
Abstract
Oomycetes, a large group of fungus-like organisms, include some destructive plant pathogens causing enormous economic damage. Phylogenetically, oomycetes belong to the kingdom Straminipila and have diverse lifestyles, including saprotrophs and both general and specialized pathogens of various eukaryotic supergroups. A rapid increase in genomic studies and next-generation sequencing technologies have led to significant progress in understanding oomycete lifestyles. However, their genetics, including transcriptional regulation, have been studied to a much lesser extent. Here, we provide a cross-species analysis of oomycete promoter for providing a first step towards elucidating gene regulation networks related to pathogenicity and life cycle stages. The clustered DNA sequences of Plasmopara halstedii transcriptome time-series expression level dataset from a preliminary study have been used as a core reference for cross-species comparisons. Using a computational pipeline, 46 potential transcription factor binding site (TFBS) motifs in 25 clusters with functionally conserved downstream genes of downy mildew and two Phytophthora species, regardless of the gene expression levels of Phytophthora transcriptomes, were found. This can now be followed up by knock-out experiments in oomycete species amenable for genetic modification.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Oomycete plant pathogens are eukaryotic, filamentous organisms of the kingdom Straminipila that exhibit obligate biotrophic, hemi-biotrophic, or necrotrophic lifestyles (Fawke et al. 2015). The most notorious and devastating oomycetes belong to the genus Phytophthora (almost 200 species) and the downy mildews (over 700 species) (Thines 2014; Scanu et al. 2021; Chen et al. 2022). Phytophthora species are hemi-biotrophic pathogens and possess two lifestyle phases, where they initially act as biotrophs and then switch to a necrotrophic phase in which they kill their host to feed on the dead matter. Plasmopara halstedii, an obligate biotroph, maintains a close interaction with sunflower hosts in which it depends on the host for its survival. Most of the obligate biotrophs show limited or very narrow host ranges, unlike many hemi-biotrophic Phytophthora species, which sometimes can parasitize members of several plant families. With respect to their importance as plant pathogens in terms of research and economical or environmental impact, the following list by Kamoun et al. (2015) provided the following ranking: at the first position Phytophthora infestans causing late blight of potatoes and tomatoes and at the fourth position Phytophthora sojae causing seed, root, and stem rot of soybean (Kamoun et al. 2015). Pl. halstedii causes downy mildew of sunflower and significant yield loss worldwide in commercial seed production (Sharma et al. 2015; Gascuel et al. 2015; Laura Martínez et al. 2021). A recent study demonstrated that several virulence mechanisms of filamentous fungi and oomycete pathogens are highly conserved and include core functions such as transport, carbohydrate metabolism, secondary metabolite synthesis, signal transduction, and amino acid metabolism (Pandaranayaka et al. 2019). However, cross-species transcriptional regulation studies to identify regulatory motifs associated with conserved gene functions are still lacking.
Both obligate biotrophic and hemi-biotrophic oomycetes use secreted effector proteins for manipulating the host immune system to their own benefit (Bozkurt et al. 2011). Of the pathogenicity-related genes or proteins, secreted apoplastic effectors are targeted to the space outside the host cytoplasm (Tian et al. 2007; Li et al. 2020). These include small cysteine-rich proteins such as cystatin-like protease inhibitors (EPIC), Nep1 (necrosis- and ethylene-inducing peptide 1)-like proteins (NLPs), toxins, and cell wall–degrading enzyme (CWDE) glycoside hydrolase families. In plant pathogenic oomycete species, a second class of effectors, the cytoplasmic effectors, is targeted to host cytoplasm, which include effectors with a recognizable N-terminal host-targeting domain such as RxLR (arginine-any-leucine-arginine)-dEER (Asp-Glu-Glu-Arg) effectors, Crinklers (CRN) with the N-terminal domains LXLFLAK (Leu-Xaa-Leu-Phe-Leu-Ala-Lys)-DWL (Asp-Trp-Leu)-HVLVVVP (recombination domain) and suppress or manipulate the host processes by manipulating targets within plant cells (Asai and Shirasu 2015; Selin et al. 2016).
Throughout the past few years, the availability of a large number of completely sequenced genomes for oomycetes became available (Sivashankari and Shanmughavel 2007; Fletcher et al. 2022; Matson et al. 2022; Cox et al. 2022). In addition, there have been rapid advances in RNA sequencing techniques leading to a growing number of studies exploring gene regulation during pathogen development and infection (Wang et al. 2009; Seidl et al. 2012). The comparative analysis of multiple genomes can provide insights into the biology of species not obtainable from singular genomes (O’Brien and Fraser 2005). For example, a study comparing human genes with genes from other organisms was able to assign functions to un-annotated genes in all genomes investigated (Sivashankari and Shanmughavel 2007).
By considering the core orthologous genes, phylogenetic analysis on deeply sequenced oomycete genomes has revealed a close relationship between downy mildews and Phytophthora (Voglmayr et al. 2004; Sharma et al. 2015; McCarthy and Fitzpatrick 2017; Bourret et al. 2018). To identify conserved gene regulatory elements, the current study compared the genome sequences of three related oomycete species, as species with close phylogenetic distance are expected to have sequence conservation and are likely to encode many identifiable orthologs (Judelson et al. 1992; McCue et al. 2002). In the current analysis, two Phytophthora species, Ph. infestans and Ph. sojae, and Pl. halstedii have been included. A study on human and mouse tissues has shown that DNA sequences controlling the expression of genes that are regulated similarly in the related species can be expected to be conserved (Sivashankari and Shanmughavel 2007). Such studies are lacking for oomycetes, so far. More recently, a de novo DNA motif-discovery study in Phytophthora predicted conserved DNA motifs by correlation with gene expression levels during infection (Seidl et al. 2012). Given the close phylogenetic relationship, it can be expected that key transcriptional regulatory networks between Phytophthora and downy mildews would be conserved [16, 17].
The present study is aimed at identifying regulatory motifs in co-regulated gene clusters that also are functionally conserved among three oomycetes species.
Datasets and methods
Pl. halstedii time-series transcriptome dataset
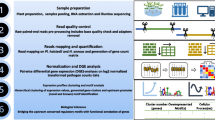
In a previous study, an expression for the life cycle of Pl. halstedii was performed (Bharti et al. 2023). For the generation of the corresponding dataset, gene expression values with false discovery rate corrected p-value or adjusted p-value (p-adj) ≤ 0.01 were clustered using hierarchical clustering, visualized in terms of Z-scores using DEGreport version 1.29.0 (Love et al. 2014; L Pantano 2019), and the 400-nt (nucleotides) upstream sequences of expressed genes were the starting point for further cross-species sequence conservation analyses. Figure 1 shows the procedural framework for motif discovery used in the current study.

The flowchart representation of methods followed in the current study. (1a) The clustered Pl. halstedii gene expression data for 16 time-points generated using hierarchical clustering and adapted as the reference information from Bharti et al. (2023). (1b) The retrieval of clustered upstream sequences up to 400 nucleotides (nt) from Pl. halstedii genome and their downstream gene sequences. (2) Independent sequence retrieval of Ph. sojae and Ph. infestans from whole genomes (Ensembl Protists release 49) and local two databases created (dbase U for upstream sequences and dbase G for downstream gene sequences). (3) The sequence alignments between the reference Pl. halstedii clustered sequences (Cl1 to Cl97) and database sequences were made using BLAST (blastn) tool with parameters e-vale < 0.01, word size 11, sequence identity > 70% for upstream, and > 50% for genes respectively. (4) The intersecting upstream sequences with their gene sequences were filtered for further motif analysis of length 4–18 nt using HMM-based MEME suite (cluster size < 50) and STREME (cluster size > 50) tool of MEME suite. In addition, the motif database created with STREME-IDs (dbase M generated from orthologous upstream sequences) using STREME tool. For the discovered MOTIF IDs (de novo + STREME) and reference hits (STREME-IDs), the motif enrichment and exact genomic location search were performed using AME and FIMO of meme suite. (5) The motifs were categorized into A (Pl. halstedii; Ph. sojae, Ph. infestans), B (Pl. halstedii; Ph. sojae), C (Ph. sojae, Ph. infestans), D (Pl. halstedii; Ph. infestans), E (any one of the three genomes). Further, the Category A motifs were interpreted for their biological significance related to virulence
Selection of other oomycete species
The key to being able to compare upstream regions between different oomycete species is the availability of their whole genome sequences. To include only high-quality data, in addition to Pl. halstedii (genome assembly GCA_900000015.1) as a reference, the well-studied oomycete Phytophthora species Ph. infestans (GCA_000142945.1) and Ph. sojae (GCA_000149755.2) were included. Whole genomes and functional annotations are retrieved from Ensembl Protists release 49 and the Joint Genome Institute (JGI) fungal portal (Grigoriev et al. 2014; Howe et al. 2020).
Upstream DNA sequence extraction and genomic alignments
Both gene sequences and their upstream 400-nt flanking regions were retrieved using an in-house python script. Initially, the local nucleotide alignments of oomycete orthologues were constructed using a command line BLAST 2.12.0 (blastn) tool (Altschul 1990; Zhang et al. 2000; Camacho et al. 2009), setting the threshold e-value to < 0.01 and the word size to 11. Subsequently, the local alignments were selected by using the criteria such as nucleotide hit length > 11 nucleotide (nt), identity of 50% or higher, and hit strand in the same direction for both upstream sequence and gene sequence alignment.
Motif discovery among aligned upstream sequences
A motif discovery approach was taken using MEME 5.2.0 or Multiple Expression Motifs (EM) for Motif Elicitation (Bailey et al. 2009) for clusters with less than 50 genes and STREME (Bailey 2021) for clusters with more than 50 genes to discover motifs between 4 and 18 nt (e-value ≤ 0.5). The following parameters were used: zero or one occurrence per sequence, binomial test based on the differential enrichment test, and p-value threshold ≤ 0.05. Furthermore, an empirical 3rd-order background Markov model based on all upstream regions of the combined backgrounds the genomes of Pl. halstedii, Ph. infestans, and Ph. sojae which accounts for single-, di-, tri-, and tetra-nucleotide distributions among the genomes, was applied. The reference genome motif database from pairwise aligned upstream regions was generated using STREME, and discovered motifs in the clustered groups of more than 50 genes were searched against this database. AME (analysis of motif enrichment) of the MEME suite detects putative motifs’ coordinates that are relatively enriched (e-value ≤ 0.5) in a set of sequences compared with reference database motifs (McLeay and Bailey 2010). By treating motifs independently, sequence sets (groups) were searched for the occurrences of known motifs using FIMO (Find Individual Motif Occurrences) (Grant et al. 2011).
Interpretation of enriched and conserved motifs associated with genes with known functions
From the published genome and time-series transcriptomic data of Pl. halstedii, genes annotated as pathogenicity related, as encoding for transcription factor (TF) proteins, as core biological pathways, as belonging to the secretome, and as clustered gene expression profiles were obtained from the supplementary data of Sharma et al. (2015) and Bharti et al. (2023). Experimentally validated oomycete effector sequences and WY domain information were retrieved from published literature and the supplementary data of EffectorO (Nur et al. 2023). For multiple protein sequence alignments in clusters, a hidden Markov model (HMM) parameter enabling for mBed-like clustering in the tool Clustal Omega 1.2.4 was used (Sievers and Higgins 2018).
Results
Alignment of upstream nucleotide and downstream gene sequences
In a previous study on the time course of sunflower infection by Pl. halstedii, gene expression analysis revealed a total of 97 expression clusters, with 8444 genes mapped successfully on 15,707 reference gene transcripts (Bharti et al. 2023). In the current study, sequence alignments of orthologous genes from the genomes of Ph. infestans and Ph. sojae could be obtained for 96 of 97 clusters. With an e-value < 0.01, identity > 70%, and hit strand in the same orientation, a similar procedure as for their downstream regions (~ 25 K gene sequences) revealed sequence identity > 50% within gene sequence alignments (Fig. 2a; Additional file 1). There was no orthologous gene found in Phytophthora spp. for the 17 gene members in cluster 22, which is the reason why no alignments for this cluster could be performed. The remaining 96 clusters contained 6279 sequences and lengths up to 400 nt from the ATG start codon (Additional file 1). A large number of orthologous gene members were assigned to cluster 11 (Additional file 3).

Sequence alignments and motif categorization in the orthologous sequences. a NCBI BLAST (blastn) tool generated the statistically significant aligned sequences and here for three of the genomes accumulatively, 96 clusters are listed such as upstream sequence alignment (no hit was found for cluster 22) and 97 clusters in gene sequences with e-value < 0.01, word size 11, sequence identity > 70%, and > 50% respectively. b The discovered motifs from upstream sequences (U <—U ∩ G) were categorized into Category A (Pl. halstedii; Ph. sojae; Ph. infestans, 112 motifs), Category B (Pl. halstedii; Ph. sojae, 76 motifs), Category C (Ph. sojae; Ph. infestans, 10 motifs), Category D (Pl. halstedii; Ph. infestans, 4 motifs), and Category E (any one of the three genomes, 43 motifs)
Reference motif database generation from orthologous oomycete genomes and motif discovery on clustered orthologous upstream regions intersected with downstream gene sequences
The tool STREME generated a reference motif database containing 15 over-represented, statistically significant motifs (p-value < 0.05). The retrieved STREME-5 motif (AGCGCGTG, 622 occurrences) out of 15 reference motifs had the highest occurrences, and the second most abundant motif was STREME-1 (CAGCGGGGCTGCCGT, 369 occurrences) (Table 1; Additional file 2). A total of 245 novel, statistically significant (p-value < 0.05) motifs were found using the tools MEME and STREME (Additional file 3). AME derived the genomic co-ordinates (p-value < 0.05) for 112 putative and cross-species conserved motifs (STREME-IDs) against the reference motif database (Additional file 3). No significantly enriched motifs were generated for 25 out of 97 expression clusters.
Motif categorization
The 245 motifs found were further divided into five categories on the basis of upstream genomic alignment conservation. The motifs conserved in all three genomes (Pl. halstedii; Ph. sojae; Ph. infestans) were categorized as Category A (112 motifs). Category B (76 motifs) was given for conserved motifs in Pl. halstedii and Ph. sojae, Category C (10 motifs) for Ph. sojae and Ph. infestans, Category D (4 motifs) for Pl. halstedii and Ph. infestans, and Category E (43 motifs) for one of the three genomes. All generated motif categories are shown in Fig. 2b, and Category A motifs with their genomic positions are shown in Figs. 3 and 4 (Additional file 3; Additional Fig. 1, 2, 3). The reference gene cluster expression profiles for Category A motifs were adapted from Bharti et al. (2023) as shown in Fig. 3.

Clustered Pl. halstedii gene expression profiles. RNA seq time-series data analysis of Pl. halstedii transcriptome among the 16 time-points (5 min, 15 min, 4 h, 8 h, 12 h, 24 h, 48 h, 72 h, 120 h, 222 h, 288 h, 290 h, 292 h, 294 h and 296 h, 296 h_prim from the infected host cotyledons and the primary leaves) covering the four infection phases (zoospore release phase, infection phase, colonization, and sporulation phase). In this figure, the adapted cluster profiles of 61 out of 97 clustered expression profiles from a previous study (Bharti et al. 2023) are shown, with an enlarged image for cluster 47 on the right side for highlighting the different stages. For the selected 61 clusters, there are upstream gene sequences conserved among Pl. halstedii, Ph. sojae, and Ph. infestans. The X-axis shows the time-points and Y-axis shows Z-score (scaled relative gene expression). Using the hierarchical clustering method, clusters with at least 15 genes per cluster were selected for further motif discovery

The conserved motifs identified and their locations relative to their parent genes. The motif discovered on the BLAST aligned upstream 400 nucleotides in a cross-species (Pl. halstedii, Ph. sojae, and Ph. infestans) comparison, using the 97 clusters of Pl. halstedii as a reference. Sequences were categorized into A (Pl. halstedii; Ph. sojae, Ph. infestans), B (Pl. halstedii; Ph. sojae), C (Ph. sojae, Ph. infestans), D (Pl. halstedii; Ph. infestans), and E (Any one of the three genomes). Thirty-two out of 112 Category A clusters shown with MOTIF IDs and logos corresponding to each gene cluster ID (number of clusters, 14) and STREME IDs listed b with logos. Enriched motif and their locations identified using AME and FIMO tool are presented in plot b
Utilizing sequence similarity-based criteria, 112 Category A motifs conserved in Pl. halstedii, Ph. sojae, and Ph. infestans were further analyzed for association with genes related to pathogenicity, exocytosis and vesicle transport, ion channels and calcium-binding proteins, plant cell wall–degrading enzymes (PCWDEs), and transcription factor (TF) proteins. None of the motifs were found associated with protease inhibitors and ion channels (Table 1).
Association of TFBS motifs with pathogenicity-related genes, exocytosis and vesicle transport, calcium-binding proteins, plant cell wall–degrading enzymes (PCWDEs), and transcription factors
Genes in 25 clusters show significant over-representation of regulatory motifs (46 motifs; p < 0.05) for genes involved in pathogenicity, exocytosis and vesicle transport, ion channels and calcium-binding proteins, plant cell wall–degrading enzymes (PCWDEs), and transcription factor (TF) proteins, which implies that genes involved in these biological functions are regulated in Pl. halstedii and two Phytophthora species in a conserved fashion in spite of their different genomic locations (Table 1).
RxLR and Crinkler (CRN) motifs and their upstream regulatory motifs
Upstream of putative RxLR-effector coding genes, the motifs found were MOTIF-60, MOTIF-61 in cluster (cl) 36, and STREME-3 in cl 90 (Additional file 4). In the cross-protein sequence similarity analysis, it was found that both motifs were associated with previously reported effectors (Fig. 4). Interestingly, two RxLR-like protein coding genes CEG44730 and PHYSODRAFT_293198 with putative regulatory motifs MOTIF-60 and 61 and the RxLR-like protein coding gene CEG41057 with STREME-3 upstream were retrieved (Sharma et al. 2015). Further, sequence similarity with the functionally characterized effectors Ph. infestans Avrblb2 and Ph. sojae avirulence homolog-5 (Avh5) revealed a putative location and an alternative RxLR motif (Fig. 4, Table 2). Interestingly, there is also protein conservation for genes within clusters containing RxLR-like proteins such the presence of radial spoke protein 11 and DnaJ heat shock protein.
In cl 61, the MOTIF-84 was found upstream of conserved CRN domain-containing proteins CEG41789 and PHYSODRAFT_316479, which had no similarity with previous experimentally validated effectors. From cl 65, three CRN-like genes, CEG44506 (crn1 and PsCRN108 like; conserved LQLFLAK domain) and PHYSODRAFT_471561 (crn2 like) with LFLAK-motif (FLAR) and with the upstream MOTIF-85, were significantly over-represented in the cluster (Fig. 5, Table 2). Two orthologous CRN-like protein coding genes with the motifs HVLVVVP had the upstream MOTIF-38; a HVLVAL motif was found downstream of MOTIF-13 (in cl 9 and cl 5, respectively) (Additional file 4). The motif STREME-5 was found upstream of cl 54, with one gene CEG44994 (Avr4/6 like) containing HVLVVVD.

Sequence similarity with experimentally validated effector sequences from literature and relationship between the upstream sequence conservation and protein sequences. The subgrouped protein sequence conservation and their identified putative upstream motifs MOTIF-60, 61 a and STREME-3 b using MEME and STREME suite for cluster 36 and 90 respectively containing RxLR-like and RxLR-dEER proteins. (2) The potential secretion RxLR-dEER motif supported with experimentally validated effectors Avrblb2 and Avh5
Other groups and the upstream regulatory motifs
Pathogenic microbes produce a variety of peptidases, which are enzymes that catalyze the breakdown of host proteins into small polypeptides to disrupt the host defense and create conditions suitable for pathogen colonization (Marshall et al. 2017; Figaj et al. 2019). In the current study, orthologous ubiquitin-specific protease, OTU (ovarian tumor)-like cysteine protease, and serine protease family were found in some clusters with the following regulatory motifs: MOTIF-15 (cl 6), MOTIF-17 (cl 7), MOTIF-86 (cl 65), MOTIF-98 (cl 81), MOTIF-103 (cl 88), and MOTIF-117 (cl 111) were found (Additional Table 1; Additional file 4).
ATP-binding cassette (ABC) transporter proteins constitute one of the largest protein families, present in both prokaryotes and eukaryotes, and transport a broad range of substances across biochemical membranes (Dassa and Bouige 2001). The regulatory motif MOTIF-78 (cl 56) was found enriched with ABC transporter proteins (Table 1; Additional file 4).
In oomycetes, cytoplasmic Ca2+ levels are controlled by calcium-binding channel proteins and channels represent key targets for anti-oomycete fungicides for pathogen control (Judelson and Blanco 2005). In the current study, the regulatory motifs MOTIF-15 (cl 6) and MOTIF-17 (cl 7) were found associated with such proteins (Table 1; Additional file 5).
Exocytosis serves for the delivery of vesicle content with enzymes such as proteases, glucanases, and callose from the pathogen cell to the host extracellular matrix and also to the plasma membrane (Leborgne-Castel and Bouhidel 2014). Rab family GTPases and transfer proteins SEC20 and SEC14 for export from endoplasmic reticulum to the Golgi apparatus were conserved in Pl. halstedii and the two Phytophthora species and associated with the motifs MOTIF-54 (cl 28), MOTIF-61 (cl 36), STREME-5 (cl 12), MOTIF-85, 86 (cl 65), and MOTIF-104 (cl 90) (Additional Table 1; Additional file 5).
Phytopathogenic oomycetes enter the plant through multiple routes, and many, including downy mildews, Ph. infestans and various Pythium species, penetrate into the host using appressoria (Judelson and Ah-Fong 2019). Genes induced in the appressorium stage by Phytophthora species include the cell wall-degrading enzymes (CWDEs) to degrade cellulose, hemicelluloses, xylan, pectin, β-1,3-glucans, and glycoproteins in the plant cell wall (Blackman et al. 2015), so the pathogen can grow into the host. Conserved glycoside hydrolase coding genes are associated with motifs MOTIF-25 (cl 12), STREME-12 (cl 12), MOTIF-82 (cl 60), MOTIF-96 (cl 75), and STREME-14 (cl 23) (Additional Table 1; Additional file 5).
Transcription factors (TFs), sequence-specific DNA-binding proteins, directly bind to regulatory regions on DNA. TFs regulate the production of virulence factors by modulating the gene expression (Charoensawan et al. 2010). Mostly, zinc finger and myb transcription factor encoding genes were found to be associated with putative upstream TFBS motifs STREME-1,13 (cl 54), MOTIF-11,12 (cl 4), STREME-1 (cl 2), STREME-14 (cl 23), MOTIF-64 (cl 41), STREME-12 (cl 3), MOTIF-121 (cl 113), MOTIF-78 (cl 56), STREME-1 (cl 3), MOTIF-60, 61 (cl 36), MOTIF-70 (cl 47), MOTIF-23 (cl 12), MOTIF-15 (cl 6), MOTIF-17 (cl 7), and MOTIF-67 (cl 45), most of which are highly GC-rich (Additional Table 1; Additional file 5).
Discussion
A previous study focused on regulatory motif discovery in upstream clustered DNA sequences from an infection time series for identifying stage-specific putative TFBS (Bharti et al. 2023). In this study, we took advantage of the previously published dataset and investigated, if there are motifs in closely related obligate biotrophs or hemi-biotroph oomycete genomes associated with similar genes and thus potentially functionally conserved. The current study investigated this by identifying the conserved regulatory sequence patterns in different oomycete species. For this, motif discovery and functional conservation analysis of clusters was investigated.
Most-represented upstream regulatory motifs
In terms of occurrences, the most represented motifs STREME-5 (AGCGCGTG; 622 sites; -34/-391 to -27/-384) and STREME-1 (CAGCGGGGCTGCCGT; 369 sites; -32/-388 to -18/-380) share similarity with conserved central CG-rich Motif-3 (5′-SGCGCS-3′) and G-box motif (5′-GDGGGG-3′), respectively, found in genome-wide upstream motif analysis of the protist parasite Cryptosporidium parvum (Oberstaller et al. 2013). Likewise, MOTIF-53 (cl 27; WCTGGCGGSYGAC; 13 nt; 4 sites; location: -103/-274 to -90/-261) has resemblance with the Motif-3 identified in C. parvum (Oberstaller et al. 2013). The second most-represented G-box-like motif reported in the present study, also found in the upstream region of C. parvum, represents a gene subset involved in DNA metabolism (Mullapudi et al. 2007). The CG-rich Motif-3 is highly similar to the binding site of the E2F-DP transcription factor, which functions as the regulators of the cell cycle and apoptosis (Zheng et al. 1999). This suggests that the motif is a major cell cycle regulator, potentially conserved throughout the SAR eukaryotic supergroup.
Biological interpretation of regulatory motifs in Category A
The findings from Roy et al. (2013) suggest that MOTIF-48 (cl 25; GGCAGCCCAA; 10 nt; 5 sites; 5 genes; location: -202/-388 to -192/-378) and MOTIF-15 (cl 6; GSCACCAASYT; 11 nt; 21 sites; 21 genes; -42/-376 to -67/-364) function as putative CCAAT box, while STREME-9 (CATTCTCCTC; 39 sites; -53/-379 to -44/-370) has similarity with the INR motif (YCAYTYY). The putative CAAT motif MOTIF-48 is associated with members of cluster 25 that contains functionally conserved genes, characterized as F-ATPase superfamily, zinc finger, and pleckstrin homology–like domain, while MOTIF-15 is associated mainly with cytoplasmic protein kinases, calcineurin-like phosphoesterase (Additional file 3). This is similar to the genes reported earlier to be associated with the CAAT motif in Phytophthora (Seidl et al. 2012; Roy et al. 2013). The FPR motif (MWTTTNC) from the study of Roy et al. (2013) was found to be similar to MOTIF-93 (cl 70; CTTCTTTCGGGMCA; 14 nt; 4 sites; 4 genes; -184/-370 to -170/-356) of the present study. STREME-9 is associated with a functionally conserved kinesin-associated protein, a DNA repair protein and protein kinases.
MOTIF-12 (cl 4; CGTACCGG; 7 sites; 6 genes; -78/-384 to -70/-376) bears similarity with Motif-9 (GTACCGGTA; 9 nt) found by Seidl et al. (2012), which was reported to be highly abundant in Phytophthora genomes (Seidl et al. 2012). The reported Motif-9 regulates a set of genes enriched in RXLR effectors and is similar to MOTIF-12, which is associated with genes coding for proteins with a pleckstrin homology–like domain and zinc finger C2H2 domain, suggesting it might be associated with regulating the expression of regulatory proteins.
In relation to expression-based cluster motif analysis performed in Plasmodium falciparum (Iengar and Joshi 2009), the motifs MOTIF-41 (cl 21; TCTGTGCAAD; 10 nt; 10 genes; -89/-359 to -79/-358), MOTIF-6 (cl 2; TCAAGTACGAGA; 11 nt; 5 sites; 5 genes; -272/-341 to -259/-346), and MOTIF-83 (cl 60; GAGSGG; 6 nt; 19 sites; 15 genes; -14/-138 to -85/-358) found in this study were observed to be similar with TGTG-motif and GAGA-motif, respectively. Both were identified in the upstream regions of the organellar translation machinery and proteasome sets in Pd. falciparum (Iengar and Joshi 2009). In addition, MOTIF-72 (cl 51; CTTCC; 5 nt; 25 sites; 20 genes; -32/-382 to -27/-377) and MOTIF-119 (cl 113; TTCC; 4 nt; 23 sites; 17 genes; -61/-387 to -57/-383) are similar to the TTCCC upstream region of a set of 15 mitochondrial genes in Pd. falciparum species. The 7-nt cold-box (GGACGAG), located upstream of transcription start site essential for PinifC3 induction during zoosporogenesis, contains the previously found GAGA-box and is similar with MOTIF-6 (TCAAGTACGAGA) of the present study (Tani and Judelson 2006) and potentially regulates genes involved in zoospore production. Similarly, the GC-rich putative core promoter element named as Downstream Promoter Element Peronosporales (DPEP) has a pattern SAASMMS, reported as well-conserved in the orthologous promoters from Ph. infestans, Phytophthora ramorum, and Ph. sojae, which bears similarity with MOTIF-6 of the present study (Roy et al. 2013) and is probably targeted by a specific family of transcription factors.
Biological interpretation of regulatory motifs in other categories
The Category B motif MOTIF-95 (cl 71; AAGGCGGAGA; 10 nt; 8 sites; genes; -97/-302 to -87/-312) contains the previously reported GAGA-motif and is similar to the 7-nt cold-box GGACGAG located upstream of transcription start site essential for PinifC3 induction during zoosporogenesis (Tani and Judelson 2006). From the same category, MOTIF-32 (cl 15; GGAAACTTG; 9 nt; 6 sites; 3 genes; -45/-185 to -36/-176) is similar to the most over-represented motif (5′-[A/C] AACTA-3′) of unknown function in the protist parasite Cryptosporidium parvum (Mullapudi et al. 2007; Oberstaller et al. 2013). MOTIF-31 (cl 14; CCCCACCAAG; 10 nt; 4 sites; 4 genes; -201/-341 to -191/-331) is similar to motif CCCCAT upstream region of a set of 15 mitochondrial genes in Pd. falciparum species (Iengar and Joshi 2009). In Category C, the part of the MOTIF-26 (cl 13; ATTGGATYGCCAAGT; 15 nt; 2 sites; 2 genes; -111/-132 to -96/-117) has some resemblance with the CCAAT box (Roy et al. 2013). The reported FPR (MWTTTNC) reported in the same study is similar to MOTIF-18 (cl 9; ACTTTATAATG; 2 sites; 2 genes; -327/-352 to -316/-341; putative FPR).
High upstream conservation for zinc finger and myb TFs coding genes
In the present study, the maximum number of genes in Category A with high upstream sequence conservation is zinc finger and myb TF coding genes. Interestingly, promoters containing one CACCT and one CACCTG known as target of the complex for the Smad-interacting protein 1 (cause a form of Hirschsprung disease in Humans) or “Sip1” from mouse embryo were found to be probably similar to the core of MOTIF-15 (cl 6; GSCACCAASYT; 11 nt; 24 sites; 21 genes; -16/-136 to -42/-376; putative CCAAT box; Remacle 1999; Postigo and Dean 2000). For the cluster 3 genes, the motif STREME-12 (TCTTCGCCAGGA; 12 sites; 11 genes; -228/-390 to -217/-379) also might be targeted by zinc-finger transcription factors (Table 1).
Similarity with experimentally validated effector sequences from literature and relationship between the upstream sequence conservation and protein sequences
Interestingly, the functionally characterized effectors Avrblb2 of pathogen Ph. infestans have a similarity with the RxLR-like protein coding gene set regulated by 16-nt-long conserved MOTIF-60 (TCAWBKNSMRKCYGRD) and the 6-nt-long MOTIF-61 (ACAAGC) of cluster 36 (Fig. 5a; Additional file 4; (Oh et al. 2009; Bozkurt et al. 2011; Sun et al. 2013). Positionally, the upstream regulatory motifs have been found to be within the location range of -58 to -383 for MOTIF-60 and range -75 to -344 for MOTIF-61 in Pl. halstedii and Ph. sojae. No positional conservation was observed within the cross-species upstream regions and the experimentally validated effector Avrblb2. A 13-nt-long motif, STREME-3 (Fig. 5b; cluster 90; CTCTGCGGCTAAA), present at 89 to 324 nucleotides upstream of genes, is conserved upstream of the functionally validated Avh5 and Avrblb2 effectors, starting at -181/-353 nt upstream of the ATG, respectively.
The orthologous, PITG_09290, CEG48632, and crn2-like (Torto et al. 2003) were found to be conserved downstream of the MOTIF-38 (AGAAKRYRATCAAGG) in cluster 19. The start position of the motif varies between -353 and -131 for a set of genes with associated with MOTIF-38 and is at position -66 upstream for the crn2-like gene of Ph. infestans. The conserved HVLVVVP positions of crn1 and crn2 genes from Torto et al. (2003) were also conserved in Pl. halstedii gene CEG48632 (Sharma et al. 2015). CEG44994, a CRN-like protein of Pl. halstedii, was found to be similar to Avr4/6 (previously Avh171) recognized by Rps4- and Rps6-containing soybean plants (Dou et al. 2010). The presence of conserved central CG-rich motif (STREME-5: AGCGCGTG), at position -170 relative to the ATG of the CRN-like gene CEG44994, is a variant of the TGC[A/G][T/G]G[C/G]GA motif implicated in the regulation of glycolysis pathway genes, in a genome-wide upstream motif analysis of the protist parasite Cryptosporidium parvum (Mullapudi et al. 2007).
The orthologous CRN-like CEG40558 and PITG_07363 sequences, associated with MOTIF-13 (Additional Fig. 5a; cl 5; GATAGTATTC; -205 to -106; GATA-like), have similarities with the experimentally validated cytoplasmic RXLRs Avrblb2 and AVR1 of Ph. infestans, as well as Avh5 of Ph. sojae (Bozkurt et al. 2011; Sun et al. 2013; Du et al. 2018). The MOTIF-13 in cluster 5 starts within a location range of -17 to -366 upstream of start codon. The cross-species conserved regulatory motif MOTIF-85 (Additional Fig. 5b; cl 65; CGTM; -252 to -152) was associated with Crinkler genes (CEG44506; crn1 and PsCRN108 like, and PHYSODRAFT_471561; crn2 like) not conserved in Pl. halstedii and Ph. sojae (Torto et al. 2003; Song et al. 2015). However, the conserved LQLFLAK domain was found in five genes associated with MOTIF-85 (Fig. 5).
Co-expression of genes, as an indicator of concerted regulation, suggests that the genes may share similar regulatory motifs. However, it is not necessarily true that the co-expressed genes must have the same or similar regulatory motifs conserved in closely related species. Even the same species with similar expression patterns might also be due to a master regulator that controls the expression of other transcription factors for various nodes, leading to coordinated expression of multiple genes. Alternatively, also the combined action of two or more transcription factors independently regulated could lead to the same expression pattern (Fig. 6).

Sequence similarity with experimentally validated effector sequences from literature and relationship between the upstream sequence conservation and protein sequences. Protein sequence conservation for genes associated with motifs MOTIF-60 and MOTIF-61 as identified using MEME and STREME on cluster 36, containing DnaJ heat shock proteins a and radial spoke protein 11 b. On the basis of protein sequence similarity search and mBed-like clustering guide tree generated using Clustal Omega, further, the protein sequences within the clusters were divided into subgroups to analyze the orthologous sequence conservation among the gene group(s) within cluster(s), where H is Pl. halstedii (CEG45488 and CEG38426); S, Ph. sojae (PHYSODRAFT_480671 and PHYSODRAFT_503617); and I, Ph. infestans (PITG_02094 and PITG_15780). The bold underline represents conserved DnaJ domain information of DnaJ heat shock protein and EF-hand domain of radial spoke protein 11; and box represents calcium ion channel domain of radial spoke protein 11
Conclusions and outlook
Genes with similar mRNA expression profiles are likely to be regulated by similar mechanisms. For testing this hypothesis, the current study focused on potential transcription factor binding motifs in the promoter regions of oomycete genes within total 46 conserved motifs associated with members of 25 expression clusters with a potential role in pathogenesis that were found. Examples for this are MOTIF 13 associated with cluster 5 and STRME-3 associated with cluster 90. The approach taken in this study could be expanded to orthologous upstream regions of other oomycete species to obtain further insights into conserved regulatory pathways. Such studies could be followed up with the identification of potential transcription factors and co-factors that bind to orthologous TFBS. The identification of highly conserved, oomycete-specific binding sites could also be potential targets for devising control strategies against oomycete pathogens.
Data availability
RNA sequencing data that support the findings of this study have been deposited in GenBank with the accession code PRJEB49134 and BMC genomics with the data object identifier or DOI (https://doi.org/10.1186/s12864-023-09214-7).
References
Altschul S (1990) Basic local alignment search tool. J Mol Biol 215:403–410. https://doi.org/10.1006/jmbi.1990.9999
Asai S, Shirasu K (2015) Plant cells under siege: plant immune system versus pathogen effectors. Curr Opin Plant Biol 28:1–8. https://doi.org/10.1016/j.pbi.2015.08.008
Bailey TL (2021) STREME: accurate and versatile sequence motif discovery. Bioinformatics 37:2834–2840. https://doi.org/10.1093/bioinformatics/btab203
Bailey TL, Boden M, Buske FA et al (2009) MEME Suite: tools for motif discovery and searching. Nucleic Acids Res 37(suppl_2):W202–W208. https://doi.org/10.1093/nar/gkp335
Bharti S, Ploch S, Thines M (2023) High-throughput time series expression profiling of Plasmopara halstedii infecting Helianthus annuus reveals conserved sequence motifs upstream of co-expressed genes. BMC Genomics 24:140. https://doi.org/10.1186/s12864-023-09214-7
Blackman LM, Cullerne DP, Torreña P et al (2015) RNA-Seq analysis of the expression of genes encoding cell wall degrading enzymes during infection of lupin (Lupinus angustifolius) by Phytophthora parasitica. PLoS ONE 10:e0136899. https://doi.org/10.1371/journal.pone.0136899
Bourret TB, Choudhury RA, Mehl HK et al (2018) Multiple origins of downy mildews and mito-nuclear discordance within the paraphyletic genus Phytophthora. PLoS ONE 13:e0192502. https://doi.org/10.1371/journal.pone.0192502
Bozkurt TO, Schornack S, Win J et al (2011) Phytophthora infestans effector AVRblb2 prevents secretion of a plant immune protease at the haustorial interface. Proc Natl Acad Sci USA 108:20832–20837. https://doi.org/10.1073/pnas.1112708109
Camacho C, Coulouris G, Avagyan V et al (2009) BLAST+: architecture and applications. BMC Bioinforma 10:1–9. https://doi.org/10.1186/1471-2105-10-421
Charoensawan V, Wilson D, Teichmann SA (2010) Genomic repertoires of DNA-binding transcription factors across the tree of life. Nucleic Acids Res 38:7364–7377. https://doi.org/10.1093/nar/gkq617
Chen Q, Bakhshi M, Balci Y et al (2022) Genera of phytopathogenic fungi: GOPHY 4. Stud Mycol 101:417–564. https://doi.org/10.3114/sim.2022.101.06
Cox MP, Guo Y, Winter DJ et al (2022) Chromosome-level assembly of the Phytophthora agathidicida genome reveals adaptation in effector gene families. Front Microbiol 13:1038444. https://doi.org/10.3389/fmicb.2022.1038444
Dassa E, Bouige P (2001) The ABC of ABCs: a phylogenetic and functional classification of ABC systems in living organisms. Res Microbiol 152:211–229. https://doi.org/10.1016/S0923-2508(01)01194-9
Dou D, Kale SD, Liu T et al (2010) Different domains of phytophthora sojae effector Avr4/6 are recognized by soybean resistance genes Rps4 and Rps6. Mol Plant Microbe Interact 23:425–435. https://doi.org/10.1094/MPMI-23-4-0425
Du Y, Weide R, Zhao Z et al (2018) RXLR effector diversity in Phytophthora infestans isolates determines recognition by potato resistance proteins; the case study AVR1 and R1. Stud Mycol 89:85–93. https://doi.org/10.1016/j.simyco.2018.01.003
Fawke S, Doumane M, Schornack S (2015) Oomycete interactions with plants: infection strategies and resistance principles. Microbiol Mol Biol Rev 79:263–280. https://doi.org/10.1128/mmbr.00010-15
Figaj D, Ambroziak P, Przepiora T, Skorko-Glonek J (2019) The role of proteases in the virulence of plant pathogenic bacteria. Int J Mol Sci 20:672. https://doi.org/10.3390/ijms20030672
Fletcher K, Shin OH, Clark KJ et al (2022) Ancestral chromosomes for family Peronosporaceae inferred from a telomere-to-telomere genome assembly of Peronospora effusa. Mol Plant Microbe Interact 35:450–463. https://doi.org/10.1094/MPMI-09-21-0227-R
Gascuel Q, Martinez Y, Boniface MC et al (2015) The sunflower downy mildew pathogen Plasmopara halstedii. Mol Plant Pathol 16:109–122. https://doi.org/10.1111/mpp.12164
Grant CE, Bailey TL, Noble WS (2011) FIMO: scanning for occurrences of a given motif. Bioinformatics 27:1017–1018. https://doi.org/10.1093/bioinformatics/btr064
Grigoriev IV, Nikitin R, Haridas S et al (2014) MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res 42(D1):D699–D704. https://doi.org/10.1093/nar/gkt1183
Howe KL, Contreras-Moreira B, De Silva N et al (2020) Ensembl Genomes 2020-enabling non-vertebrate genomic research. Nucleic Acids Res 48:D689–D695. https://doi.org/10.1093/nar/gkz890
Iengar P, Joshi NV (2009) Identification of putative regulatory motifs in the upstream regions of co-expressed functional groups of genes in Plasmodium falciparum. BMC Genomics 10(1):1–21. https://doi.org/10.1186/1471-2164-10-18
Judelson HS, Ah-Fong AMV (2019) Exchanges at the plant-oomycete interface that influence disease. Plant Physiol 179:1198–1211. https://doi.org/10.1104/pp.18.00979
Judelson HS, Blanco FA (2005) The spores of Phytophthora: weapons of the plant destroyer. Nat Rev Microbiol 3:47–58. https://doi.org/10.1038/nrmicro1064
Judelson HS, Tyler BM, Michelmore RW (1992) Regulatory sequences for expressing genes in oomycete fungi. MGG Mol Gen Genet 234:138–146. https://doi.org/10.1007/BF00272355
Kamoun S, Furzer O, Jones JDG et al (2015) The Top 10 oomycete pathogens in molecular plant pathology. Mol Plant Pathol 16:413–434. https://doi.org/10.1111/mpp.12190
L Pantano (2019) DEGreport: report of DEG analysis. R package version 1(8): 10.18129. http://lpantano.github.io/DEGreport/
Laura Martínez A, José Quiroz F, Delia Carrera A (2021) Detection of Plasmopara halstedii in sunflower seeds: a case study using molecular testing. J Saudi Soc Agric Sci 20:395–400. https://doi.org/10.1016/j.jssas.2021.04.007
Leborgne-Castel N, Bouhidel K (2014) Plasma membrane protein trafficking in plant-microbe interactions: a plant cell point of view. Front Plant Sci 5:735. https://doi.org/10.3389/fpls.2014.00735
Li Y, Han Y, Qu M et al (2020) Apoplastic cell death-inducing proteins of filamentous plant pathogens: roles in plant-pathogen interactions. Front Genet 11:661. https://doi.org/10.3389/fgene.2020.00661
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12):1–21. https://doi.org/10.1186/s13059-014-0550-8
Marshall NC, Finlay BB, Overall CM (2017) Sharpening host defenses during infection: proteases cut to the chase. Mol Cell Proteomics 16:S161–S171. https://doi.org/10.1074/mcp.O116.066456
Matson MEH, Liang Q, Lonardi S, Judelson HS (2022) Karyotype variation, spontaneous genome rearrangements affecting chemical insensitivity, and expression level polymorphisms in the plant pathogen Phytophthora infestans revealed using its first chromosome-scale assembly. PLoS Pathogens 18:e1010869. https://doi.org/10.1371/journal.ppat.1010869
McCarthy CGP, Fitzpatrick DA (2017) Phylogenomic reconstruction of the oomycete phylogeny derived from 37 genomes. mSphere 2(2):10–1128. https://doi.org/10.1128/msphere.00095-17
McCue LA, Thompson W, Steven Carmack C, Lawrence CE (2002) Factors influencing the identification of transcription factor binding sites by cross-species comparison. Genome Res 12:1523–1532. https://doi.org/10.1101/gr.323602
McLeay RC, Bailey TL (2010) Motif enrichment analysis: a unified framework and an evaluation on ChIP data. BMC Bioinforma 11:1–11. https://doi.org/10.1186/1471-2105-11-165
Mullapudi N, Lancto CA, Abrahamsen MS, Kissinger JC (2007) Identification of putative cis-regulatory elements in Cryptosporidium parvum by de novo pattern finding. BMC Genomics 8:13. https://doi.org/10.1186/1471-2164-8-13
Nur MJ, Wood KJ, Michelmore RW (2023) EffectorO: motif-independent prediction of effectors in oomycete genomes using machine learning and lineage-specificity. Mol Plant-Microbe Interactions®. https://doi.org/10.1094/MPMI-11-22-0236-TA
O’Brien SJ, Fraser CM (2005) Genomes and evolution: the power of comparative genomics. Curr Opin Genet Dev 15:569–571
Oberstaller J, Joseph SJ, Kissinger JC (2013) Genome-wide upstream motif analysis of Cryptosporidium parvum genes clustered by expression profile. BMC Genomics 14:516. https://doi.org/10.1186/1471-2164-14-516
Oh SK, Young C, Lee M et al (2009) In planta expression screens of Phytophthora infestans RXLR effectors reveal diverse phenotypes, including activation of the solanum bulbocastanum disease resistance protein Rpi-blb2. Plant Cell 21:2928–2947. https://doi.org/10.1105/tpc.109.068247
Pandaranayaka EPJ, Frenkel O, Elad Y et al (2019) Network analysis exposes core functions in major lifestyles of fungal and oomycete plant pathogens. BMC Genomics 20:1–15. https://doi.org/10.1186/s12864-019-6409-3
Postigo AA, Dean DC (2000) Differential expression and function of members of the zfh-1 family of zinc finger/homeodomain repressors. Proc Natl Acad Sci USA 97:6391–6396. https://doi.org/10.1073/pnas.97.12.6391
Remacle JE, Kraft H, Lerchner W et al (1999) New mode of DNA binding of multi-zinc finger transcription factors: δEF1 family members bind with two hands to two target sites. EMBO J 18:5073–5084. https://doi.org/10.1093/emboj/18.18.5073
Roy S, Poidevin L, Jiang T, Judelson HS (2013) Novel core promoter elements in the oomycete pathogen Phytophthora infestans and their influence on expression detected by genome-wide analysis. BMC Genomics 14(1):1–15. https://doi.org/10.1186/1471-2164-14-106
Scanu B, Jung T, Masigol H et al (2021) Phytophthora heterospora sp. nov., a new pseudoconidia-producing sister species of P. palmivora. J Fungi 7:870. https://doi.org/10.3390/jof7100870
Seidl MF, Wang RP, van den Ackerveken G et al (2012) Bioinformatic inference of specific and general transcription factor binding sites in the plant pathogen Phytophthora infestans. PLoS ONE 7(12):e51295. https://doi.org/10.1371/journal.pone.0051295
Selin C, de Kievit TR, Belmonte MF, Fernando WGD (2016) Elucidating the role of effectors in plant-fungal interactions: progress and challenges. Front Microbiol 7:600. https://doi.org/10.3389/fmicb.2016.00600
Sharma R, Xia X, Cano LM et al (2015) Genome analyses of the sunflower pathogen Plasmopara halstedii provide insights into effector evolution in downy mildews and Phytophthora. BMC Genomics 16:1–23. https://doi.org/10.1186/s12864-015-1904-7
Sievers F, Higgins DG (2018) Clustal Omega for making accurate alignments of many protein sequences. Protein Sci 27:135–145. https://doi.org/10.1002/pro.3290
Sivashankari S, Shanmughavel P (2007) Comparative genomics - a perspective. Bioinformation 1:376–378. https://doi.org/10.6026/97320630001376
Song T, Ma Z, Shen D et al (2015) An oomycete CRN effector reprograms expression of plant HSP genes by targeting their promoters. PLoS Pathogens 11:e1005348. https://doi.org/10.1371/journal.ppat.1005348
Sun F, Kale SD, Azurmendi HF et al (2013) Structural basis for interactions of the Phytophthora sojae RxLR effector Avh5 with phosphatidylinositol 3-phosphate and for host cell entry. Mol Plant Microbe Interact 26:330–344. https://doi.org/10.1094/MPMI-07-12-0184-R
Tani S, Judelson H (2006) Activation of zoosporogenesis-specific genes in Phytophthora infestans involves a 7-nucleotide promoter motif and cold-induced membrane rigidity. Eukaryot Cell 5:745–752. https://doi.org/10.1128/EC.5.4.745-752.2006
Thines M (2014) Phylogeny and evolution of plant pathogenic oomycetes-a global overview. Eur J Plant Pathol 138:431–447. https://doi.org/10.1007/s10658-013-0366-5
Tian M, Win J, Song J et al (2007) A Phytophthora infestans cystatin-like protein targets a novel tomato papain-like apoplastic protease. Plant Physiol 143:364–377. https://doi.org/10.1104/pp.106.090050
Torto TA, Li S, Styer A et al (2003) EST mining and functional expression assays identify extracellular effector proteins from the plant pathogen Phytophthora. Genome Res 13:1675–1685. https://doi.org/10.1101/gr.910003
Voglmayr H, Riethmüller A, Göker M et al (2004) Phylogenetic relationships of Plasmopara, Bremia and other genera of downy mildew pathogens with pyriform haustoria based on Bayesian analysis of partial LSU rDNA sequence data. Mycol Res 108:1011–1024. https://doi.org/10.1017/S0953756204000954
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63
Zhang Z, Schwartz S, Wagner L, Miller W (2000) A greedy algorithm for aligning DNA sequences. J Comput Biol 7:203–214
Zheng N, Fraenkel E, Pabo CO, Pavletich NP (1999) Structural basis of DNA recognition by the heterodimeric cell cycle transcription factor E2F-DP. Genes Dev 13:666–674. https://doi.org/10.1101/gad.13.6.666
Acknowledgements
We would like to thank Sebastian Ploch for his support. We also thank LOEWE for providing the computational resources. We gratefully acknowledge Dr. Claus Weiland for assisting in the establishment of the computational server access.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by Deutscher Akademischer Austauschdienst (DAAD) under the grants (Research Grants-Doctoral Programmes in Germany, 2017/18). SB has received support under the grant number funding term-57299294 from DAAD. MT is supported by LOEWE in the framework of the Centre for Translational Biodiversity Genomics (TBG), funded by the government of Hessen.
Author information
Authors and Affiliations
Contributions
MT designed the experiment. SB designed the computational analyses with support from MT. SB performed the computational analyses and interpreted the results. SB and MT wrote the manuscript. Both authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval
Not applicable.
Consent for publication
The publisher Springer has the authors’ permission to publish research findings.
Competing interests
MT is an Editor-in-Chief of Mycological Progress journal. However, he did not participate in the handling of this manuscript or influenced any editorial decisions. SB was supported for carrying out research under the DAAD doctoral program.
Disclaimer
The funder had no role in study design, data collection and interpretation, decision to submit the work for publication, or preparation of the manuscript.
Additional information
Section Editor: Tanay Bose
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bharti, S., Thines, M. Conservation of putative transcription factor binding sites of co-expressed Plasmopara halstedii genes in two Phytophthora species. Mycol Progress 22, 63 (2023). https://doi.org/10.1007/s11557-023-01911-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11557-023-01911-7