
Abstract
Downy mildews are the most species-rich group of oomycetes, with more than 700 known species. The relationships within the main downy mildew lineages (i.e. the downy mildews with pyriform haustoria, the downy mildews with coloured conidia, and the brassicolous downy mildews) are increasingly well resolved, and 20 well-characterised monophyletic genera have been described. However, their relationships to each other, the various lineages of graminicolous downy mildews, and to the species subsumed in Phytophthora are still unresolved. Recent phylogenomic studies have suggested a polyphyly of the downy mildews, but with a limited taxon sampling within Phytophthora. As taxon sampling is crucial for inferring relationships between large groups, we have conducted a multigene analysis with a set of 72 Phytophthora species and included all known downy mildew lineages. In addition, we performed approximately unbiased (AU) testing as an additional approach to evaluate major nodes. Our analyses resolve the downy mildews as a monophyletic assemblage in all phylogenetic algorithms used. We thus conclude that the evolution of the obligate biotrophy characteristic of downy mildews was a singular event and that all downy mildew pathogens can be traced to a single ancestor.
Similar content being viewed by others

Introduction
Downy mildews are obligate biotrophic pathogens parasitic to a broad number of angiosperm hosts (Thines and Choi 2016). Traditionally, they have been placed in a family of its own, the Peronosporaceae, which was usually opposed to the cultivable Pythiaceae, which members have a similar sexual reproduction, but less differentiated sporangiophores (Dick 2001). The genus Phytophthora was included within the latter family, as the vast majority of species can be cultivated and most do not produce highly differentiated sporangiophores. However, already early phylogenetic investigations (Cooke et al. 2000; Riethmüller et al. 2002) have revealed a very close relationship of Phytophthora and downy mildews and multigene phylogenies have demonstrated that the downy mildews are indeed nested within the genus Phytophthora (Göker et al. 2007), rendering the genus Phytophthora paraphyletic (Runge et al. 2011a). Thus, the classification of the Peronosporaceae was revised to include the genus Phytophthora and other related genera (Calycofera, Halophytophthora, Globisporangium, Nothophytophthora, Phytopythium) to avoid an inflation of ill-delineated taxa on the family level within the Peronosporales (Hulvey et al. 2010; Beakes and Thines 2017).
To delineate genera in downy mildews, sporangiophore and haustorium morphology are key features that have enabled the identification of monophyletic lineages (Constantinescu 1989, 1998; Constantinescu and Fatehi 2002; Göker et al. 2003; Voglmayr et al. 2004; Constantinescu et al. 2005; Thines et al. 2006, 2007, 2015; Telle and Thines 2012). All of the 20 currently accepted genera of downy mildews are monophyletic and well characterised (Telle and Thines 2012; Thines et al. 2015; Thines and Choi 2016).
Downy mildew diseases are characterised by an interaction, in which the plant usually shows some chlorotic discolouration as a result of infestation, and which remains biotrophic throughout the entire asexual life cycle, from the establishment of the infection to the sporulation of the pathogen through the stomata of the host. Many species of downy mildew can cause severe economic losses, such as Bremia lactucae in lettuce (Lebeda et al. 2008; Wroblewski et al. 2007), Peronospora belbahrii in basil (Belbahri et al. 2005; Thines et al. 2009), Peronospora effusa in spinach (Choi et al. 2015), Plasmopara destructor in balsamines (Görg et al. 2017), Plasmopara viticola in grapes (Salinari et al. 2006; Fontaine et al. 2013), and Pseudoperonospora cubensis in cucurbits (Lebeda and Cohen 2011; Runge et al. 2011b).
Species currently classified in the genus Phytophthora establish necrotrophic to hemibiotrophic interactions with their hosts (Erwin and Ribeiro 1996; Lee and Rose 2010; Jupe et al. 2013). Several of these species are causing destructive diseases, such as potato late blight caused by Phytophthora infestans (Haas et al. 2009, Yoshida et al. 2013) and sudden oak death caused by Phytophthora ramorum (Rizzo et al. 2002; Grünwald et al. 2019). Of special concern are some species with broad host ranges that affect a variety of crops, such as Phytophthora capsici (Hausbeck and Lamour 2004; Lamour et al. 2012), Phytophthora cinnamomi (Burgess et al. 2017), and Phytophthora palmivora (Brasier and Griffin 1979; Ali et al. 2017). While it seems clear that Phytophthora is paraphyletic or polyphyletic with respect to the downy mildews (Göker et al. 2007; Runge et al. 2011a; Sharma et al. 2015), the relationships of various well-defined monophyletic clades are still not fully resolved (Kroon et al. 2004; Blair et al. 2008; Martin et al. 2014; Yang et al. 2017), and the search for synapomorphies that could be used for delineation from other clades is difficult, none of the characters previously used for delineating Phytophthora groups (Waterhouse 1963) fully coincide with the phylogenetic structure of the genus (Cooke et al. 2000; Kroon et al. 2004; Blair et al. 2008). Thus, while it is clear that Phytophthora requires taxonomic revision (Runge et al. 2011a, b), only a minor step towards this has been taken until now (Thines 2023), since the infrageneric relationships of Phytophthora are unresolved (Martin et al. 2014; Yang et al. 2017).
The question of whether the downy mildews are monophyletic or not has been the subject of much dispute over the past years, as phylogenomic approaches with a limited taxon sampling in Phytophthora have often, but not always, supported downy mildew polyphyly (McCarthy and Fitzpatrick 2017; Yang et al. 2017; Bourret et al. 2018; Fletcher et al. 2019).
As there are still rather few genomes of Phytophthora species and downy mildew pathogens available, it was the aim of the current study to test the hypothesis that downy mildews are monophyletic by thorough multigene phylogenetic reconstructions. For this, we included all major oomycete lineages and in addition Kawakamia cyperi, an obligate biotrophic pathogen that causes downy mildew-like symptoms on various sedge species (Miyabe 1904), yet had been usually included in Phytophthora (Erwin and Ribeiro 1996). Furthermore, we also included Peronophythora litchi, a unique pathogen that is cultivable and had been included in Phytophthora (Göker et al. 2007), yet forms sporangiophores with considerable complexity (Chen 1961; Ye et al. 2016). To add another line of evidence, we performed AU analyses in addition to phylogenetic reconstructions to test the topology of major associations found in this study.
Materials and methods
Oomycete material and microscopy
Material of Kawakamia cyperi from Cyperus esculentus was collected from fields in Idaho in August 2012. Samples were dried between tissue paper and deposited in the herbarium Senckenbergianum (FR) under the accession number FR-0046142.
For microscopy, specimens were hydrated with 5% chloral hydrate solution and sporangia and sporogenous hyphae carefully scraped off the surface using a scalpel blade. The oomycete material was transferred to 5% chloral hydrate solution on a slide and covered with coverslips. Microscopic observations were made in DIC at × 400 magnification using a Zeiss Imager2 (Zeiss, Oberkochen, Germany) equipped with a Zeiss Axiocam with 3.2 Mp resolution. Measurements were done from images taken after calibration with the Zeiss Axiovision software. Measurements are reported as (maximum –) mean minus standard deviation – mean – mean plus standard deviation (– maximum).
DNA, extraction, PCR, and sequencing
DNA of Kawakamia cyperi was extracted using the innuPREP Plant DNA Kit (Analytik Jena GmbH, Jena, Germany) as outlined in Telle and Thines (2008). Seven genes (COX2, nrLSU, HSP90, EF1A, 60S, ENOLASE, and ß-TUB) were amplified and sequenced using the primers given in Table 1. For four genes new primers were designed. PCR reactions were conducted using MangoTaq reaction buffer (colourless), 200 µM dNTPs, 2 mM MgCl2, 0.75 U MangoTaq polymerase (Bioline, Luckenwalde, Germany), 0.8 mg/mL BSA (bovine serum albumin), and 400 µM of each primer (Merck KGaA, Darmstadt, Germany) in an Eppendorf mastercycler ProS with vapo.protect lid (Eppendorf, Hamburg, Germany) using the programs given in Table 2. Because the qualities of the sequences of three genes (HSP90, ENOLASE, and ß-TUB) were poor, the PCR products were cloned in Escherichia coli using the CloneJET PCR cloning kit (ThermoFisherScientific, Waltham, USA) following the instructions provided with the kit. The inserts were amplified and sequenced using the provided plasmid primers. All PCR products were sequenced at the Senckenberg Biodiversity and Climate Research Centre (SBiK-F). For the cloned genes, forward and reverse sequences of at least three clones were used to assemble a consensus sequence to avoid PCR artefacts.
Phylogenetic reconstructions
The reference sequences for all Pythium and Phytophthora specimens were obtained from the study of Blair et al. (2008). Furthermore, the alignment was complemented by the addition of sequences extracted from the published genomes of Peronophythora litchii (Ye et al. 2016, referred therein as Phytophthora litchii) and Phytophthora podocarpi (Studholme et al. 2015), as well as the sequences of Ph. arenaria (CBS 127950) and Ph. alticola (CBS 121939) available as individual genes on NCBI (https://www.ncbi.nlm.nih.gov/). The sequence data of Pseudoperonospora cubensis originate from Runge et al. (2011a, b); those of Hyaloperonospora thlaspeos-perfoliati were extracted from preliminary genome data; and those of the downy mildews Sclerospora graminicola (Kobayashi et al. 2017), Plasmopara halstedii (Sharma et al. 2015), Plasmopara muralis and Plasmopara viticola (Dussert et al. 2019), Peronospora effusa (Klein et al. 2020), Peronospora belbahrii (Thines et al. 2020), and Peronospora tabacina (Derevnina et al. 2015) were extracted from published genomes. A summary of the data used and the corresponding accession numbers is provided in Supplementary File 1. Despite the availability of additional genomes, full-length high-quality (i.e. without frameshift mutations in core genes or a high number of ambiguities) data of all genes used in this analysis could not be obtained, which is why those data were not included. The seven genes present for all species included were aligned separately using the Mafft webserver applying the G-INS-i algorithm for the protein-coding genes and Q-INS-i for the nrLSU gene. All further parameters were set to default (https://mafft.cbrc.jp/alignment/server/; Katoh et al. 2019). The seven alignments were manually trimmed to remove leading and trailing gaps and subsequently merged to a final alignment with a length of 5240 positions. A partitioning file was prepared including codon-based gene partitioning for all genes except for the non-protein coding nrLSU gene resulting in 19 partitions. This was optimized using PartitionFinder 2 (Lanfear et al. 2016) applying the greedy search algorithm (Lanfear et al. 2012) and setting the branch length parameter to unlinked. Phylogenetic reconstructions were done using the maximum likelihood (ML) algorithm as well as minimum evolution (ME). Maximum likelihood analyses was conducted using IQ-TREE (version 2.2, Nguyen et al. 2015) applying a partitioned analysis (Chernomor et al. 2016) with the best scheme from PartitionFinder 2. All other parameters were set to default and bootstrapping (Felsenstein 1985) was performed with 1000 non-parametric replicates. Minimum evolution analysis was conducted with MEGA (version 7, Kumar et al. 2016) applying the minimum evolution method (Rzhetsky and Nei 1992) with 1000 bootstraps (Felsenstein 1985) and the Tamura-Nei algorithm (Tamura and Nei 1993). The gamma distribution shape parameter was estimated by the tool provided by MEGA and set to 0.2695. The patterns among lineages parameter were set to different (heterogenous).
In order to test the phylogenetic affiliations of the downy mildew subclades with itself and with the clades of Phytophthora, the approximately unbiased (AU) test (Shimodaira 2002) was applied (Table 3). For this, the site-wise log-likelihoods for 94 associations were calculated using IQtree (Nguyen et al. 2015) with a reduced alignment containing 35 specimens including one downy mildew species from every genus and representatives of the clades and subclades of Phytophthora, according to Yang et al. (2017). The AU-test using the TREEASS program of the CONSEL package (Shimodaira and Hasegawa 2001) was conducted as described in Runge et al. (2011a, b) with Ph. boehmeriae set as outgroup. All files used in the analyses are provided in Supplementary File 2.
To satisfy a request during review, we also downloaded the alignment of Bourret et al. (2018) for analysis. From that dataset, we removed leading and trailing ends, sequences with large amounts of missing data, and gapped sites. This filtering was done to remove the larger part of noise incited by bad quality sequences, and the resulting alignment can be retrieved from Supplementary File 3. Subsequently, the alignment was subjected to ME analysis in MEGA7, using default parameters, except for choosing the Tamura-Nei substitution model (most complex standard model offered by MEGA7) and 500 bootstrap replicates. In addition, ML and ME were done in FastTree2 and RAxML, as implemented on the TrEase webserver (www.thines-lab.senckenberg.de/trease), using 500 and 1000 bootstrap replicates, respectively. This set was meant to be used only for a rough checking, if the signal found in our detailed analyses would also be retrieved from the dataset of Bourret et al. (2018), if only a rather simple quality enhancement would be done.
Results
Morphology
Kawakamia cyperi was observed to have irregular sporangiophores with a slightly thickened wall (Fig. 1 A–D). Sporangia were often clustered (Fig. 1 M), similar to Sclerophthora, with a conspicuous papilla that was visible already early during sporangial development (Fig. 1 D, E) and mostly with a broad pedicel, tapering towards the ultimate branchlet (Fig. 1 F–J). Sporangia (n = 100) were (24.5–)31.5–36.5–41.5(–48) µm long and (16.2–)19.5–22.5–25.5(–28.5) µm broad, with a length to breadth ratio of (1.33–)1.46–1.63–1.8(–2.09). Haustoria were pyriform to broadly lobed, often more than one per host cell (Fig. 1 K, L). Antheridia were diclinous or monoclinous, mostly paragynous. Oospores (n = 14) were pale ochre to pale brown, thick walled, aplerotic (Fig. 1 N–Q), (23–)25.5–28–30.5(–32.5) µm in outer diameter, inner diameter (17–)19–21.5–23(–25.5) µm, the ratio of the outer to the inner diameter was (1.22–)1.26–1-32–1.38(–1.5), and the wall thickness was (2.5–)3–3.5–4(–4.5) µm.

Microscopic features of Kawakamia cyperi on Cyperus esculentum. A–C sporangiophores; D, E developing sporangia; F–J mature sporangia with variable pedicels; K, L haustoria; M clustered sporangia; N–Q oogonia with oospores. Bar = 5 µm in all pictures, except for N–Q, where they are 10 µm
Phylogeny
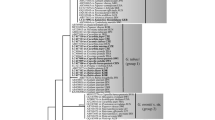
The phylogeny of the individual loci (COX2, nrLSU, HSP90, EF1A, 60S, ENOLASE, and ß-TUB) did not yield support for conflicting topologies and, thus, all loci were concatenated for phylogenetic analysis, resulting in an alignment of 5240 positions. Phylogenetic reconstructions in maximum likelihood (ML) and minimum evolution (ME) resulted in largely congruent topologies with no support for conflicting topologies. Thus, only the tree from the ML analysis is shown in Fig. 2, with bootstrap support (BS) from ML and ME inference added on the branches in the respective order.

Phylogenetic reconstruction in maximum likelihood, with support values from maximum likelihood and minimum evolution, in the respective order. A minus sign denotes lack of support for the displayed or a conflicting topology. Thickened branches indicate maximum support in both analyses
In all phylogenetic analyses, the downy mildews, including Kawakamia cyperi, formed a monophyletic clade with maximum support. Within this clade the Plasmopara specimens and Bremia lactucae were grouped together with strong support in both analyses. Together with Sclerospora graminicola and Kawakamia cyperi, they formed a subclade that received some support in ML. Pseudoperonospora cubensis, belonging to one of the two genera with coloured conidia, was inferred as the sister lineage to all other downy mildew genera, even though without support.
Clade 4 of Phytophthora s.l., including Peronophythora litchii, the pathogen causing litchi downy blight in East Asia, was resolved as monophyletic with considerable support (80% and 97% BS) and found to be the sister group to clade 12 with no to moderate support (89% BS in ME). Phytophthora clade 1, which contains Phytophthora infestans, the type species of the genus, was resolved with strong support (99% and 92% BS). The monophyly of clade 2 also received strong support (99% and 94% BS). While the relationships of clades 1, 2, and 4, and the downy mildews could not be resolved, they collectively formed a monophyletic group with no to moderate support (84% BS in ME).
Clades 3, 5, and 6 received maximum support for their monophyly, while the monophyly of clade 7 received maximum to high support (100% and 99% BS). Collectively, these formed an unsupported group together with Phytophthora podocarpi. The remaining groups were clade 8 and a group consisting of species of clades 9 and 10, which both received good support for their monophyly (99% and 81% BS, as well as 100% and 99% BS, respectively). The relationships of the monophyletic groups to each other could not be resolved.
Phylogenetic analyses of the dataset of Bourret et al. (2018) after removing some missing data and bad quality sequences resulted in resolving the downy mildews as monophyletic in ME analyses, with maximum support in the reconstruction with FastTree2, and moderate support in the reconstruction done with MEGA7 (Supplementary File 4). However, in ML the DM were split into two clades (see discussion of the signal erosion artefact known from ML analyses).
Approximately unbiased (AU) testing
As the multigene phylogeny with high sampling from existing sequence information in both Phytophthora and the downy mildews revealed downy mildew monophyly in contrast to all phylogenomic investigations with low taxon sampling, except Ye et al. (2016), AU testing was carried out. In the AU testing the topology found in the current multigene phylogeny was tested against diverging topologies found in phylogenomic studies with limited sampling in both Phytophthora and downy mildews. These were a splitting of downy mildews, as observed in Sharma et al. (2015), McCarthy and Fitzpatrick (2017), Kobayashi et al. (2017), Bourret et al. (2018), Fletcher et al. (2019), Dussert et al. (2019), and Klein et al. (2020), an association of Plasmopara (here in DMPH) with clade 1 (Sharma et al. 2015; McCarthy and Fitzpatrick 2017; Bourret et al. 2018; Fletcher et al. 2019; Dussert et al. 2019; Klein et al. 2020), an association of some downy mildews with clade 5 of Phytophthora (Bourret et al. 2018), an association of clade 1 with Hyaloperonospora (here in DM2), and a grouping of clade 4 not with downy mildews (Ye et al. 2016). The AU testing conducted in this study (Table 3) refuted all associations that would not result in a monophyly of the downy mildews or a grouping of all downy mildews with clades 1 or 4 (Table 3).
Taxonomy
A new combination for Phytophthora cyperi-bulbosi in Kawakamia is made here, as all morphological features and the obligate biotrophy of the pathogen support this placement.
Kawakamia cyperi-bulbosi (Seethal. & K. Ramakr.) Thines, comb. nov., MycoBank MB847402
Basionym: Phytophthora cyperi-bulbosi Seethal. & K. Ramakr., Curr. Sci. 22(5): 150 (1953)
Discussion
Evolution of downy mildews
Even though there is now a general agreement that the genus Phytophthora is closely related to the downy mildews and thus needs to be treated in the Peronosporaceae instead of the Pythiaceae (Cooke et al. 2000; Riethmüller et al. 2002; Göker et al. 2003, 2007; Thines 2009; Hulvey et al. 2010; Beakes and Thines 2017), the relationship between downy mildews and members of the genus Phytophthora s.l. has been elusive. While some studies inferred a monophyly of the downy mildews (Göker et al. 2007; Ye et al. 2016), others have concluded that the downy mildews are polyphyletic (Sharma et al. 2015; McCarthy and Fitzpatrick 2017; Bourret et al. 2018; Dussert et al. 2019; Fletcher et al. 2019; Klein et al. 2020). In this study, which included a broad sampling of Phytophthora based on Blair et al. (2008) and all downy mildew lineages, the downy mildews formed a monophyletic clade with maximum support in all analyses. This result was confirmed by the AU analyses, which were not performed in previous studies inferring a polyphyly of downy mildews, and which rejected all topologies observed in previous studies that suggested a potential polyphyly of the downy mildews.
That phylogenomic studies yielded topologies inconsistent with a monophyly of the downy mildews has been seen as evidence that obligate biotrophy and downy mildew characteristics have evolved at least twice in the Peronosporaceae. However, high bootstrap support only means that the same topology is produced when alignment columns are randomly sampled, which is to be expected in case of extremely large datasets, as the randomisation effect is low. The problem with phylogenomic investigations is that due to the very large amount of data, phylogenetic signal heterogeneity is too equally distributed to be readily picked up by bootstrapping resulting in very high support values for almost any given branch. For example, this can be observed in a recent study by Winkworth et al. (2022), where the topology observed by Bourret et al. (2018) and characterised as discordance between mitochondrial and nuclear loci is highly supported based on bootstrapping of a dataset with complete mitochondrial genomes. Quartet sampling, which is less prone to saturation artefacts, is a potential alternative to bootstrapping, but is still not widely used (Klötzl and Haubold 2016; Pease et al. 2018). However, the topologies of such phylogenies can change significantly, if additional taxa are included, especially in a situation where deep branching is difficult to resolve and incomplete lineage sorting at major splits might have occurred (Mishra et al. 2018). This means that broad taxon sampling is crucial in phylogenomic analyses, which is complicated by the still low amount of genomes available. In addition, especially when including diverse lineages with highly divergent mutation rates, alignments are often not straightforward, leading to incorrect assignment of homology. A way to avoid this is to focus on highly conserved genes that can be unambiguously aligned, and to exclude alignment parts where homology assignment is low (Philippe et al. 2000; Kück et al. 2010, 2014; Wu et al. 2012; Mishra et al. 2018). This approach will lead to a massive reduction in data, but avoids various artefacts, thus providing more credible phylogenetic reconstructions. It is notable that a strict filtering of genomic data will result in datasets with rather few high confidence genes, not too far from “normal” multigene analyses. Thus, multigene phylogenies using well-characterised and alignable loci on a comprehensive taxon sampling are probably better suited for inferring relationships difficult to disentangle than phylogenomic analyses using a large amount of ill-characterised loci on a comparatively small dataset. However, once comprehensive amounts of genomes of downy mildews and all Phytophthora lineages have become available, the approaches mentioned above are likely to provide a more detailed picture of the relationships between downy mildew and Phytophthora s.l. lineages.
It should be noted that for an unknown reason, the addition of Ph. podocarpi to the dataset is a major cause of disturbance in the phylogenetic signal. Without its inclusion, the monophyly of the downy mildews and their sister-group relationship to clade 4 is supported with maximum and high support values (data not shown). With its inclusion, the monophyly of downy mildew still received maximum support, but the support for grouping the different clades of Phytophthora and to position of the monophyletic downy mildews within them becomes unresolved. We assume that the cause of this might be incomplete lineage sorting due to a very quick radiation of Phytophthora lineages at the time downy mildews emerged. However, this clearly needs further careful evaluation once even more genomes become available. In any case, phylogenetic reconstructions based on lineages with largely differing mutation rates, such as the downy mildews and Phytophthora, can lead to significant artefacts in likelihood analyses (Struck et al. 2014; Redmond and McLysaght 2021; Susko and Roger 2021), even when a correct model is assumed (Kück et al. 2012), a problem which, alongside various other assumptions (Bromham 2019), has so far not been widely taken into consideration. One of the major issues with the likelihood assumption is that signal erosion can lead to a shifting of long branches to the inclusion of various short branches in between (Kück et al. 2012) or a grouping of long branches with short branches sharing some sequence similarities (Fleming et al. 2023). Thus, as both morphology and minimum evolution phylogenetic reconstructions support a monophyly of the downy mildews, also when using a quality-filtered dataset from Bourret et al. (2018), we assume that the splitting of the downy mildews in ML observed when the dataset of Bourret et al. 2018, but not in the current dataset, is the result of a branch-length heterogeneity artefact.
It is noteworthy that Kawakamia cyperi, introduced by Miyabe (1904), and characterised by sporangia with a unique pedicel, somewhat differentiated sporangiophores and thick-walled oospores, was found to be nested within the downy mildews with strong support. The genus was not accepted widely, and both Waterhouse (1963) and Ho (1990) considered Kawakamia cyperi to be a member of Phytophthora. The conspicuous pedicel that broadens towards the sporangium was the key feature Miyabe (1904) based the description of the genus on. However, Ho and Chang (1992) did not observe this feature in the type specimen, suggesting that it might not preserve well. In the current study, the pedicel as described by Miyabe was clearly visible. Kawakamia has mostly not been accepted as a genus independent from Phytophthora, even though it is obligate biotrophic and causes symptoms resembling downy mildew infections. In line with the assumed obligate biotrophic nature (Seethalakshmi 1953; Erwin and Ribeiro 1996), trials to cultivate the species failed an all media tried (CMA, OMA, PCA, PDA, SAM, V8; Thines, unpublished results), supporting previous reports that the phytophthora-like downy mildew species from Cyperaceae are not cultivable (Seethalakshmi 1953; Erwin and Ribeiro 1996). Thus, phylogenetic relationships, lack of cultivability, and the unique morphology all support treating Kawakamia as an independent genus. If the several species of Kawakamia described on Cyperaceae belong to several species or just one or two needs to be clarified in future studies. However, on the basis of its tuberculatae oogonia, Kawakamia cyperi-bulbosi is distinct from the other species described form Cyperus (Seethalakshmi 1953; Seethalakshmi and Ramakrishnan 1953). Kawakamia cyperi was also reported from Digitaria (Ho et al. 2004), which would extend the host range of the genus to Poaceae. However, the strongly plerotic oospores and the lack of the characteristic pedicel suggest that this was a different species. In any case, the presence of another phytophthora-like graminicolous downy mildew provides further support for the hypothesis that the downy mildews might have evolved from hosts in Poaceae (Thines 2009).
The concept of what exactly constitutes a downy mildew probably needs to be updated in the light of the current study. Already Thines (2009) emphasised that some downy mildew genera on Poaceae exhibit some features that could be seen as phytophthora-like, e.g. the continuous outgrowth of the sporangiophores in Viennotia, die somewhat successive formation of sporangia and the lack of well-differentiated sporangiophores in Sclerophthora, and the intracellular mycelium observed in Poakatesthia (Thines 2009). Even though it can currently not be unambiguously clarified, if the rather undifferentiated sporangiophores observed in Kawakamia and Sclerophthora represent an ancestral state or a derived reduction, it is noteworthy that in the clades probably most closely related to downy mildews (clades 1 and 4), there are several species with rather complex sporangiophores, suggesting that a reduction is the more likely scenario. Considering this, the most clear-cut synapomorphy of downy mildews with respect to Phytophthora seems to be their obligate biotrophic nature, while the complex sporangiophores are a secondary trait that is variable and has probably faced reduction in Kawakamia and Sclerophthora.
In line with a previous study (Ye et al. 2016), the downy mildews were found to be monophyletic and associated with Phytophthora s.l. clades 1 and 4 in this study. Clade 4 also contains Peronophythora litchi, a pathogen causing downy blight of litchi. Members of clade 4 produce great amounts of aerial, papillate sporangia that are borne on somewhat differentiated sporangiophores and usually produce aplerotic oospores. The highest degree of differentiation is found in Peronophythora litchii, where sporangiophores superficially resemble sporangiophores of Peronospora (Chen 1961; Ye et al. 2016). A first step to resolving the polyphyly of Phytophthora has been taken by Thines (2023), who transferred the members of clade 4 to the genus Peronophythora.
The evidence that downy mildews had evolved more than once from within phytophthora-like ancestors (which would render Phytophthora polyphyletic, not paraphyletic in the logically concise definition given by Nelson (1971)) is not strong and probably the result of a branch-heterogeneity artefact (Kück et al. 2012; Fleming et al. 2023). It is refuted by the current study that includes only complete data (using a matrix without missing data) of high quality, a balanced selection of taxa within downy mildews, and AU testing as a second line of evidence. In the AU analysis only the monophyly of the downy mildews, the sister-group relationship of downy mildews with Peronophythora and clade 1, as well as this group with clade 1, received meaningful support. In contrast, none of the previously inferred scenarios for a splitting of the downy mildews (e.g. Bourret et al. 2018; Fletcher et al. 2019) received any support. Thus, it seems highly likely that obligate biotrophy, the hallmark of downy mildew physiology, has evolved just once in Phytophthora, giving rise to the highly diverse downy mildew lineage. As Brasier et al. (2022) emphasised that already phytophthora-like species are diverse and species-rich. This is even surpassed by downy mildews, which have a much higher degree of morphological differentiation, e.g. spanning the complete spectrum from phytophthora-like to highly complex sporangiophores, and are likely to be much more species-rich than phytophthora-like groups (Thines and Choi 2016). It will be an interesting subject for future studies which evolutionary innovation has triggered this differentiation from the ancestral lineage, which possibly gave rise to all downy mildew lineages currently recognised.
Taxonomic considerations arising from the findings of this study
Recently, Brasier et al. (2022) proposed that Phytophthora should remain as a paraphyletic genus. However, several of their considerations are debatable. For example, the statement is incorrect that the genus concept in downy mildews would be in a way that some genera were more closely related to each other than closely related Phytophthora species. In fact, the genetic distances between downy mildew genera and species are considerable (e.g. Göker et al. 2007; Runge et al. 2011a, b; Bourret et al. 2018; this study). Only the genera Bremia, Novotelnova, and Protobremia are rather closely related and probably will need to be merged into Bremia to avoid an inflation of genera (Choi and Thines 2015). It is noteworthy that the several major clades of Phytophthora have remained stable entities over the past two decades, as highlighted by Brasier et al. (2022), and which seem to render it possible to achieve a stable classification with establishing new genera to resolve the paraphyly of Phytophthora. As the relationships of the various potential genus-level groups to each other are not fully resolved, we refrain from formally introducing new genera or reactivating old genus names, such as Phloeophthora (Klebhahn 1906), with its type species Phloephthora syringae for clade 8, as the largest possible groups should be named to avoid unnecessary taxonomic changes among species now included in Phytophthora. Also it seems to be recommendable to use names relatable to Phytophthora when introducing new genus names. Apart from the already described names, Peronophythora and Phloeophthora, this could be names like Paraphytophthora, Pseudophytophthora, or Phytophthoropsis, to keep the communications barrier low, as also recommended by Brasier et al. (2022). The phylogenetic reconstruction presented in this study suggests that it might be necessary to introduce or reactivate five genus names for clade 12; clade 2; clades 3, 5, 6, and 7; clade 8; and clades 9 and 10, while clade 1 with the type species, Phytophthora infestans, would retain the name Phytophthora. However, before formal taxonomic changes can be made, it needs to be ascertained that clade 12 remains isolated and that clades 3, 5, 6, and 7 form a monophylum. Therefore, broadly sampled phylogenomic studies, including also some genera closely related to Phytophthora, are warranted for this.
The rather general and descriptive terms used so far to characterise phytophthora-like species have their use in identifying species, but are hardly informative for grouping phylogenetically related species (Brasier et al. 2022). This is not surprising, considering that many of the widely recognised characters rather refer to ecological adaptations, host range, or mode of dissemination, which will be relevant to the adaptation to a certain environment, but are unlikely to carry a phylogenetic signal. Thus, the pairings of unrelated phytophthora-like species with similar phenotypic traits as presented by Brasier et al. (2022) cannot be used as an argumentation against recognising genus-level groups as genera. Their conclusion that there are “often considerable biological and phylogenetic distances between Phytophthora and the DMs [downy mildews]” (Brasier et al. 2022) is refuted by other parts of the discussion in which they state that are some downy mildew genera that have retained phytophthora-like traits and that clade 4 has several downy-mildew-like features. This is in line with this study and previous considerations by Thines (2009), who showed that the traditional divide between Phytophthora and downy mildews is artificial, and that obligate biotrophy is probably the uniting synapomorphy of the downy mildew genera.
Accepting paraphyly is not representing the scientific consensus. In fact, much progress in understanding evolutionary processes has been due to recognising lineages in previously paraphyletic taxa as independent, e.g. in the case of Reptilia that are paraphyletic with respect to birds (e.g. Card et al. 2023). Even with abandoning the formal use of “Reptilia”, the informal designation “reptiles” to refer to groups that share some plesiomophic traits is still useful for communication. The same applies also to the term “phytophthora-like”, for organisms with traits that before were thought to be characteristic of a monophylum. In this conjunction, it should also be noted that there are several other phytophthora-like groups that have been previously separated from Phytophthora, e.g. Halophytophthora and the earlier-diverging genera Salisapilia and Halophytophthora (Hulvey et al. 2010; Bennett et al. 2018; Bennett and Thines 2019), often primarily triggered by on phylogenetic. Together with the phylogenetic investigations, a re-evaluation of morphological characters was also done, leading to the appraisal of seemingly minor morphological traits, such as the “cigarette-ash-like” plugs typical for Salisapilia (Hulvey et al. 2010; Bennett and Thines 2019) as synapomorphic features. Even in the absence of morphological features, it seems to be reasonable to recognise the genus-level groups currently represented by clades as genera, to reflect evolutionary relationships. This has also been done by Jung et al. (2017) when formally describing Nothophytophthora with the statement “Nothophytophthora is morphologically similar to Phytophthora and phylogenetically constitutes a monophyletic sister genus of Phytophthora”. The same argument is applicable also for the other lineages of phytophthora-like organisms.
Availability of data and materials
Sequence data have been deposited in GeneBank.
References
Ali SS, Shao J, Lary DJ, Kronmiller B, Shen D, Strem MD, Amoako-Attah I, Akrofi AY, Begoude BA, Ten Hoopen GM, Coulibaly K, Kebe BI, Melnick RL, Guiltinan MJ, Tyler BM, Meinhardt LW, Bailey BA (2017) Phytophthora megakarya and Phytophthora palmivora, closely related causal agents of cacao black pod rot, underwent increases in genome sizes and gene numbers by different mechanisms. Genome Biol Evol 9:536–557
Beakes GW, Thines M (2017) Hyphochytriomycota and Oomycota. In: Archibald J, Simpson A, Slamovits C (eds) Handbook of the Protists, 2nd edn. Springer International Publishing, Cham, pp 435–505
Belbahri L, Calmin G, Pawlowski J, Lefort F (2005) Phylogenetic analysis and real time PCR detection of a presumably undescribed Peronospora species on sweet basil and sage. Mycol Res 109:1276–1287
Bennett RM, Thines M (2019) Revisiting Salisapiliaceae. Fungal Syst Evol 3:171–185
Bennett RM, Devanadera MK, Dedeles GR, Thines M (2018) A revision of Salispina, its placement in a new family, Salispinaceae (Rhipidiales), and description of a fourth species, S. hoi sp. nov. IMA Fungus 9:259–269
Blair JE, Coffey MD, Park SY, Geiser DM, Kang S (2008) A multi-locus phylogeny for Phytophthora utilizing markers derived from complete genome sequences. Fungal Genet Biol 45:266–277
Bourret TB, Choudhury RA, Mehl HK, Blomquist CL, McRoberts N, Rizzo DM (2018) Multiple origins of downy mildews and mito-nuclear discordance within the paraphyletic genus Phytophthora. PloS ONE 13:e0192502
Brasier CM, Griffin MJ (1979) Taxonomy of ’Phytophthora palmivora’ on cocoa. Trans Br Mycol Soc 72:111–143
Brasier C, Scanu B, Cooke D, Jung T (2022) Phytophthora: an ancient, historic, biologically and structurally cohesive and evolutionarily successful generic concept in need of preservation. IMA Fungus 13:12
Bromham L (2019) Six impossible things before breakfast: assumptions, models, and belief in molecular dating. Trends Ecol Evol 34:474–486
Burgess TI, Scott JK, Mcdougall KL, Stukely MJ, Crane C, Dunstan WA, Brigg F, Andjic V, White D, Rudman T, Arentz F, Ota N, Hardy GE (2017) Current and projected global distribution of Phytophthora cinnamomi, one of the world’s worst plant pathogens. Global Chang Biol 23:1661–1674
Card DC, Jennings WB, Edwards SV (2023) Genome evolution and the future of phylogenomics of non-avian reptiles. Animals 13:471
Chen CC (1961) A species of Peronophythora gen. nov. parasitic on litchi fruit in Taiwan. Spec Publ Coll Agric Natl Taiwan Univ 10:1–37
Chernomor O, von Haeseler A, Minh BQ (2016) Terrace aware data structure for phylogenomic inference from supermatrices. Syst Biol 65:997–1008
Choi YJ, Thines M (2015) Host jumps and radiation, not co-divergence drives diversification of obligate pathogens. A case study in downy mildews and Asteraceae. PLoS ONE 10:e0133655
Choi Y-J, Klosterman SJ, Kummer V, Voglmayr H, Shin H-D, Thines M (2015) Multi-locus tree and species tree approaches toward resolving a complex clade of downy mildews (Straminipila, Oomycota), including pathogens of beet and spinach. Mol Phylogenet Evol 86:24–34
Constantinescu O (1989) Peronospora complex on Compositae. Sydowia 41:79–107
Constantinescu O (1998) A revision of Basidiophora (Chromista, Peronosporales). Nova Hedwigia 66:251–265
Constantinescu O, Fatehi J (2002) Peronospora-like fungi (Chromista, Peronosporales) parasitic on Brassicaceae and related hosts. Nova Hedwigia 74:291–338
Constantinescu O, Voglmayr H, Fatehi J, Thines M (2005) Plasmoverna gen. nov., and the taxonomy and nomenclature of Plasmopara (Chromista, Peronosporales). Taxon 54:813–821
Cooke DEL, Drenth A, Duncan JM, Wagels G, Brasier CM (2000) A molecular phylogeny of Phytophthora and related oomycetes. Fungal Genet Biol 30:17–32
Derevnina L, Chin-Wo-Reyes S, Martin F, Wood K, Froenicke L, Spring O, Michelmore R (2015) Genome sequence and architecture of the tobacco downy mildew pathogen Peronospora tabacina. Mol Plant Microbe Interact 28:1198–1215
Dick MW (2001) Staminipilous fungi: systematics of the Peronosporomycetes including accounts of the marine straminipilous protists, the plasmodiophorids and similar organisms. Kluwer Academic Publishers, Dordrecht
Dussert Y, Mazet ID, Couture C, Gouzy J, Piron MC, Kuchly C, Bouchez O, Rispe C, Mestre P, Delmotte F (2019) A high-quality grapevine downy mildew genome assembly reveals rapidly evolving and lineage-specific putative host adaptation genes. Genome Biol Evol 11:954–969
Erwin DC, Ribeiro OK (1996) Phytophthora diseases worldwide. APS Press, St. Paul, MN
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Fleming JF, Valero-Gracia A, Struck TH (2023) Identifying and addressing methodological incongruence in phylogenomics: a review. Evol Appl 16:1087–1104
Fletcher K, Gil J, Bertier LD, Kenefick A, Wood KJ, Zhang L, Reyes-Chin-Wo S, Cavanaugh K, Tsuchida C, Wong J, Michelmore R (2019) Genomic signatures of heterokaryosis in the oomycete pathogen Bremia lactucae. Nat Commun 10:2645
Fontaine MC, Austerlitz F, Giraud T, Labbé F, Papura D, Richard-Cervera S, Delmotte F (2013) Genetic signature of a range expansion and leap-frog event after the recent invasion of Europe by the grapevine downy mildew pathogen Plasmopara viticola. Mol Ecol 22:2771–2786
Göker M, Voglmayr H, Riethmüller A, Weiß M, Oberwinkler F (2003) Taxonomic aspects of Peronosporaceae inferred from Bayesian molecular phylogenetics. Can J Bot 81:672–683
Göker M, Voglmayr H, Riethmüller A, Oberwinkler F (2007) How do obligate parasites evolve? A multi-gene phylogenetic analysis of downy mildews. Fungal Genet Biol 44:105–122
Görg M, Ploch S, Kruse J, Kummer V, Runge F, Choi Y-J, Thines M (2017) Revision of Plasmopara (Oomycota, Peronosporales) parasitic to impatiens. Mycol Prog 16:791–799
Grünwald NJ, LeBoldus JM, Hamelin RC (2019) Ecology and evolution of the sudden oak death pathogen Phytophthora ramorum. Ann Rev Phytopathol 57:301–321
Haas BJ, Kamoun S, Zody MC, Jiang RH, Handsaker RE, Cano LM, Grabherr M, Kodira CD, Raffaele S, Torto-Alalibo T, Bozkurt TO, Ah-Fong AM, Alvarado L, Anderson VL, Armstrong MR, Avrova A, Baxter L, Beynon J, Boevink PC, Bollmann SR, Bos JI, Bulone V, Cai G, Cakir C, Carrington JC, Chawner M, Conti L, Costanzo S, Ewan R, Fahlgren N, Fischbach MA, Fugelstad J, Gilroy EM, Gnerre S, Green PJ, Grenville-Briggs LJ, Griffith J, Grünwald NJ, Horn K, Horner NR, Hu CH, Huitema E, Jeong DH, Jones AM, Jones JD, Jones RW, Karlsson EK, Kunjeti SG, Lamour K, Liu Z, Ma L, Maclean D, Chibucos MC, McDonald H, McWalters J, Meijer HJ, Morgan W, Morris PF, Munro CA, O’Neill K, Ospina-Giraldo M, Pinzón A, Pritchard L, Ramsahoye B, Ren Q, Restrepo S, Roy S, Sadanandom A, Savidor A, Schornack S, Schwartz DC, Schumann UD, Schwessinger B, Seyer L, Sharpe T, Silvar C, Song J, Studholme DJ, Sykes S, Thines M, van de Vondervoort PJ, Phuntumart V, Wawra S, Weide R, Win J, Young C, Zhou S, Fry W, Meyers BC, van West P, Ristaino J, Govers F, Birch PR, Whisson SC, Judelson HS, Nusbaum C (2009) Genome sequence and comparative analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 461:393–398
Hausbeck MK, Lamour KH (2004) Phytophthora capsici on vegetable crops: research progress and management challenges. Plant Dis 88:1292–1303
Ho HH (1990) Taiwan Phytophthora. Bot Bull Acad Sin 31:89–106
Ho HH, Chang HS (1992) A re-evaluation of Phytophthora species described by K. Sawada in Taiwan. Mycotaxon 43:297–316
Ho HH, Zheng FC, Zeng HC (2004) Phytophthora cyperi on Digitaria cillaris in Hainan Province of China. Mycotaxon 90:431–435
Hudspeth DSS, Stenger D, Hudspeth MES (2003) A cox2 phylogenetic hypothesis for the downy mildew and white rusts. Fungal Divers 13:47–57
Hulvey J, Telle S, Nigrelli L, Lamour K, Thines M (2010) Salisapiliaceae–a new family of oomycetes from marsh grass litter of southeastern North America. Persoonia 25:109–116
Jung T, Scanu B, Bakonyi J, Seress D, Kovács GM, Durán A, Sanfuentes von Stowasser E, Schena L, Mosca S, Thu PQ, Nguyen CM, Fajardo S, González M, Pérez-Sierra A, Rees H, Cravador A, Maia C, Horta Jung M (2017) Nothophytophthora gen. nov., a new sister genus of Phytophthora from natural and semi-natural ecosystems. Persoonia 39:143–174
Jupe J, Stam R, Howden AJ, Morris JA, Zhang R, Hedley PE, Huitema E (2013) Phytophthora capsici-tomato interaction features dramatic shifts in gene expression associated with a hemi-biotrophic lifestyle. Genome Biol 14:R63
Katoh K, Rozewicki J, Yamada KD (2019) MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinforma 20:1160–1166
Klebhahn H (1906) Eine neue Pilzkrankheit der Syringen. Centralblatt für Bakteriologie, Parasitenkunde und Infektionskrankheiten, Zweite Abteilung 15:335–336
Klein J, Neilen M, van Verk M, Dutilh BE, Van den Ackerveken G (2020) Genome reconstruction of the non-culturable spinach downy mildew Peronospora effusa by metagenome filtering. PLoS ONE 15:e0225808
Klötzl F, Haubold B (2016) Support values for genome phylogenies. Life 6:11
Kobayashi M, Hiraka Y, Abe A, Yaegashi H, Natsume S, Kikuchi H, Takagi H, Saitoh H, Win J, Kamoun S, Terauchi R (2017) Genome analysis of the foxtail millet pathogen Sclerospora graminicola reveals the complex effector repertoire of graminicolous downy mildews. BMC Genomics 18:897
Kroon LPNM, Bakker FT, Van Den Bosch GBM, Bonants PJM, Flier WG (2004) Phylogenetic analysis of Phytophthora species based on mitochondrial and nuclear DNA sequences. Fungal Genet Biol 41:766–782
Kück P, Meusemann K, Dambach J, Thormann B, von Reumont BM, Wägele JW, Misof B (2010) Parametric and non-parametric masking of randomness in sequence alignments can be improved and leads to better resolved trees. Front Zool 7:10
Kück P, Mayer C, Wägele JW, Misof B (2012) Long branch effects distort maximum likelihood phylogenies in simulations despite selection of the correct model. PLoS ONE 7:e36593
Kück P, Meid SA, Groß C, Wägele JW, Misof B (2014) AliGROOVE–visualization of heterogeneous sequence divergence within multiple sequence alignments and detection of inflated branch support. BMC Bioinforma 15:294
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Lamour KH, Stam R, Jupe J, Huitema E (2012) The oomycete broad-host-range pathogen Phytophthora capsici. Mol Plant Pathol 13:329–337
Lanfear R, Calcott B, Ho SY, Guindon S (2012) PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol 29:1695–1701
Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B (2016) PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol 34:772–773
Lebeda A, Cohen Y (2011) Cucurbit downy mildew (Pseudoperonospora cubensis)—biology, ecology, epidemiology, host-pathogen interaction and control. Eur J Plant Pathol 129:157–192
Lebeda A, Sedlářová M, Petřivalský M, Prokopová J (2008) Diversity of defence mechanisms in plant–oomycete interactions: a case study of Lactuca spp. and Bremia lactucae. Eur J Plant Pathol 122:71–89
Lee SJ, Rose JK (2010) Mediation of the transition from biotrophy to necrotrophy in hemibiotrophic plant pathogens by secreted effector proteins. Plant Signal Behav 5:769–772
Martin FN, Blair JE, Coffey MD (2014) A combined mitochondrial and nuclear multilocus phylogeny of the genus Phytophthora. Fungal Genet Biol 66:19–32
McCarthy CG, Fitzpatrick DA (2017) Phylogenomic reconstruction of the oomycete phylogeny derived from 37 genomes. mSphere 2:e00095-17
Mishra B, Choi Y-J, Thines M (2018) Phylogenomics of Bartheletia paradoxa reveals its basal position in Agaricomycotina and that the early evolutionary history of basidiomycetes was rapid and probably not strictly bifurcating. Mycol Prog 17:333–341
Miyabe K (1904) Kawakamia, a new genus belonging to Peronosporaceae. Shokubutsugaku Zasshi 17(202):306–307
Moncalvo JM, Wang HH, Hseu RS (1995) Phylogenetic relationships in Ganoderma inferred from the internal transcribed spacers and 25S ribosomal DNA sequences. Mycologia 87:223–238
Nelson G (1971) Paraphyly and polyphyly: re-definitions. Syst Zool 20:471–472
Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274
Pease JB, Brown JW, Walker JF, Hinchliff CE, Smith SA (2018) Quartet sampling distinguishes lack of support from conflicting support in the green plant tree of life. Am J Bot 105:385–403
Philippe H, Lopez P, Brinkmann H, Budin K, Germot A, Laurent J, Moreira D, Müller M, Le Guyader H (2000) Early–branching or fast–evolving eukaryotes? An answer based on slowly evolving positions. Proceedings of the Royal Society in London. Series B 267(1449):1213–1221
Redmond AK, McLysaght A (2021) Evidence for sponges as sister to all other animals from partitioned phylogenomics with mixture models and recoding. Nat Commun 12(1):1783
Riethmuller A, Voglmayr H, Goker M, Weiß M, Oberwinkler F (2002) Phylogenetic relationships of the downy mildews (Peronosporales) and related groups based on nuclear large subunit ribosomal DNA sequences. Mycologia 94:834–849
Rizzo DM, Garbelotto M, Davidson JM, Slaughter GW, Koike ST (2002) Phytophthora ramorum as the cause of extensive mortality of Quercus spp. and Lithocarpus densiflorus in California. Plant Dis 86:205–214
Runge F, Telle S, Ploch S, Savory E, Day B, Sharma R, Thines M (2011a) The inclusion of downy mildews in a multi-locus-dataset and its reanalysis reveals a high degree of paraphyly in Phytophthora. IMA Fungus 2:163–171
Runge F, Choi Y-J, Thines M (2011b) Phylogenetic investigations in the genus Pseudoperonospora reveal overlooked species and cryptic diversity in the P. cubensis species cluster. Eur J Plant Pathol 129:135–146
Rzhetsky A, Nei M (1992) A simple method for estimating and testing minimum evolution trees. Mol Biol Evol 9:945–967
Salinari F, Giosue S, Rettori A, Rossi V, Tubiello FN, Spanna F, Gullino ML (2006) Downy mildew (Plasmopara viticola) epidemics on grapevine under climate change. Glob Chang Biol 12:1299–1307
Seethalakshmi KV (1953) Blight of Cyperus rotundus L. and C. bulbosus Vahl. Indian Phytopathol 6:57–62
Seethalakshmi KV, Ramakrishnan TS (1953) Phytophthora cyperi-bulbosi, sp. nov. on Cyperus bulbosus Vahl. Curr Sci 22(5):149–150
Sharma R, Xia X, Cano LM, Evangelisti E, Kemen E, Judelson H, Oome S, Sambles C, Van den Hoogen DJ, Kitner M, Klein J, Meijer HJG, Spring O, Win J, Zipper R, Bode HB, Govers F, Kamoun S, Schornack S, Studholme DJ, Van den Ackerveken G, Thines M (2015) Genome analyses of the sunflower pathogen Plasmopara halstedii provide insights into effector evolution in downy mildews and Phytophthora. BMC Genomics 16:741
Shimodaira H (2002) An approximately unbiased test of phylogenetic tree selection. Syst Biol 51:492–508
Shimodaira H, Hasegawa M (2001) CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics 17:1246–1247
Struck TH, Wey-Fabrizius AR, Golombek A, Hering L, Weigert A, Bleidorn C, Klebow S, Iakovenko N, Hausdorf B, Petersen M, Kück P, Herlyn H, Hankeln T (2014) Platyzoan paraphyly based on phylogenomic data supports a noncoelomate ancestry of Spiralia. Mol Biol Evol 31:1833–1849
Studholme DJ, McDougal RL, Sambles C, Hansen E, Hardy G, Grant M, Ganley RJ, Williams NM (2015) Genome sequences of six Phytophthora species associated with forests in New Zealand. Genomics Data 7:54–56
Susko E, Roger AJ (2021) Long branch attraction biases in phylogenetics. Syst Biol 70:838–843
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10:512–526
Telle S, Thines M (2008) Amplification of cox2 (~ 620 bp) from 2 mg of up to 129 years old herbarium specimens, comparing 19 extraction methods and 15 polymerases. PLoS One 3:e3584
Telle S, Thines M (2012) Reclassification of an enigmatic downy mildew species on lovegrass (Eragrostis) to the new genus Eraphthora, with a key to the genera of the Peronosporaceae. Mycol Prog 11:121–129
Thines M (2009) Bridging the gulf: Phytophthora and downy mildews are connected by rare grass parasites. PLoS ONE 4:e4790
Thines M, Choi Y-J (2016) Evolution, diversity, and taxonomy of the Peronosporaceae, with focus on the genus Peronospora. Phytopathology 106:6–18
Thines M, Göker M, Spring O, Oberwinkler F (2006) A revision of Bremia graminicola. Mycol Res 110:646–656
Thines M, Göker M, Oberwinkler F, Spring O (2007) A revision of Plasmopara penniseti, with implications for the host range of the downy mildews with pyriform haustoria. Mycol Res 111:1377–1385
Thines M, Telle S, Choi Y-J, Tan Y-P, Shivas RG (2015) Baobabopsis, a new genus of graminicolous downy mildews from tropical Australia, with an updated key to the genera of downy mildews. IMA Fungus 6:483–491
Thines M, Sharma R, Rodenburg SYA, Gogleva A, Judelson HS, Xia X, van den Hoogen J, Kitner M, Klein J, Neilen M, de Ridder D, Seidl MF, van den Ackerveken G, Govers F, Schornack S, Studholme DJ (2020) The genome of Peronospora belbahrii reveals high heterozygosity, a low number of canonical effectors, and TC-rich promotors. Mol Plant Microbe Interact 33:742–753
Thines M (2023) Nomenclatural novelties. Index Fungorum 548
Voglmayr H, Riethmüller A, Göker M, Weiss M, Oberwinkler F (2004) Phylogenetic relationships of Plasmopara, Bremia and other genera of downy mildew pathogens with pyriform haustoria based on Bayesian analysis of partial LSU rDNA sequence data. Mycol Res 108:1011–1024
Waterhouse GM (1963) Key to the species of Phytophthora de Bary. Commonw Mycol Inst Kew UK Mycol Pap 92:22
Winkworth RC, Neal G, Ogas RA, Nelson BCW, McLenachan PA, Bellgard SE, Lockhart PJ (2022) Comparative analyses of complete Peronosporaceae (Oomycota) mitogenome sequences—insights into structural evolution and phylogeny. Genome Biol Evol 14:evac049
Wroblewski T, Piskurewicz U, Tomczak A, Ochoa O, Michelmore RW (2007) Silencing of the major family of NBS–LRR-encoding genes in lettuce results in the loss of multiple resistance specificities. Plant J 51:803–818
Wu M, Chatterji S, Eisen JA (2012) Accounting for alignment uncertainty in phylogenomics. PloS ONE 7:e30288
Yang X, Tyler BM, Hong C (2017) An expanded phylogeny for the genus Phytophthora. IMA Fungus 8:355–384
Ye W, Wang Y, Shen D, Li D, Pu T, Jiang Z, Zhang Z, Zheng X, Tyler BM, Wang Y (2016) Sequencing of the litchi downy blight pathogen reveals it is a Phytophthora species with downy mildew-like characteristics. Mol Plant Microbe Interact 29:573–583
Yoshida K, Schuenemann VJ, Cano LM, Pais M, Mishra B, Sharma R, Lanz C, Martin FN, Kamoun S, Krause J, Thines M, Weigel D, Burbano HA (2013) The rise and fall of the Phytophthora infestans lineage that triggered the Irish potato famine. eLife 2:e00731
Acknowledgements
Ram Sampangi and Krishna Moham are gratefully acknowledged for providing specimens of Kawakamia cyperi. We thank Ricarda Prinz for providing literature. This manuscript was first submitted to and revised according to the recommendations of two anonymous reviewers and an editor. It was retracted from that journal to submit it to the special issue on Phytophthora in Mycological Progress. We are indebted and grateful to the reviewers and editor on that submission, as they helped greatly in improving the manuscript and analyses. In addition, we would also like to express our thankfulness to the reviewers that evaluated the manuscript for Mycological Progress and helped improving it. Evi Weber is gratefully acknowledged for meticulous editing.
Funding
Open Access funding enabled and organized by Projekt DEAL. This study was supported by the government of Hesse in the Framework of the cluster for Integrative Fungal Research (IPF) and the Centre for Translational Biodiversity Genomics (TBG).
Author information
Authors and Affiliations
Contributions
MT conceived the study; BM analyzed the unpublished genome of Hyaloperonospora thalspeos-perfoliati and provided sequence data; SP conducted the phylogenetic reconstructions and AU analysis; MT did the microscopy work; MT wrote the manuscript with major contributions from SP; all authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
MT is the Editor-in-Chief of Mycological Progress, but was not involved in the editorial processes associated with handling this manuscript.
Additional information
Section Editor: Tanay Bose
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the “Topical collection—since de Bary: Progress in Phytophthora research
Supplementary Information
Below is the link to the electronic supplementary material.
11557_2023_1932_MOESM1_ESM.docx
Supplementary file1 (DOCX 57 KB) Supplementary File 1. This file contains a summary of the data used and the corresponding accession numbers.
11557_2023_1932_MOESM2_ESM.docx
Supplementary file2 (DOCX 1562 KB) Supplementary File 2. This file contains the alignments used for the phylogenetic reconstructions and AU analysis, as well as the corresponding partitioning file.
11557_2023_1932_MOESM3_ESM.fas
Supplementary file3 (FAS 448 KB) Supplementary File 3. This file contains a dataset based on Bourret et al. (2018) with gapped sites and taxa with incomplete sequence information removed.
11557_2023_1932_MOESM4_ESM.docx
Supplementary file4 (DOCX 54 KB) Supplementary File 4. Phylogenetic reconstructions in Minimum Evolution and Maximum Likelihood analyses using a quality-filtered dataset based on Bourret et al. (2018).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thines, M., Mishra, B. & Ploch, S. Multigene analyses with a broad sampling in Phytophthora and related genera provide evidence for the monophyly of downy mildews. Mycol Progress 22, 82 (2023). https://doi.org/10.1007/s11557-023-01932-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11557-023-01932-2