.1 Material, Heat Treatments, and Hardness Measurements
The steel used in this study was a soft annealed 1.1-mm-thick plate with the chemical composition given in Table I. Samples of size 6 × 6 × 1.1 mm3 were cut and austenitized at 880 °C for 20 minutes in a tube furnace under argon atmosphere followed by quenching in a Bi-Sn metal bath at 300 °C. The samples were isothermally held at that temperature for different durations in order to allow for complete transformation to bainite and tempering before quenching in brine.
Dilatometry testing was conducted in a BÄHR DIL 805 A/D using a heating rate of 5 K/s up to the austenitization temperature of 880 °C, holding for 20 minutes, and thereafter cooling to 300 °C using a cooling rate of about 300 K/s to avoid any other transformations. Quench-hardening was also performed to achieve a fully martensitic microstructure. In that case, a cooling rate of 300 K/s was maintained as far as possible down towards room temperature. The dilatometry samples were cut by wire electrical discharge machining into a size of 10 × 4 × 1.1 mm3.
Hardness measurements were performed using a semi-automatic micro-hardness tester Future-Tech FM. Fifteen Vickers hardness indents using a load of 1 kg were performed per sample. Indentations were conducted on a mechanically polished surface.
2.2 Microscopy
Samples for microstructural characterization were mechanically polished in steps until final polishing using colloidal silica with a particle size of 0.02 μm. Prior to light optical microscopy (LOM) and secondary electron (SE) imaging by scanning electron microscopy (SEM), samples were etched with either 2 vol pct nital or 2 mass pct picric acid dissolved in ethanol. For SEM characterization using backscattered electrons (BSE), electron channeling contrast imaging (ECCI), and electron backscatter diffraction (EBSD), non-etched samples were used. LOM and SEM were conducted using a Leica DM RM microscope and a JEOL JSM-7800F, operating at 12 kV with a working distance of 7 mm for imaging, respectively.
Thickness measurements of bainite plates were performed from images using ECCI. When measuring the thickness of the individual bainite plates, the mean linear intercept method was used as shown in Figure 1. A stereological correction was applied on the randomly selected sections of the packets to estimate the true thickness. Eq. [1] was used, where \( \bar{L}_{\text{T}} \) is the measured thickness and t is the true thickness.[14]

Illustration of plate thickness measurements on SEM ECCI images
For EBSD, a step size of 50 nm was applied, the SEM was operating at 12 kV with a working distance of 20 mm. Measurements and post-processing of data were conducted using the software Bruker QUANTAX CrystAlign.
The sizes of the cementite and niobium carbide particles were measured on carbon extraction replica samples prepared by mechanically polishing samples and etching with 2 pct picric acid followed by sputtering a 100 to 200 Å thick carbon layer on the etched surface. The carbon film was thereafter cut in a pattern of about 1x1 mm2 pieces. The sample was then submerged in 7 pct perchloric acid to release the carbon film from the sample surface. The films were cleaned two times in ethanol and finally in distilled water before put on a circular Cu-grid with a diameter of 3 mm. The samples were analyzed in a JEOL JEM-2100F transmission electron microscope (TEM) in scanning mode (STEM) operating at 200 kV. The niobium carbides were identified using energy dispersive X-ray spectroscopy (EDS), measured with an Oxford spectrometer.
TEM was also used in dark-field mode to investigate possible presence of retained austenite at inter-plate boundaries in the bainitic ferrite. The samples for dark-field imaging were prepared using a focused ion beam SEM (FIB-SEM), FEI Nova Nanolab 600, operating at 30 kV for rough ion milling and 5 kV for final ion polishing.
Samples for atom probe tomography (APT) were prepared using FIB-SEM lift-out in an FEI Versa 3D, finished with annular milling. The rough milling was conducted using an operating voltage of 30 kV and final polishing was performed at 5 kV. The APT experiments were performed using a LEAP 3000X HR (Imago Scientific Instruments) in laser pulse mode with a pulse energy of 0.3 nJ. The sample temperature was set to 50 K and the evaporation rate was 0.5 pct. The acquired data were analyzed using IVAS 3.4.12 (Cameca), and reconstructions were made using voltage mode with an image compression factor of 1.65, a field factor of 4.0 and an evaporation field of 25 V/nm. The carbon content in the matrix was estimated by evaluating the carbon content in cubes of 10 × 10 × 10 nm3 and then averaged. These cubes were placed in regions of the matrix where no obvious clustering could be detected from the 3D atom maps.
2.3 XRD
Samples for XRD were polished with a final mechanical polishing step using 1 μm diamond suspension, followed by electro-polishing in 7 vol pct perchloric acid at 6 V for approximately 30 seconds to remove any mechanically induced strain and/or martensite. The XRD measurements were conducted using a Bruker D8 Discover with Cu-Kα radiation and a LynxEye 1D energy dispersive detector. Data were recorded from 40 to 140 deg 2θ with angular intervals of 0.02 deg and a counting time per step of 8 seconds. The Cu-Kα2 was subtracted from the data using the software DIFFRAC.EVA V4.0, before further analyses of the peaks using the software LIPRAS.[15] The peaks were least-square fitted using a pseudo-Voigt function, see Figure 2. The instrumental peak broadening was determined using an Al2O3 reference sample and then subtracted from the measured peak widths by using the closest reference peak with respect to 2θ.

XRD data fitted using a pseudo-Voigt function, the intensity is given in an arbitrary unit
The carbon content in the bainitic ferrite was evaluated from the d-spacing of the (222) peak and Eq. [2].[16] It should be noted that there are several alternative equations in the literature. The soft annealed condition was used as a reference, assuming ferrite has an equilibrium carbon content in that sample, and the change in d-spacing was then solely related to the change of carbon content in the bainitic ferrite under the assumption that no substitutional element diffusion occurs at the tempering temperature that would affect the change in the lattice parameter. For austenite, Scott and Drillet have shown that the evaluated carbon content can vary widely depending on the equation applied.[17] In this study, it was estimated using Eq. [3] proposed by Dyson and Holmes.[18]
The parameters a0α and a0γ are the experimentally measured lattice parameter [Å] in both Eqs. [2] and [3], aFe is the lattice parameter for pure iron, and alloying contents are given in wt pct.
The total amount of retained austenite was also estimated (trying to adhere to the procedure in ASTM E975-13[19] when possible). However, only one austenite peak was observed and included in the analysis, the 022 γ-peak. On the other hand, six ferrite peaks could be included in the analysis of the volume fraction of austenite. Cementite was accounted for using the equilibrium values calculated using Thermo-Calc software.[20,21]
The full width at half maximum (FWHM) was used to calculate the dislocation density applying the Williamson–Hall method[22,23] where peak broadening ΔK (Eq. [4]) is plotted as a function of the diffraction vector, K (Eq. [5]).
λ is the X-ray wavelength and \( \theta \) is the Bragg angle in radians. By linearly fitting the peak broadening with respect to the diffraction vector, the slope will correspond to the mean square microstrain ε as
The dislocation density ρ could then be calculated as
where k is a constant set to 14.4 for a BCC metal with the Burger’s vector (b) along [111].[23] F is the interaction parameter which can be assumed to be 1 and \( b = \frac{\sqrt 3 }{2}\,*\,2.87\,{\text{to}}\,2.5\,{\AA} \) where 2.87 Å is the lattice parameter of pure BCC Fe.[22,23]
2.4 Thermodynamic Calculations and Kinetic Simulations
Thermodynamic calculations were performed using the Thermo-Calc software and the TCFE9 database to evaluate equilibrium conditions.[20,21] Coarsening was simulated using the DICTRA software and the TCFE9 and MOBFE4 databases. A spherical symmetry of the particle and surrounding matrix was assumed and the simulations were performed in 1D. The particle size was set to the smallest of the experimentally measured particles to simulate the case where the driving force of coarsening is the highest. The interfacial energy was varied between 0.1 and 0.7 Jm−2. In addition, TC-PRISMA was used to simulate precipitation and growth, using the same databases.
3 Results and Discussion
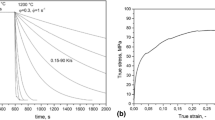
The isothermal transformation time to obtain a fully bainitic structure was determined to about 8 minutes using dilatometry. The dilatometry curve can be seen in Figure 3(a); it takes about 350 seconds to reach 95 pct transformation and 499 seconds to reach 99 pct transformation, evaluated as a ratio with respect to the maximum length change. That complete transformation had occurred after 8 minutes was confirmed by interrupted isothermal treatments in the metal bath. The martensite start temperature (MS) was also determined with dilatometry for two samples (see example in Figure 3(b)), and the MS was found to be 259 °C ± 2 °C. The MS was also calculated to be 263 °C using a thermodynamic-based model.[24] Hence, both experiments and calculations provide an MS which is well below the isothermal heat treatment temperature, and the material studied can therefore be considered to be fully bainitic.

Dilatometry curves for the imposed bainitic heat treatment. (a) The austenitization time is 20 min followed by rapid cooling and isothermal holding for 1 h at 300 °C. 95 pct of the maximum elongation is reached after 350 seconds and 99 pct is reached after 499 seconds. (b) The elongation for heating and austenitization directly followed by quenching to determine the MS temperature
To study tempering of bainite, isothermal heat treatments were performed in the time interval from 10 minutes to 24 hours. A hardness reduction during the extended isothermal tempering at 300 °C was observed as shown in Figure 4. For the same process, Figure 5 shows that there is no obvious microstructural difference between the initial sample tempered for 10 minutes and the final sample tempered for 24 hours. However, it was observed that samples isothermally tempered for 24 hours were more easily etched than samples tempered for only 10 minutes.

Hardness as function of heat treatment time at 300 °C, note the broken x-axis before the last time of 1440 min (24 h)

SEM micrographs after isothermal heat treatment at 300 °C for 10 min, (a) and 24 h, (b). Both samples were etched with picric acid
In order to explain the hardness decrease, it will be necessary to isolate all governing factors by careful characterization. We assume that the following factors should contribute to the hardness (strength) of the material: grain boundary strengthening, precipitation strengthening, dislocation strengthening, and solid solution strengthening.
3.1 Grain Boundary Strengthening
In bainite, grain boundary strengthening is related to the plate and packet size of the bainitic ferrite since their boundaries are possible obstacles for dislocation movement and the mean free path of the dislocations determines the strengthening effect. In the EBSD micrographs using inverse pole figure (IPF) coloring, Figure 6, the packets, which are separated by high-angle boundaries, are clearly seen by the difference of color. However, the smaller plates, separated by lower angle misorientations, within the packets cannot be distinguished. The mean free path for dislocations is evaluated as the mean size of packets, i.e., mean distance between high-angle boundaries. This evaluation was performed by analyzing the mean distance between high-angle boundaries considering different misorientation angle criteria. The results from this analysis are presented in Figure 7 for misorientation angle threshold varied from 2 to 20 deg. The plates, on the other hand, are aligned parallel to each other and build up the packets, as shown in Figure 1. The boundaries between the plates have much lower misorientation angles and these are more difficult to detect using EBSD, both because of the low misorientation but also because of their smaller size approaching the resolution limit of the measurements. The bainite plate thickness were therefore measured using BSE images and are summarized in Table II.

EBSD micrographs using IPF coloring after heat treatment at 300 °C for 10 min, (a) and 24 h, (b)

Mean free path between the packets as a function of the misorientation angle chosen for the analysis after heat treatment at 300 °C for 10 min and 24 h, respectively
From the microstructural analyses, it may be concluded that the changes in plate and packet thickness during tempering are negligible as presented in Figure 7 and Table II and could thus not explain the observed softening. It should be noted though that high-angle boundaries, as between packets, should be the main obstacle to dislocation movement, while low-angle boundaries, as those between plates, will pose a much smaller obstacle and the dislocations may pass through them with relative ease, according to the literature.[25,26,27] It should be emphasized that if films of austenite separate the plates they are likely to be more important obstacles to dislocation movements. Here, retained austenite was not observed between plates, as further discussed in Section III–B.
The prior austenite grain size is assumed to be constant, since it is only affected by the austenitization step, which was the same for all samples and, hence, its effect on packet and plate size can be neglected.
3.2 Softening Due to Retained Austenite
As previously mentioned, retained austenite has often been attributed a significant contribution to the softening of bainitic steels. In the material investigated in this work, the retained austenite content is very low and difficult to detect using LOM and SEM. Nonetheless, even if the content is low it could have an effect depending on how it is distributed, e.g., if it is in the plate boundaries and is therefore analyzed.
Only one austenite peak could be identified from the XRD measurement indicating that there is only a very small amount present. The uncertainty in the measurements is thus high and close to the amount detected. Thus, no value can be given with certainty. It was also noted that the peak broadening in the 10-minute sample makes it harder to separate the peak from the background than the peak measured in the 24-hour sample. In both samples, the retained austenite was estimated to be around 1 to 3 pct and no statistical difference between them could be determined. In addition to the XRD data, the phase distribution was determined using EBSD as shown in the phase map in Figure 8, indicating that the retained austenite is located at the high-angle boundaries between the packets.

Phase fractions from EBSD data (a, b) and the corresponding area from the IPF maps (c, d). It can be seen that the austenite (red) is located in the high-angle boundaries of ferrite (blue). For color images, the reader is referred to the web version of the paper (Color figure online)
The width and location of the austenite was further studied with TEM in the 10-minute sample, see Figure 9. The thickness of the austenite films is estimated to be in the range 5 to 20 nm and the total amount observed is very low, and in reasonable agreement with the XRD and EBSD. No austenite could be detected between the plates. This strengthens the conclusion above that the plates will pose a minimal obstacle to the movement of the dislocations. The TEM results also show that the thickness of the austenite film is less than the resolution of the EBSD measurements ~50 nm. Therefore, the phase fraction of austenite that was obtained from EBSD is highly uncertain and will not be considered further. By combining the different methods, it can be concluded that the total amount of retained austenite is limited to a maximum of a few percent in both samples and is located in the high-angle boundaries. It can thus be concluded that the austenite only could have a limited effect on the overall softening of the material.

TEM images of sample heat treated at 300 °C for 10 min confirming the presence of austenite in the high-angle boundaries. The left image is a bright-field TEM image where different crystallographic orientations of the ferrite and defects give rise to diffraction contrast, the middle is the same area in dark field, and the right image is a zoom-in at the arrow. No films could be detected in the low-angle boundaries between the bainitic plates
3.3 Precipitation Strengthening
Due to the large amount of cementite in the material, coarsening or spheroidization of the cementite during tempering would lower the hardness and is an important factor to consider.
STEM was conducted on carbon extraction replicas to determine the size of the cementite. Several hundreds of particles were measured and the result is presented in Table III. The standard deviation of the mean size in the measurements is large due to the large size distribution presented in Figure 10. Some variation is observed between 10 minutes and 24 hours but the differences are too small to confirm coarsening. The particle diameter, d, was calculated as an average from both the measured length and width of each particle. DICTRA simulations were performed showing that coarsening of the cementite is negligible after 24 hours at 300 °C, in agreement with the STEM measurement. Both simulation and experiments show that changes in size and shape of the cementite particles cannot be excluded but are very small and thus the corresponding change in hardness is most likely negligible in comparison to other effects.

Size distribution of the length of the cementite particles measured. The data were fitted with a probability density function so that both sets of data are comparable in the histogram
Micrometer-sized niobium carbides were detected in LOM and SEM and nanometer-sized niobium carbides on carbon replicas were detected in TEM. Most of the nano-sized niobium carbides were in the size range 3 to 9 nm. However, neither simulations using DICTRA or TC-Prisma nor experimental results indicate any coarsening after tempering at 300 °C and the same size of the niobium carbides was also detected in carbon replicas from the as-received material indicating that they are fairly stable through the whole process. Furthermore, equilibrium calculations using Thermo-Calc showed that the dissolution of the niobium carbides mainly takes place between 1000 °C and 1280 °C where 95 pct is dissolved. Considering that an austenitization temperature of 880 °C is used in this work, only a very small fraction of the niobium carbides will be dissolved and thus precipitation of niobium carbides will be limited and the contribution to the change in hardness at 300 °C should be negligible. It could also be noted that the total amount of niobium carbides is very low due to the low niobium content in the alloy.
3.4 Solid Solution Strengthening
Solid solution strengthening is a result of both interstitially and substitutionally dissolved alloying elements and a redistribution during tempering could have an impact on the hardness.
3.4.1 Substitutional alloying elements
DICTRA calculations show that the approximate diffusion distance at 300 °C for substitutional alloying elements is so short that it for times considered here could be described as a frozen-in condition, even when considering the higher mobility in grain boundaries and interfaces. The temperature has to be increased to above 400 °C before grain boundary and interface diffusion of substitutional elements could be of importance. APT results presented in Figure 11 support the calculations as no clustering of the substitutional elements can be observed. This is also supported by experimental data from the literature at similar temperatures.[28,29,30,31,32]

Elemental maps of different substitutional elements from the APT measurement on the 24-h sample
3.4.2 Interstitial alloying elements
The carbon content in the austenite was measured using XRD and was evaluated using Eq. [3]. It is very high as presented in Table IV, but in line with observations in the literature using APT measurements on thin-film austenite where it is shown that the carbon content tends to be higher in thin austenitic films than in blocky austenite.[33,34,35,36,37] Thus, given the thin thickness of the retained austenite films in the current work, see Figure 9, and the demonstrated stability during the quenching and isothermal tempering, the values presented in Table IV seem reasonable. However, the error is presumably larger than indicated by the standard deviation due to the very small amount of austenite and the fact that only three samples were measured for each time; there are also additional uncertainties from the equation itself. In this study, no peak splitting of the austenite peaks could be observed; however, small effects from residual stresses cannot be excluded. This has frequently been observed in prior studies where the peak corresponding to a higher carbon content has been attributed to the thin-film austenite and the peak corresponding to a lower carbon content peak to the blocky austenite.[38,39,40] This is expected since no blocky austenite was observed in the studied material.
Carbon within the bainitic ferrite could plausibly account for parts of the strength in bainitic steels. Thus, it is important to investigate if the content of carbon differs between the 10-minute and 24-hour samples. The location of the carbon is also important for the evaluation, if it is dissolved in the matrix or if it is bound to dislocations as Cottrell atmospheres. The carbon content in the bainitic ferrite was determined using both XRD and APT, where APT also indicates the distribution of carbon. Equation [2] was used to calculate the carbon content from the XRD results[16] and is presented in Table IV. The results from the 10-minute and 24-hour samples are somewhat overlapping and the difference in the average composition is less than 0.05 wt pct.
There is a large spread in the APT measurements presented in Figure 12 and Table IV but the result does however indicate that there is a decrease of the carbon content with increasing time. Comparing the APT results for the 10-minute and 24-hour samples in Figure 12, it can be seen that the 10-minute sample has more clustered carbon atoms in the matrix than the 24-hour sample. These clusters could very well be Cottrell atmospheres located at the vicinity of dislocations. The clusters could also be a result of other defects or small carbides or a combination of the above. However, any clear conclusion regarding the carbon is hard to make due to the very limited volume and number of samples investigated. This also results in a high standard deviation for both APT and XRD measurements of the carbon content in the bainitic ferrite. The observation from the APT, if assuming that carbon is clustering at the vicinity of dislocations, correlates well with the decrease in dislocation density with increasing tempering time which is described in more detail in the next section.

APT elemental maps of carbon for samples heat treated for 10 min and 24 h at 300 °C. A few large cementite particles are seen together with some minor enrichments, either at dislocations or very small carbides
Thus, by combining the results from APT and XRD it is indicated that carbon will have some effect on the solid solution strengthening. Further, since much of the carbon in the ferrite probably is located in Cottrell atmospheres, all carbon detected in the ferrite matrix will not contribute to solid solution strengthening as if it would have been randomly distributed in the matrix. Both APT and XRD indicate that the amount of carbon found in the ferrite matrix exceeds the equilibrium value for the temperature in both samples.
It may further be assumed that the carbon in the matrix is closely related to the dislocations and it is therefore difficult to separate the contributions from the two strengthening mechanisms.[10]
3.5 Effect of Dislocations and Internal Stresses on Hardness
The dislocation density is calculated from XRD data and plotted as function of isothermal heat treatment time in Figure 13. It is evident that the dislocation density decreases during the whole tempering time but at a higher rate in the beginning. The initial value for the 10-minute sample seems to be rather high when compared to results in the literature,[6,10,41,42] as presented in Table V. However, for the longer heat treatment times the dislocation density does not deviate as much from the values reported in the literature. It should be emphasized that other studies do not account for the change over time after the materials have been fully transformed to bainite as reported in this work; instead one value at each temperature is reported. It is therefore difficult to make a direct comparison since the isothermal transformation time evidently is an important factor. The transformation times will also vary depending on alloy composition which is not always clearly stated, e.g., bainitic steels containing high amount of Si have a rather long heat treatment time to be fully transformed. However, it is here shown a clear decrease in dislocation density with tempering time which will have an impact on the hardness.

Dislocation densities calculated from the XRD data as function of isothermal heat treatment time at 300 °C
4 Contributions to Hardness Decrease
It has within this work been shown that the change in hardness due to tempering could be strongly related to the change in dislocation density. It is therefore of interest to estimate the hardness decrease due to the decrease in dislocation density to compare with the experimentally observed decrease in hardness. There are several values and equations[8,12,43] in the literature to estimate the strength contribution \( \sigma_{\text{d}} \) due to dislocations. Bhadeshia[12] suggested
where ρd is the dislocation density, and this equation will be used here. The standard DIN 50150 was used to convert the calculated strength contribution from dislocation density into a hardness value. The calculated hardness contribution is compared with the measured loss in hardness in Figure 14. By plotting the total hardness on the left y-axis and the calculated hardness from the dislocations density on the right y-axis using the same scale of both y-axes, a direct comparison can be made between the two curves and a good correlation is shown.

Comparison of the measured hardness (left y-axis) and the calculated hardness contribution from the dislocation density (right y-axis). Note that the scale on the two y-axes has the same increment
When considering all the different possible contributions to the loss in hardness, it is clear that there is a more significant effect from the dislocations compared to the other contributions discussed above. The theoretical estimate also shows that the decrease may represent the total loss over time, see Figure 14. The dislocation density may be overestimated at the start due to other contributions affecting the FWHM used to calculate the dislocation density. For example, Hutchinson et al.[44,45] have shown that peak broadening in martensite also can be related to internal stresses and not only to dislocation density. The same principles could also be valid for bainite and could thus explain the relatively high dislocation densities calculated in bainitic steels.[44,45] However, the equations previously used to estimate the dislocation density in bainite have not taken other contributions, such as stresses, into account, thus assuming that the changes in FWHM originate from the dislocation density only. So in order to use previously derived equations and make a fair comparison of the amount of dislocations, the calculation in this work has been conducted in the same way.
Contributions from other strengthening mechanisms cannot be excluded and some contribution from the carbon is expected as solid solution strengthening; however, this contribution is hard to separate from the contribution of the dislocations. The carbon in the ferrite matrix could also presumably cause some tetragonality of the bainitic ferrite, and this could have an impact on the dislocation measurement; however, this topic is a separate and still ongoing discussion.[46,47,48] The other possible contributions presented in this study can only contribute to minor changes in the overall hardness decrease at longer times and cannot explain the rapid hardness decrease at shorter tempering times.
5 Conclusions
The microstructural evolution during tempering of bainite in the present work can be summarized as follows:
-
1.
No or very limited change in the amount of retained austenite occurred.
-
2.
No redistribution of substitutional elements in the bainite was observed. This is in accordance with simulations and previously presented APT data in the literature.
-
3.
A decrease of the carbon content in the matrix was indicated and it is assumed to be closely related to dislocations where carbon is found as Cottrell atmospheres initially.
-
4.
No significant coarsening or spheroidization of cementite or niobium carbides was observed. This is in accordance with kinetic simulations.
-
5.
No structural changes or coarsening on any hierarchic level of the bainitic ferrite structure could be observed.
-
6.
The dislocation density decreases over the whole tempering time, but the change is most significant during the first hour.
Theoretical estimations of the hardness contribution from the dislocations indicate that it is large enough to account for the observed decrease in hardness. Further separating the contributions from the carbon in the matrix and at dislocations was not possible in this work, since the carbon is assumed to be in the vicinity of the dislocations as Cottrell atmospheres, and thus the two effects are likely intertwined. Since no other structural features show any measurable change, it is concluded that the main cause of the decrease in hardness during tempering should be attributed to the decrease in dislocation density.
References
A. Hultgren: A Metallographic Study on Tungsten Steels, Wiley, New York, 1920.
A. Borgenstam, M. Hillert and J. Ågren: Acta Mater., 2009, Vol. 57, 3242-52.
J. Yin, M. Hillert and A. Borgenstam: Metall. Mater. Trans. A, 2017, Vol. 48A, 4006-24.
J. Yin, M. Hillert and A. Borgenstam: Metall. Mater. Trans. A, 2017, Vol. 48A, 1444-58.
A. S. Podder, PhD thesis, University of Cambridge, Cambridge, UK, 2011.
M. J. Peet, PhD thesis, University of Cambridge, Cambridge, UK, 2010.
M. J. Peet, S. S. Babu, M. K. Miller and H. Bhadeshia: Metall. Mater. Trans. A, 2017, Vol. 48A, 3410-18.
S. He, B. He, K. Zhu and M. Huang: Acta Mater., 2017, Vol. 135, 382-89.
C. Garcia-Mateo, M. Peet, F. Caballero and H. Bhadeshia: Mater. Sci. Technol., 2004, Vol. 20, 814-18.
C. Garcia-Mateo and F. Caballero: ISIJ Int., 2005, Vol. 45, 1736-40.
C. Garcia-Mateo, F. G. Caballero, and H.K.D.H. Bhadeshia: ISIJ Int., 2003, Vol. 43, 1821-25.
H. K. D. H. Bhadeshia: Bainite in Steels: Theory and Practice, Institute of Materials, Minerals & Mining, Wakefield, 2015.
M. A. Santajuana, R. Rementeria, M. Kuntz, J. A. Jimenez, F. G. Caballero and C. Garcia-Mateo: Metall. Mater. Trans. A, 2018, Vol. 49A, 2026-36.
L. Chang and H. Bhadeshia: Mater. Sci. Technol., 1995, Vol. 11, 874-82.
G. Esteves, K. Ramos, C. M. Fancher, and J. L. Jones: https://www.researchgate.net/publication/316985889_LIPRAS_Line-Profile_Analysis_Software, 2017.
H. Bhadeshia, S. David, J. Vitek and R. Reed: Mater. Sci. Technol., 1991, Vol. 7, 686-98.
C. Scott and J. Drillet: Scr. Mater., 2007, Vol. 56, 489-92.
D. Dyson: J. Iron Steel Inst., 1970, Vol. 208, 469-74.
ASTM, Standard Practice for X-ray Determination of Retained Austenite in Steel with Near Random Crystallographic Orientation, ASTM, West Conshohocken, 2013.
J.-O. Andersson, T. Helander, L. Höglund, P. Shi and B. Sundman: Calphad, 2002, Vol. 26, 273-312.
A. Borgenstam, L. Höglund, J. Ågren and A. Engström: J. Phase Equilib., 2000, Vol. 21, 269.
G. Williamson and W. Hall: Acta Metall., 1953, Vol. 1, 22-31.
G. Williamson and R. Smallman: Philos. Mag., 1956,Vol. 1, 34-46.
A. Stormvinter, A. Borgenstam and J. Ågren: Metall. Mater. Trans. A, 2012, Vol. 43, 3870-79.
C. Du, J. Hoefnagels, R. Vaes and M. Geers: Scr. Mater., 2016, Vol. 116, 117-21.
T. Ohmura, A. Minor, E. Stach and J. Morris: J. Mater. Res., 2004, Vol. 19, 3626-32.
A. Shibata, T. Nagoshi, M. Sone, S. Morito and Y. Higo: Mater. Sci. Eng. A, 2010, Vol. 527, 7538-44.
G. Miyamoto, J. Oh, K. Hono, T. Furuhara and T. Maki: Acta Mater., 2007, Vol. 55, 5027-38.
W. Song, P.-P. Choi, G. Inden, U. Prahl, D. Raabe and W. Bleck: Metall. Mater. Trans. A, 2014, Vol. 45, 595-606.
C. Zhu, X. Xiong, A. Cerezo, R. Hardwicke, G. Krauss and G. Smith: Ultramicroscopy, 2007, Vol. 107, 808-12.
F. G. Caballero, M. K. Miller, S. S. Babu and C. Garcia-Mateo: Acta Mater., 2007, Vol. 55, 381-90.
F. G. Caballero, M. K. Miller, C. Garcia-Mateo, C. Capdevila and S. S. Babu: Acta Mater., 2008, Vol. 56, 188-99.
F. G. Caballero, M. K. Miller, A. Clarke and C. Garcia-Mateo: Scr. Mater., 2010, Vol. 63, 442-45.
F. G. Caballero, C. Garcia-Mateo, M. Santofimia, M. K. Miller and C. G. De Andrés: Acta Mater., 2009, Vol. 57, 8-17.
F. G. Caballero, M. K. Miller and C. Garcia-Mateo: Acta Mater., 2010, Vol. 58, 2338-43.
F. G. Caballero, M. K. Miller and C. Garcia-Mateo: Mater. Sci. Technol., 2010, Vol. 26, 889-98.
C. Garcia-Mateo, F. G. Caballero, M. K. Miller and J. A. Jiménez: J. Mater. Sci., 2012, Vol. 47, 1004-10.
H. Stone, M. Peet, H. Bhadeshia, P. Withers, S. S. Babu, and E. D. Specht: Proc. R. Soc. A, 2008, Vol. 464, 1009-27.
L. Guo, H. Bhadeshia, H. Roelofs and M. Lembke: Mater. Sci. Technol., 2017, Vol. 33, 2147-56.
A. S. Podder, I. Lonardelli, A. Molinari and H. Bhadeshia: Proc. R. Soc. A, 2011, Vol. 467, 3141-56.
M. Fondekar, A. Rao and A. Mallik: Metall. Trans., 1970, Vol. 1, 885-90.
S. He, B. He, K. Zhu and M. Huang: Acta Mater., 2018, Vol. 149, 46-56.
S. Jonsson: Mechanical Properties of Metals and Dislocation Theory from an Engineer´S Perspective, The Royal Institute of Technology, Department of Materials Science and Engineering, Stockholm, 2010.
B. Hutchinson, D. Lindell and M. Barnett: ISIJ Int., 2015, Vol. 55, 1114-22.
B. Hutchinson, P. Bate, D. Lindell, A. Malik, M. Barnett and P. Lynch: Acta Mater., 2018, Vol. 152, 239-47.
C. Garcia-Mateo, J.A. Jiménez, H.-W. Yen, M. K. Miller, L. Morales-Rivas, M. Kuntz, S.P. Ringer, J.R. Yang, and F. G. Caballero: Acta Mater., 2015, Vol. 91, 162-73.
C. N. Hulme-Smith, M. J. Peet, I. Lonardelli, A. C. Dippel and H. K. D. H. Bhadeshia: Mater. Sci., 2015, Vol. 31, 254-56.
C.N. Hulme-Smith, I. Lonardelli, A.C. Dippel, H.K.D.H. Bhadeshia, Scr. Mater., 2013, Vol. 69, 409-12.
Acknowledgments
The authors are grateful to Dr. Prasath Babu, KTH for help with lift-out TEM sample preparation and TEM investigations, Dr. Fredrik Lindberg, Swerim, for help with STEM investigations, Dr. Kristina Lindgren, Chalmers, for lift-out APT sample preparation, and Dr. Ziyong Hou, KTH, for help with preparing carbon extraction replica samples. The financial support was provided by Husqvarna AB and the European Union’s Horizon 2020 research and innovation program under grant agreement No 686135. Open access funding was provided by Royal Institute of Technology.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Manuscript submitted May 5, 2020; accepted September 9, 2020.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ståhlkrantz, A., Hedström, P., Sarius, N. et al. Effect of Tempering on the Bainitic Microstructure Evolution Correlated with the Hardness in a Low-Alloy Medium-Carbon Steel. Metall Mater Trans A 51, 6470–6481 (2020). https://doi.org/10.1007/s11661-020-06030-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11661-020-06030-6