
Abstract
Poly(ADP-ribose) polymerase (PARP) inhibitors (PARPis) have transformed the ovarian cancer (OC) treatment landscape. This narrative review provides a comprehensive overview of data for the PARPis olaparib, niraparib, and rucaparib in patients with OC and discusses their role in disease management, with a focus on the use of PARPis as maintenance therapy in the United States (US). Olaparib was the first PARPi to be approved as first-line maintenance monotherapy in the US, with maintenance niraparib subsequently approved in the first-line setting. Data also support the efficacy of rucaparib as first-line maintenance monotherapy. PARPi maintenance combination therapy (olaparib plus bevacizumab) also provides benefit in patients with newly diagnosed advanced OC whose tumors tested positive for homologous recombination deficiency (HRD). Biomarker testing is critical in the newly diagnosed setting to identify patients most likely to benefit from PARPi maintenance therapy and guide treatment decisions. Clinical trial data support the use of PARPis (olaparib, niraparib, rucaparib) as second-line or later maintenance therapy in patients with platinum-sensitive relapsed OC. Although distinct differences in tolerability profile were observed between PARPis, they were generally well tolerated, with the majority of adverse events managed by dose modification. PARPis had no detrimental effect on patients’ health-related quality of life. Real-world data support the use of PARPis in OC, although some differences between PARPis are apparent. Data from trials investigating novel combination strategies, such as PARPis plus immune checkpoint inhibitors, are awaited with interest; the optimal sequencing of novel therapies in OC remains to be established.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
We provide a comprehensive overview of data for the poly(ADP-ribose) polymerase (PARP) inhibitors olaparib, niraparib, and rucaparib in patients with ovarian cancer (OC) and discuss their role in disease management. |
In the newly diagnosed OC setting, PARP inhibitors provide the greatest clinical benefit in patients with a BRCA1 and/or BRCA2 mutation (BRCAm) or whose tumors test positive for homologous recombination deficiency, meaning biomarker testing is critical to identify those patients most likely to benefit from PARP inhibitor maintenance therapy and guide treatment decisions. |
Although there are distinct differences in tolerability profile between PARP inhibitors, they are generally well tolerated, with the majority of adverse events managed by dose modification. |
1 Introduction
Ovarian cancer (OC) is often diagnosed at an advanced stage and is associated with poor prognosis. Until recently, first-line treatment for advanced (International Federation of Gynecology and Obstetrics [FIGO] stage II–IV) OC included debulking surgery combined with platinum-based chemotherapy [1]. Despite exquisite sensitivity to platinum-based therapy in the front line, most patients relapse within 3 years despite treatment [2] and are often retreated with multiple courses of therapy, including further cytoreductive surgery and chemotherapy [1].
The OC treatment landscape has evolved with the development of targeted therapies, such as anti-angiogenic agents and poly(ADP-ribose) polymerase (PARP) inhibitors (PARPis). The anti-angiogenic agent bevacizumab was the first targeted therapy to be approved in the United States (US) for use in OC [3]. Bevacizumab has demonstrated efficacy in patients with newly diagnosed [4,5,6,7], platinum-sensitive relapsed (PSR) [8,9,10], and platinum-resistant relapsed [11] OC. Given its efficacy in these settings, treatment guidelines include bevacizumab-containing regimens as options in first-line and later-line settings, with maintenance bevacizumab recommended in patients in response to platinum-based regimens incorporating bevacizumab [1, 2, 12].
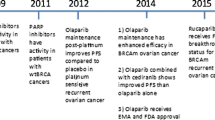
More recently, PARPis have emerged as important new therapies in OC, with three PARPis, olaparib, niraparib, and rucaparib, currently approved by the US FDA as maintenance therapy for patients with OC (Fig. 1 and Table 1) [13,14,15]. As more treatment options become available, determining the best therapy for patients can be challenging. The authors undertook a comprehensive narrative review of evidence supporting the use of PARPis in OC, including topics not easily captured by systematic reviews. This review focuses on data from Phase III trials and trials that led to the approval of PARPis in OC, highlighting the current and future treatment landscape in OC, including the role of biomarker testing and adverse event (AE) management strategies.

US approval of PARP inhibitors for use in patients with OC*. *The trial(s) on which approval was based is shown in parentheses. 1L first-line, 3L+ third-line or later, 4L+ fourth-line or later, BRCAm BRCA1 and/or BRCA2 mutation, g germline, HRD homologous recombination deficiency, OC ovarian cancer, PARP poly(ADP-ribose) polymerase, PSR platinum-sensitive relapsed, +ve positive
2 Methods
Literature searches for the narrative review were initially conducted in PubMed for papers published up to 6 September 2021, using the following search terms: ‘(PARP inhibitor OR olaparib OR veliparib OR niraparib OR rucaparib) AND (ovarian cancer)’. A search alert in PubMed was used to capture additional articles published between 6 September 2021 and 9 January 2023. Searches were restricted to ‘Humans’, ‘Clinical Trial’, ‘Clinical Study’, and ‘Research support, non-U.S. Gov’t’.
Databases of the American Society of Clinical Oncology, European Society of Gynaecological Oncology, European Society for Medical Oncology, International Gynecologic Cancer Society, and Society of Gynaecological Oncology were also searched for congress abstracts from 2019 to 2022.
Articles retrieved from the above searches were included if they were Phase III clinical trials or trials that led to the approval of PARPis in OC and key trials conducted thereafter. Clinical studies of PARPis not approved for use in the US or novel treatments, preclinical studies, in vitro studies, and review articles were excluded.
In addition, reference lists of retrieved papers were hand-searched for relevant studies, and key papers were included based on the authors’ clinical experience and knowledge of the field.
3 Efficacy in Ovarian Cancer
3.1 First-Line Maintenance Monotherapy
Olaparib was the first PARPi to be approved as first-line maintenance monotherapy in the US based on the results of the Phase III SOLO1 trial [16], with niraparib subsequently approved in the first-line setting based on the results of the Phase III PRIMA trial [17]. Results of the Phase III ATHENA-MONO trial evaluating rucaparib as first-line maintenance monotherapy are also included for completeness [18]. All three studies included patients with stage III–IV, high-grade serous or endometrioid OC, primary peritoneal cancer, and/or fallopian tube cancer who had clinical complete response (CR) or partial response (PR) after platinum-based chemotherapy. SOLO1 enrolled patients with tumors with a BRCA1 and/or BRCA2 mutation (BRCAm) [16], whereas PRIMA [17] and ATHENA-MONO [18] enrolled patients regardless of tumor biomarker status. Although all patients with newly diagnosed advanced OC are at high risk of disease progression, PRIMA only enrolled patients considered at higher clinical risk (patients with FIGO stage III disease and no residual macroscopic disease after upfront surgery were excluded from the study) [17], whereas SOLO1 [16] and ATHENA-MONO [18] included patients irrespective of their clinical risk.
SOLO1 randomized 391 patients to receive maintenance olaparib tablets or placebo for up to 2 years or until disease progression (Table 2); patients with ongoing evidence of disease at 2 years could continue to receive study treatment at the investigators’ discretion (at the time of the primary analysis, 10% of patients randomized to olaparib and 2% of those randomized to placebo had continued treatment beyond 2 years) [16, 19]. After a median follow-up of ≈41 months, a statistically significant improvement in the primary endpoint of investigator-assessed progression-free survival (PFS) was observed with olaparib versus placebo (median not reached vs. 13.8 months), with a hazard ratio (HR) of 0.30 (95% confidence interval [CI] 0.23–0.41; P < 0.001) [Table 2]. At 3 years, 60% of patients in the olaparib group versus 27% in the placebo group were free of PFS events [16]. Exploratory subgroup analyses showed that the risk of disease progression or death was significantly reduced with olaparib versus placebo in patients with both higher-risk disease (FIGO stage III with upfront surgery and residual disease or neoadjuvant chemotherapy, or FIGO stage IV; HR 0.34, 95% CI 0.24–0.48) and lower-risk disease (FIGO stage III with upfront surgery and no residual disease; HR 0.33, 95% CI 0.20–0.52) [20].
An updated, post hoc analysis showed that the PFS benefit derived from 2 years’ maintenance therapy with olaparib was sustained beyond the end of treatment [21]. After a median follow-up of ≈5 years, median PFS was 56.0 months with maintenance olaparib compared with 13.8 months with placebo (HR 0.33, 95% CI 0.25–0.43). Consistent PFS benefit was observed in both the higher-risk (median PFS 40.6 vs. 11.1 months; HR 0.34, 95% CI 0.24–0.49) and lower-risk (median PFS not reached vs. 21.9 months; HR 0.38, 95% CI 0.25–0.59) subgroups in an exploratory analysis (Table 2) [21].
A prespecified descriptive analysis conducted after 7 years of follow-up showed a clinically meaningful improvement in overall survival (OS) with olaparib versus placebo (median OS not reached vs. 75.2 months; HR 0.55, 95% CI 0.40–0.76; P = 0.0004 [P < 0.0001 required to declare statistical significance]) in SOLO1 (Table 2) [22]. At 7 years, 67% of patients in the olaparib group versus 46.5% of patients in the placebo group were alive, and 45.3% versus 20.6% were alive and had not received a first subsequent treatment [22].
In PRIMA, 733 patients were randomized to receive maintenance niraparib or placebo for 36 months or until disease progression (Table 2); patients were eligible regardless of biomarker status. A subsequent protocol amendment permitted the use of an individualized starting dose (ISD) of niraparib based on baseline weight and platelet levels, because of increased risk of thrombocytopenia [17]. The primary endpoint was PFS as assessed by real-time blinded independent central review (BICR) in patients with homologous recombination deficiency (HRD)-positive tumors (defined as a BRCAm and/or genomic instability [genomic instability score ≥ 42]; MyChoice® CDx test [Myriad Genetic Laboratories, Inc., Salt Lake City, UT, US]) and in the overall population. After a median follow-up of 13.8 months, median PFS was significantly longer with maintenance niraparib than with placebo, both in patients whose tumors tested positive for HRD (21.9 vs. 10.4 months; HR 0.43; 95% CI 0.31–0.59; P < 0.001) and in the overall population (13.8 vs, 8.2 months; HR 0.62; 95% CI 0.50–0.76; P < 0.001) (Table 2) [17]. In prespecified exploratory analyses, a PFS benefit was seen with maintenance niraparib versus placebo in patients with a BRCAm, as well as patients without a BRCAm, patients whose tumors tested positive for HRD without BRCAm, and patients whose tumors tested negative for HRD (Table 2) [17, 23]. At the time of the primary analysis, no difference was observed between the fixed starting dose (HR 0.59; 95% CI 0.46–0.76) and ISD (HR 0.69; 95% CI 0.48–0.98) groups in terms of PFS benefit with niraparib versus placebo [24] (Table 2; an updated analysis of investigator-assessed PFS [25] is also shown in Table 2). However, in a non-analytical analysis reported in the European Medicines Agency (EMA) assessment report in patients receiving an ISD of niraparib 200 mg based on bodyweight and platelet count, while a significant PFS benefit was observed with niraparib in patients whose tumors tested positive for HRD, there was no significant difference between niraparib and placebo in the overall population or in patients whose tumors tested negative for HRD (Table 2) [26]. OS data were immature at the time of the primary analysis [17].
An updated analysis showed that the PFS benefit was maintained after a median 3.5 years of follow-up [27]. HRs for investigator-assessed PFS with niraparib versus placebo were 0.66 (95% CI 0.56–0.79) in the intent-to-treat (ITT) population, 0.52 (95% CI 0.40–0.68) in patients whose tumors tested positive for HRD, and 0.65 (95% CI 0.49–0.87) in patients whose tumors tested negative for HRD (Table 2).
Additionally, the Phase III PRIME study evaluated niraparib (ISD) versus placebo as first-line maintenance therapy in 384 Chinese patients with newly diagnosed advanced OC. Treatment continued for up to 3 years or until disease progression or unacceptable toxicity. Like PRIMA, PRIME enrolled patients regardless of biomarker status, but included patients with or without residual disease after primary debulking surgery, and the assay used to test tumor HRD status differed between PRIME (BGI assay; BGI Genomics, Shenzhen, China) and PRIMA (MyChoice® CDx). After a median follow-up of 27.5 months, a statistically significant PFS benefit was observed with the niraparib ISD regimen versus placebo in the ITT population (HR 0.45; 95% CI 0.34–0.60; P < 0.001) and across prespecified subgroups, including groups based on biomarker or postoperative residual disease status (Table 2) [28].
ATHENA-MONO randomized 538 patients to receive maintenance rucaparib or placebo for up to 2 years or until disease progression, death, or unacceptable toxicity (Table 2) [18]. Patients were eligible regardless of biomarker status and were stratified according to HRD status using the FoundationOne CDx™ next-generation sequencing assay (Foundation Medicine, Inc., Cambridge, MA, US). The primary endpoint was investigator-assessed PFS in patients with HRD-positive tumors (defined as a BRCAm and/or a high genomic loss of heterozygosity [LOH] score [≥ 16%]) and in the overall population. After a median follow-up of ≈26 months, median PFS was significantly longer with maintenance rucaparib than with placebo both in patients whose tumors tested positive for HRD (28.7 vs. 11.3 months; HR 0.47; 95% CI 0.31–0.72; P = 0.0004) and in the overall population (20.2 vs. 9.2 months; HR 0.52; 95% CI 0.40–0.68; P < 0.0001) (Table 2). In prespecified exploratory analyses, a PFS benefit was seen with maintenance rucaparib versus placebo in patients with a BRCAm, patients with non-BRCAm/LOH high tumors, and patients whose tumors tested negative for HRD (Table 2) [18]. PFS benefit was seen with rucaparib over placebo regardless of clinical risk (Table 2) [29]. OS data were immature at the time of the primary analysis [18].
Taken together, findings from SOLO1 [16], PRIMA [17], and ATHENA-MONO [18] indicate that PARPi maintenance therapy provides the greatest benefit in the first-line setting in patients with a BRCAm [16,17,18] (prespecified exploratory analyses in PRIMA [17] and ATHENA-MONO [18]) or whose tumors test positive for HRD [17, 18]. A PFS benefit was also seen in the overall PRIMA and ATHENA-MONO populations regardless of biomarker status. The limited benefit seen with niraparib or rucaparib in patients whose tumors tested negative for HRD highlights the importance of testing for HRD status. Benefit in SOLO1 and ATHENA-MONO was seen regardless of clinical risk. Longer-term follow-up in SOLO1 indicated an OS benefit with olaparib versus placebo and that maintenance olaparib provides long-term remission in some patients; factors predicting which patients will experience long-term benefit from PARPi maintenance therapy remain to be identified. Maintenance therapy with a PARPi should be considered in all patients with newly diagnosed advanced OC regardless of their clinical risk. Data strongly support the first-line use of maintenance PARPi therapy in patients with a BRCAm or whose tumors test positive for HRD, with maintenance therapy with bevacizumab alone remaining an option for some patients, including some patients whose tumors test negative for HRD, a population with high unmet need [30].
3.2 First-Line Maintenance Combination Therapy
Results of the Phase III PAOLA-1 study led to the US approval of olaparib in combination with bevacizumab for the maintenance treatment of patients with advanced OC who are in response to first-line platinum-based chemotherapy and whose tumors tested positive for HRD [31]. Results of the Phase II OVARIO trial evaluating niraparib plus bevacizumab as first-line maintenance combination therapy in patients with newly diagnosed advanced OC are also included for completeness [32].
PAOLA-1 randomized 806 patients with newly diagnosed, stage III–IV, high-grade serous or endometrioid OC, primary peritoneal cancer, and/or fallopian tube cancer who had no evidence of disease or a clinical CR or PR after platinum-based chemotherapy plus bevacizumab. Patients were eligible irrespective of biomarker status or clinical risk. Following randomization, patients received maintenance olaparib tablets or placebo for up to 24 months or until disease progression or unacceptable toxicity; all patients received bevacizumab for up to 15 months in total (Table 3).
After a median follow-up of 22.9 months, the primary endpoint of investigator-assessed PFS was significantly longer with olaparib plus bevacizumab than with placebo plus bevacizumab (median 22.1 vs. 16.6 months; HR 0.59; 95% CI 0.49–0.72; P < 0.001) [31]. Results of subgroup analyses showed a substantial PFS benefit with maintenance olaparib plus bevacizumab versus bevacizumab alone in patients with a tumor BRCAm (tBRCAm) (HR 0.31; 95% CI 0.20–0.47) and in patients whose tumors tested positive for HRD (HR 0.33; 95% CI 0.25–0.45; (MyChoice® CDx test) (Table 3). Patients whose tumors tested negative for HRD did not show a PFS benefit with olaparib plus bevacizumab maintenance versus bevacizumab alone (HR 1.00; 95% CI 0.75–1.35) [31]. An exploratory analysis showed that PFS was substantially improved with olaparib plus bevacizumab versus bevacizumab alone in both higher-risk patients (FIGO stage III with upfront surgery and residual disease or neoadjuvant chemotherapy, or FIGO stage IV; HR 0.60; 95% CI 0.49–0.74) and lower-risk patients (FIGO stage III with upfront surgery and no residual disease; HR 0.46; 95% CI 0.30–0.72), with the greatest PFS benefit observed in the tBRCAm and HRD-positive subgroups [33] (Table 3). Results of the main time to second progression or death (PFS2) analysis are also shown in Table 3 [34].
Final OS analysis after approximately 5 years of follow-up showed a median OS of 56.5 months with olaparib plus bevacizumab versus 51.6 months with placebo plus bevacizumab in the ITT population (HR 0.92; 95% CI 0.76–1.12; P = 0.4118) (Table 3) [35]. Clinically meaningful OS improvements were seen with maintenance olaparib plus bevacizumab versus bevacizumab alone in patients with a tBRCAm (HR 0.60; 95% CI 0.39–0.93; 73% of patients in the olaparib plus bevacizumab group vs. 54% of patients in the placebo plus bevacizumab group were alive at 5 years) and in patients whose tumors tested positive for HRD (HR 0.62; 95% CI 0.45–0.85; 66% of patients in the olaparib plus bevacizumab group versus 48% of patients in the placebo plus bevacizumab group were alive at 5 years) (Table 3). No survival benefit was seen in patients whose tumors tested negative for HRD (HR 1.19; 95% CI 0.88–1.63) [35].
OVARIO enrolled 105 patients with newly diagnosed, stage III–IV, high-grade serous or endometrioid OC, primary peritoneal cancer and/or fallopian tube cancer who had no evidence of disease or a clinical CR or PR after platinum-based chemotherapy plus bevacizumab [32]. Patients with high-grade serous or endometrioid histology were enrolled irrespective of biomarker status; additionally, other epithelial non-mucinous OC patients were allowed to enroll if they had a germline BRCAm (gBRCAm). With maintenance niraparib plus bevacizumab, the 18-month PFS rate (primary endpoint) in the ITT population was 62%, with a median PFS of 19.6 months (95% CI 16.5–25.1) (Table 3). Subgroup analysis found that 18-month PFS rates were highest (and median PFS was longest) in patients whose tumors tested positive for HRD or who had a BRCAm (Table 3) [32].
Based on the results of PAOLA-1, maintenance combination therapy with olaparib plus bevacizumab should be considered in patients with newly diagnosed advanced OC whose tumors test positive for HRD, regardless of their clinical risk. Interestingly, the greatest impact may be in patients who have historically been defined to have “lower-risk” advanced stage disease (stage III, upfront surgery with no residual disease).
3.3 Second-Line or Later Maintenance Monotherapy
Olaparib, niraparib, and rucaparib are approved in the US as second-line or later maintenance monotherapy in patients in response to platinum-based chemotherapy. The approval of olaparib was based on the results of the Phase II Study 19 [36] and Phase III SOLO2 [37] studies and the approvals of niraparib and rucaparib were based on the Phase III NOVA [38] and ARIEL3 [39] studies, respectively. Subsequent studies included two olaparib studies, Phase IIIb OPINION [40] and Phase IV ORZORA [41], as well as the Phase III NORA study of niraparib [42]. All studies included patients with PSR OC (PSROC) who were in response to platinum-based chemotherapy and had received two or more prior platinum-based regimens (Table 4).
In Study 19, 265 patients were randomized to receive maintenance olaparib capsules or placebo until disease progression (Table 4) [36]. After a median follow-up of 5.6 months, the primary endpoint of PFS in the overall population was significantly longer with maintenance olaparib than with placebo (median PFS 8.4 vs. 4.8 months; HR 0.35; 95% CI 0.25–0.49; P < 0.001; Table 4) [36, 43]. A retrospective, preplanned, subgroup analysis demonstrated a PFS benefit with maintenance olaparib versus placebo in both patients with a BRCAm (HR 0.18; 95% CI 0.10–0.31) and patients without a BRCAm (HR 0.54; 95% CI 0.34–0.85), with a greater PFS benefit seen in patients with a BRCAm (Table 4) [43]. At the time of final analysis (79% data maturity), although the predefined threshold (P < 0.0095) for statistical significance was not met, an apparent OS advantage was observed with olaparib versus placebo in the overall population (HR 0.73; 95% CI 0.55‒0.95; nominal P = 0.02138) with the greatest benefit seen in patients with a BRCAm (HR 0.62; 95% CI 0.42–0.93; nominal P = 0.02140) and an HR favoring olaparib also seen in patients without a BRCAm (HR 0.84; 95% CI 0.57–1.25) [44]. Crossover of placebo patients (12% of ITT placebo patients and 23% of BRCAm placebo patients crossed over) to a PARPi following disease progression may have confounded the OS results [45]. Fifteen patients (11%) were taking olaparib for >6 years, suggesting a durable response [44].
In SOLO2, 295 patients with a gBRCAm were randomized to receive maintenance olaparib tablets or placebo until disease progression (Table 4) [37]. After a median follow-up of ≈22 months, the primary endpoint of investigator-assessed PFS was significantly longer with olaparib than with placebo (median 19.1 vs. 5.5 months; HR 0.30; 95% CI 0.22–0.41; P < 0.0001). PFS rates at 24 months were 43% and 15%, respectively [37]. Maintenance olaparib provided a clinically meaningful OS benefit of 12.9 months over placebo at final OS analysis. After a median-follow-up of ≈65 months, median OS was 51.7 months with olaparib compared with 38.8 months with placebo (HR 0.74; 95% CI 0.54–1.00; P = 0.054), unadjusted for the 38% of patients in the placebo group who received subsequent PARPi therapy [46]. The OS benefit was also apparent in a prespecified exploratory OS analysis adjusted for subsequent PARP therapy in the placebo group (HR 0.56; 95% CI 0.35–0.97) (Table 4). Cumulative exposure of ≥5 years was seen in 22% of patients in the olaparib group (vs. 9% of patients in the placebo group), indicating a durable response to maintenance olaparib in this subgroup of patients [46].
Subsequent studies support the use of maintenance olaparib in patients without a gBRCAm (OPINION [40, 47]; Table 4) and in patients with a tBRCAm of somatic and/or germline origin as well as in an exploratory non-BRCA homologous recombination repair (HRR) mutation (HRRm) cohort (ORZORA [41, 48]; Table 4).
In the NOVA study, 553 patients with (n = 203) and without (n = 350) a gBRCAm were randomized to receive maintenance niraparib or placebo until disease progression, unacceptable toxicity, death, withdrawal of consent, or loss to follow-up (Table 4) [38]. After a median follow-up of 16.9 months, PFS (primary endpoint) was significantly longer with maintenance niraparib than with placebo in the three efficacy populations, patients with a gBRCAm (median 21.0 vs. 5.5. months; HR 0.27; 95% CI 0.17–0.41; P < 0.001), patients without a gBRCAm (median 9.3 vs. 3.9 months; HR 0.45; 95% CI 0.34–0.61; P < 0.001), and patients whose tumors tested HRD-positive without a gBRCAm (median 12.9 vs. 3.8 months; HR 0.38; 95% CI 0.24–0.59; P < 0.001) [38]. Preplanned exploratory analyses found consistent PFS benefit with niraparib versus placebo in patients whose tumors tested HRD-positive with a somatic BRCAm (sBRCAm) (HR 0.27; 95% CI 0.08–0.90), patients whose tumors tested HRD-positive without a BRCAm (HR 0.38; 95% CI 0.23–0.63), and patients whose tumors tested HRD-negative (HR 0.58; 95% CI 0.36–0.92) (Table 4) [38]. Additionally, a retrospective exploratory analysis showed that in addition to patients with BRCAm and other HRRm, clinical benefit with niraparib was also observed in patients whose tumors tested HRD-negative without HRRm (Table 4) [49]. After a median follow-up of 5.5 years (data cut-off [DCO] 1 October 2020), median OS with maintenance niraparib versus placebo was 43.6 versus 41.6 months in the gBRCAm cohort (HR 0.93; 95% CI 0.63–1.36), 31.1 versus 36.5 months in the non-gBRCAm cohort (HR 1.10; 95% CI 0.83–1.46), and 37.3 versus 41.4 months in the non-gBRCAm, HRD-positive cohort (HR 1.32; 95% CI 0.84–2.06) (Table 4) [50, 51]. Although NOVA was not powered to evaluate between-group differences in OS, these results bring into question whether there could be an OS detriment to patients in the non-gBRCAm and the non-gBRCAm, HRD-positive subgroups who received maintenance niraparib compared with placebo [50, 51]. It should be noted that results may be confounded by crossover (46% of placebo patients in the gBRCAm cohort and 13% in the non-gBRCAm cohort received subsequent PARPi therapy) and missing data (OS data missing in 14% of patients in both the gBRCAm and non-gBRCAm cohorts) [50, 51]. In an updated OS analysis (DCO 31 March 2021), which accounted for missing survival data, median OS with maintenance niraparib versus placebo was 40.9 versus 38.1 months in the gBRCAm cohort (HR 0.85; 95% CI 0.61–1.20), 31.0 versus 34.8 months in the non-gBRCAm cohort (HR 1.06; 95% CI 0.81–1.37), and 35.6 versus 41.4 months in the non-gBRCAm, HRD-positive cohort (HR 1.29; 95% CI 0.85–1.95) (Table 4) [52]. Based on these results, maintenance therapy with niraparib has been restricted to patients with PSROC who have a gBRCAm in the US [14].
The Phase III NORA study in 265 Chinese patients demonstrated the efficacy of a niraparib ISD regimen as maintenance therapy, as evidenced by a significant reduction in the risk of disease progression and death with niraparib versus placebo after a median follow-up of 15.8 months (HR 0.32; 95% CI 0.23–0.45; P < 0.0001; Table 4) [42].
In ARIEL3, 564 patients were randomized to receive maintenance rucaparib or placebo until disease progression, death, or other reason for discontinuation [39]. The study met its primary endpoint with significantly longer investigator-assessed PFS seen with maintenance rucaparib than with placebo in patients with a BRCAm (median 16.6. vs. 5.4 months; HR 0.23; 95% CI 0.16–0.34; P < 0.0001), in patients whose tumors tested HRD-positive (median 13.6 vs. 5.4 months; HR 0.32; 95% CI 0.24–0.42; P < 0.0001), and in the ITT population (median 10.8 vs. 5.4 months; HR 0.36; 95% CI 0.30–0.45; P < 0.0001) (Table 4) [39]. A post hoc exploratory analysis assessed the clinical and molecular characteristics of patients with exceptional PFS benefit, where exceptional benefit was defined as double the median PFS (≥2 years) in the ITT population [53]. Overall, 21.1% of patients in the rucaparib group and 2.1% of patients in the placebo group showed exceptional benefit, with PFS of ≥2 years (Table 4); 13.9% and 6.9% of patients in the rucaparib group had PFS of ≥3 and ≥4 years, respectively. Results showed that exceptional benefit was more common in, but not exclusive to, patients with favorable clinical characteristics (including no measurable disease at baseline, longer penultimate platinum-free interval and CR to last platinum therapy) and known mechanisms of PARPi sensitivity (including BRCA1, BRCA2, RAD51C, and RAD51D alterations and genome-wide LOH) [53]. In the final OS analysis of ARIEL3, (median follow-up of 6.4 years), median OS with maintenance rucaparib versus placebo was 45.9 versus 47.8 months in the BRCAm cohort (HR 0.83; 95% CI 0.58–1.19), 40.5 versus 47.8 months in the HRD-positive cohort (HR 1.01; 95% CI 0.77–1.32), and 36.0 versus 43.2 months in the ITT population (HR 1.00; 95% CI 0.81–1.22) (Table 4). [54] Approximately 45% of patients in the placebo group received subsequent PARPi therapy [54]. ARIEL3 was not powered to evaluate between-group differences in OS; however, based on these results, maintenance rucaparib has been restricted to patients with PSROC who have a BRCAm in the US [15].
To summarize, PFS data from olaparib, niraparib, and rucaparib studies support the use of PARPi maintenance therapy in patients with PSROC, regardless of biomarker status. OS data from SOLO2 also support the use of maintenance olaparib in the relapsed disease setting in patients with BRCAm and Study 19 demonstrated an apparent OS advantage for olaparib over placebo in the overall population of patients with or without a BRCAm. In patients with PSROC, as requested by the FDA, maintenance niraparib is restricted to those with a gBRCAm, based on final OS data from NOVA, and maintenance rucaparib is restricted to those with a BRCAm, based on final OS data from ARIEL3, in the US; it should be noted that neither study was powered to assess between-group differences in OS. It is clear that the outcomes in platinum-sensitive patients who respond to a platinum doublet are quite poor without maintenance therapy with PFS of ≤5.5 months. A subset of patients will derive exceptional benefit from PARPi maintenance therapy in the relapsed disease setting; as well as HRD status, clinical factors such as platinum sensitivity seem to be important predictors of response to PARPi maintenance therapy.
3.4 Later-Line Treatment
In patients with relapsed advanced OC, olaparib was approved in the US as later-line treatment in patients with a gBRCAm, niraparib in patients whose tumors tested HRD-positive and rucaparib in patients with a BRCAm; however, these treatment indications have been voluntarily withdrawn (Fig. 1 and Table 1). The studies leading to the approval of these PARPis and subsequent studies are discussed briefly below and are shown in Table 5.
An early Phase II study (NCT00664781) [55], the three-part Phase I/II Study 10 [56, 57], and the two-part Phase II ARIEL2 study [57, 58] evaluated rucaparib treatment in OC. The approval of rucaparib treatment for patients with relapsed OC and a BRCAm who had received two or more prior chemotherapies was based on an integrated analysis of data from Study 10 Part 2A (n = 42) and ARIEL2 Parts 1 and 2 (n = 64) [57] (Table 5).
The Phase III ARIEL4 study subsequently evaluated rucaparib in patients with relapsed high-grade OC who had a gBRCAm or sBRCAm and had received two or more prior platinum or non-platinum chemotherapy regimens (Table 5) with a planned crossover to rucaparib for those who progressed on the chemotherapy arm (72% underwent crossover to a PARPi as first subsequent therapy) [59]. At the final OS analysis, a possible detriment in OS was observed with rucaparib versus chemotherapy (median OS 19.4 vs. 25.4 months; HR 1.31; 95% CI 1.00–1.73; P = 0.0507), driven by results in the subgroup of patients with platinum resistance (Table 5) [60]. It is important to note that an unusually high number of patients in the rucaparib arm did not receive any subsequent therapy after progressing on rucaparib compared with those who received chemotherapy (43% vs. 24% in the platinum-resistant subgroup; 38% vs. 16% in the partially platinum-sensitive subgroup; and 46% vs. 15% in the fully platinum-sensitive subgroup) [60, 61]. Based on these results, rucaparib has been voluntarily withdrawn in the US for the treatment of patients with BRCAm OC who have received two or more prior lines of chemotherapy [61].
The approval of olaparib in the treatment of patients with PSROC and a gBRCAm who have received three or more prior lines of chemotherapy was based on the results of the Phase II Study 42 trial (Table 5) [62].
The Phase III SOLO3 study in patients with gBRCAm PSROC who had received two or more prior lines of platinum-based chemotherapy confirmed and extended the results of Study 42 in the treatment setting (Table 5) [63]. At final analysis, OS (HR 1.07; 95% CI 0.76–1.49) and PFS2 (HR 0.80; 95% CI 0.56–1.15) did not significantly differ between the olaparib and chemotherapy groups (Table 5) [64]. A subsequent post hoc analysis found favorable OS for olaparib versus chemotherapy in the subgroup of patients who had received two prior lines of chemotherapy and a potential detrimental effect in patients who had received three or more prior lines of chemotherapy (Table 5) [65]. Based on these results, olaparib has been voluntarily withdrawn in the US for the treatment of patients with gBRCAm OC who have received three or more prior lines of chemotherapy [66].
The Phase II LIGHT study evaluated olaparib treatment in patients with PSROC and known BRCAm and HRD status who had received one or more prior lines of platinum-based chemotherapy (Table 5) [67]. Subgroup analyses found that ORR and median PFS were generally similar in patients with one or two or more prior lines of chemotherapy in the BRCAm cohorts and the HRD-positive non-BRCAm cohort (Table 5) [67]. At final OS analysis, the 18-month OS rate was 60–88% (Table 5) [68].
The approval of niraparib in the later-line treatment of patients with HRD-positive advanced OC who had been treated with three or more previous chemotherapy regimens was based on the results of the Phase II QUADRA study (Table 5) [69]. After a median follow-up of 12.2 months, the median OS was 17.2 months in 456 patients with measurable disease who had received three or more previous therapies (modified per-protocol population) (Table 5) [69]. A decision was made to voluntarily withdraw niraparib in the US for the treatment of patients with advanced OC whose tumors are associated with an HRD-positive status and who have received three or more prior lines of chemotherapy based on a totality of information from PARPis in the later-line treatment setting in OC [70].
In summary, olaparib, niraparib, and rucaparib are no longer indicated in the US for later-line treatment in patients with relapsed OC. It should be noted that neither SOLO3 nor ARIEL4 were powered to assess between-group differences in OS.
4 Role of Biomarker Testing in Optimal Therapeutic Decisions
Regardless of the PARPi administered, HRD testing is critical in the newly diagnosed setting to identify which patients may experience the greatest benefit from PARPi maintenance therapy and guide treatment decisions [1, 71]. As discussed previously, PARPi maintenance therapy showed the greatest benefit in newly diagnosed OC patients with a BRCAm or who tested positive for HRD in clinical trials.
Myriad MyChoice® CDx and FoundationOne CDx™ are US-approved companion diagnostics in OC. Clinical trials in the newly diagnosed setting have used MyChoice® CDx, which tests for the presence of a BRCAm and/or genomic instability (LOH, telomeric allelic imbalance, and large-scale state transitions) [17, 31], and FoundationOne CDx™, which tests for the presence of a BRCAm and LOH [18].
Laboratory-developed tests (e.g. the Geneva HRD test [72]) that can be deployed in a clinical laboratory may provide a viable alternative to commercial assays for determining HRD status.
In terms of testing for BRCAm, both germline and tumor testing are warranted [1]. Current guidelines recommend germline testing of all patients with epithelial OC at diagnosis, and tumor testing for sBRCAm for patients in whom a gBRCAm is not detected [73, 74].
Notably, tumor testing reliably identified BRCAm that are germline in origin in clinical trial settings [75, 76]. The availability of a reliable tumor test for use in clinical practice may permit more flexibility in the approach to testing, with initial tumor testing followed by genetic testing of patients in whom a tBRCAm is detected.
Germline testing remains essential if a tBRCAm is identified so the patient is aware of their personal risk of other cancers (e.g. breast cancer) and first- or second-degree blood relatives can be offered genetic risk evaluation, counseling, and testing [73].
Interestingly, as compared with the newly diagnosed setting, results of Phase III trials [38, 39] suggest that the benefit of HRD testing is impactful but not as profound in PSROC. The dominant factors are clinical, with platinum sensitivity being the dominant predictor of response in this setting.
The predictive potential of non-BRCA HRRm (e.g. mutations in RAD51B, RAD51C, RAD51D, BRIP1, PALB2, NBN, ATM, CHK1, CHK2, CDK12) has been evaluated in an exploratory fashion. In PAOLA-1, HRRm gene panels (excluding BRCA) did not predict the efficacy of maintenance olaparib plus bevacizumab in the newly diagnosed setting [77]. Using a 13-gene panel, the HR for PFS in patients with HRRm excluding tBRCAm (n = 54), was 0.95 (95% CI 0.49–1.94); on expansion of this panel to include five additional genes (n = 72), the HR for PFS was 1.01 (95% CI 0.55–1.95). Consistent results were also observed in patients with HRRm excluding tBRCAm using three other HRR gene panels [77]. However, in a post hoc analysis of ARIEL2 in patients with relapsed OC, RAD51C and RAD51D mutations predicted response to treatment with rucaparib, similar to BRCAm; the median PFS in patients with RAD51C/RAD51D-mutated OC (n = 7) was similar to that in patients with BRCAm (n = 138) (11.0 vs. 7.8 months; HR 1.52; 95% CI 0.67–3.44; P = 0.32) [78].
5 Safety and Tolerability of Poly(ADP-Ribose) Polymerase (PARP) Inhibitors
Although similarities are evident in the tolerability profiles of the different PARPis (with AEs such as anemia, neutropenia, nausea, vomiting, and fatigue considered class effects), distinct differences are also observed, requiring customization of monitoring and/or dosing regimens. When considered individually, the AE profile of each PARPi was generally consistent when administered as monotherapy in maintenance or treatment settings.
The safety profile of combination therapy with olaparib plus bevacizumab in PAOLA-1 was generally consistent with that observed with olaparib monotherapy [16, 37], with the exception of hypertension, which is commonly associated with bevacizumab [31]. Adding maintenance olaparib to bevacizumab did not increase bevacizumab-associated AEs, with a numerically lower incidence of hypertension with olaparib plus bevacizumab than with bevacizumab alone [31].
The most commonly reported hematologic and non-hematologic AEs generally occurred early in patients receiving PARPis. For olaparib, the median time to first onset was 1.94 months for anemia, 1.77 months for neutropenia, 2.83 months for thrombocytopenia, 0.13 months for nausea, 0.72 months for fatigue/asthenia, and 1.46 months for vomiting in SOLO1 [19, 79]. For niraparib, the incidence of thrombocytopenia, nausea, vomiting, diarrhea, fatigue, insomnia, and hypertension was highest during the first month of therapy and declined thereafter, whereas the incidence of anemia and neutropenia peaked in months 3 and 2, respectively, of maintenance niraparib therapy in NOVA [38, 80]. For rucaparib, the median time to first onset was 56 days for anemia, 52 days for thrombocytopenia, 5–15 days for nausea, vomiting, fatigue/asthenia, dysgeusia, and increased alanine aminotransferase (ALT)/aspartate aminotransferase (AST) levels, 22–29 days for decrease appetite, constipation, and diarrhea, and 45 days for abdominal pain in an integrated analysis of trial data [81].
When considering the safety and tolerability profiles of the PARPis, it is important to note that the duration of maintenance therapy was capped in newly diagnosed OC (2 years for olaparib [16, 31] and rucaparib [18] and 3 years for niraparib [17]), whereas olaparib [36, 37, 63, 79], niraparib [17, 38, 69], and rucaparib [39, 82] were continued until disease progression in the second-line or later maintenance and later-line treatment settings. For example, the median duration of maintenance olaparib therapy was 24.6 months in SOLO1 [22], whereas cumulative exposure of ≥5 years was seen in 22% of olaparib patients in the final analysis of SOLO2 [46]. The duration of study treatment in the key PARPi trials is shown in Table 6. Other factors, such as prior treatment and the duration of follow-up (shown in Tables 2–5), should also be considered when interpreting safety findings. Importantly, no new safety signals were identified with olaparib [22, 34, 46], niraparib [27, 50, 80], or rucaparib [82, 83] during longer-term follow-up.
Comparisons across trials should be made with caution because of differences in patient populations and trial methods between the studies. It should also be noted that only studies in which patients received the tablet formulation of olaparib are discussed in this section, given it is the only formulation currently marketed in the US for all olaparib indications [13].
5.1 Hematologic Adverse Events
Anemia and neutropenia are class effects of PARPis, with anemia being one of the most commonly reported treatment-emergent AEs reported with olaparib, niraparib, or rucaparib in randomized, placebo-controlled maintenance therapy studies, and randomized, controlled later-line treatment trials (Online Resource Table S1) [14, 17, 18, 22, 34, 38, 46, 59, 63, 83].
Grade ≥3 anemia was the most common grade ≥3 treatment-emergent AE reported in olaparib patients in SOLO1 [22], SOLO2 [46], and SOLO3 [63], and in rucaparib patients in ATHENA-MONO [18], ARIEL3 [83], and ARIEL4 [59] (Online Resource Table S1).
Although thrombocytopenia is seen with all PARPis [14, 17, 22, 34, 38, 46, 59, 63, 83], there is an increased risk of thrombocytopenia with niraparib (66% of niraparib patients vs. 5% of placebo patients in PRIMA [14] and 61% vs. 6%, respectively, in NOVA [38]), including grade ≥3 thrombocytopenia (39% vs. <1%, respectively, in PRIMA [14], and 34% vs. 1%, respectively, in NOVA [38]) (Online Resource Table S1).
The PRIMA protocol was amended to incorporate an ISD [17] because of the safety benefit observed with the lower starting dose in a retrospective analysis of NOVA [84]. In NOVA, the study protocol mandated an interruption of treatment for patients with specific hematologic and non-hematologic AEs, with resumption of treatment at a lower dose. The subsequent retrospective analysis suggested that patients with baseline body weight of <77 kg or baseline platelets of <150,000/µL may benefit from a lower starting dose of 200 mg/day [84], which is now the recommended dose for patients with a baseline body weight of <77 kg or baseline platelets of <150,000/µL who are receiving maintenance niraparib in the newly diagnosed setting [14]. The lower starting dose approved for use in patients receiving first-line maintenance therapy who have a lower bodyweight or baseline platelet count helps ameliorate thrombocytopenia [14]. In PRIMA, the incidence of grade ≥3 thrombocytopenia was 22% in patients whose starting dose of niraparib was based on baseline body weight or platelet count [14], which is closer to the rates of grade ≥3 thrombocytopenia reported in SOLO1 (1%) [22] and ATHENA-MONO (7%) [18].
The incidence of hematologic AEs reported in patients receiving niraparib also decreased over time [50, 80]. In NOVA, the incidence of grade ≥3 thrombocytopenia in niraparib patients decreased from 34% at year 1 to 3% at years 2–3 [50].
In terms of managing bone marrow suppression, US prescribing information recommends that PARPi therapy should not commence until hematologic toxicity caused by prior chemotherapy has recovered to grade ≤1 [13,14,15].
In patients receiving olaparib [13] or rucaparib [15], the complete blood count (CBC) should be monitored at baseline and monthly thereafter. Dose modification of olaparib [13] or rucaparib [15] may be required to manage prolonged hematologic toxicities. If blood counts do not recover, the patient should be referred to a hematologist for further investigation [13, 15].
In patients receiving niraparib, the CBC should be monitored weekly for the first month, and with dosage changes related to hematologic toxicity, monthly for the next 11 months and then periodically thereafter [14]. If prolonged hematologic toxicities persist despite dose interruption of niraparib, discontinue niraparib and refer the patient to a hematologist for further investigation [14].
Myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) have been reported in patients receiving olaparib, niraparib and rucaparib and are included in the warnings and precautions section of the US prescribing information for all three PARPis [13,14,15]. Collection of data pertaining to these AEs differed across trials.
A low incidence of MDS/AML was reported in patients receiving first-line maintenance olaparib with or without bevacizumab. In SOLO1, MDS/AML was reported in 1% of patients receiving maintenance olaparib (vs. 0% of placebo patients) at the primary PFS analysis [16] and in 2% of olaparib patients (vs. 1% of placebo patients) with longer-term follow-up [22]. In PAOLA-1, MDS/AML/aplastic anemia was reported in 1% of patients receiving maintenance olaparib plus bevacizumab (vs. 0.4% of placebo plus bevacizumab patients) at the time of the primary PFS analysis [31] and in 2% of olaparib patients (vs. 2% of placebo patients) at the final OS analysis [35].
In the relapsed disease setting in SOLO2, MDS/AML was reported in 2% of patients receiving maintenance olaparib (vs. 4% of placebo patients) at the primary PFS analysis [37] and in 8% of olaparib patients (vs. 4% of placebo patients) at the final OS analysis [46]. The imbalance in MDS/AML seen between olaparib and placebo patients with longer-term follow-up should be considered in the context of potential baseline risk factors (e.g. prior chemotherapy with DNA-damaging agents), the late onset of these events, and the survival benefit seen with olaparib in SOLO2 (patients with longer survival may have more time to develop late-onset toxicities).
During third-line or later treatment with olaparib in SOLO3, MDS/AML was reported in 2% of patients receiving olaparib treatment (vs. 4% of non-platinum chemotherapy patients) at the primary analysis [63].
In PRIMA, one case of MDS was reported in patients receiving first-line maintenance niraparib (0.3%; no cases of MDS/AML were reported in placebo patients) at the primary PFS analysis [17], and in 1.2% of niraparib patients (vs. 1.2% of placebo patients) with longer-term follow-up [27]. In the relapsed disease setting in the NOVA trial, the incidence of MDS/AML was 1% in maintenance niraparib patients and 1% in placebo patients at the primary PFS analysis [38] and 4% versus 2% at the final OS analysis [50].
At the primary PFS analysis in ATHENA-MONO, MDS/AML was reported in two patients in the rucaparib group (0.5%) and no patients in the placebo group [18]. In the relapsed disease setting in the ARIEL3 trial, the incidence of MDS/AML was 1% in patients receiving maintenance rucaparib, with no cases reported in the placebo group at the primary PFS analysis [39], and 4% versus 3% at the final OS analysis [54].
PARPi therapy should be discontinued in any patient in whom MDS/AML is confirmed [13,14,15], and the risk should be discussed with each patient prescribed a PARPi.
5.2 Non-Hematologic Adverse Events
Gastrointestinal AEs (e.g. nausea, vomiting, diarrhea, constipation, abdominal pain, dysgeusia, decreased appetite) are among the most common treatment-emergent AEs reported with PARPis in randomized, placebo-controlled maintenance therapy studies and randomized, controlled later-line treatment trials (Online Resource Table S2) [14, 17, 18, 22, 34, 38, 46, 59, 63, 83]. The vast majority of these AEs were mild to moderate.
Patients receiving rucaparib in ATHENA-MONO [18], ARIEL3 [83], and ARIEL4 [59] also experienced increased ALT/AST levels (Online Resource Table S2). Elevations in ALT/AST occurred within the first few weeks of rucaparib therapy and were reversible, rarely associated with increases in bilirubin, and not felt to be clinically meaningful [39, 82].
Gastrointestinal AEs are usually manageable with supportive therapy (e.g. antinausea/antiemetic therapy, antidiarrheal medication, laxatives, and dietary modification as appropriate) and/or dose modification [85,86,87].
Fatigue was commonly reported in patients receiving olaparib, niraparib, or rucaparib in randomized, placebo-controlled maintenance therapy studies and randomized, controlled later-line treatment trials (Online Resource Table S2) [14, 17, 18, 22, 34, 38, 46, 59, 63, 83]. Most fatigue events were mild to moderate. Fatigue/asthenia can usually be managed using supportive care (e.g. strategies to conserve energy) and dose modification [85,86,87].
Pneumonitis/interstitial lung disease (ILD) has rarely been reported with PARPis in clinical trials. Various confounding factors may contribute to development of pneumonitis (e.g. pneumonitis is a recognized adverse effect of many anticancer therapies [88]). Pneumonitis is included in the warnings and precautions section of the US prescribing information for olaparib, with 0.8% of 2901 olaparib patients reported as developing pneumonitis across various tumor types [13]. In placebo-controlled maintenance therapy studies, pneumonitis/ILD was reported in 2% of olaparib patients versus 0% of placebo patients in SOLO1 [16], pneumonitis/ILD/bronchiolitis was reported in 1% of olaparib plus bevacizumab patients versus 0% of placebo plus bevacizumab patients in PAOLA-1 [34], and pneumonitis was reported in 2% of olaparib patients versus 0% of placebo patients in SOLO2 [46]. Olaparib should be interrupted in patients presenting with new or worsening respiratory symptoms (e.g. dyspnea, cough, fever) [13], signs (e.g. hypoxia), or radiological abnormalities [13] and the source of the symptoms assessed [13]. Olaparib should be discontinued and appropriate treatment initiated if pneumonitis is confirmed. Pneumonitis has also been reported with niraparib during post-marketing experience [14, 88].
Hypertension is included in the warnings and precautions section of the US prescribing information for niraparib, as both hypertension and hypertensive crisis have been reported in patients receiving this PARPi [14]. In placebo-controlled maintenance therapy studies, hypertension was reported in 17% of niraparib patients versus 7% of placebo patients in PRIMA [17] and in 19% versus 4% of patients, in NOVA [38], with grade ≥3 hypertension reported in 6% versus 1% of patients, in PRIMA [17] and in 8% versus 2% of patients, in NOVA [38]. In patients receiving niraparib, blood pressure (BP) and heart rate should be monitored regularly and patients with cardiovascular disorders should be monitored closely [14]. Hypertension should be managed with antihypertensives and the niraparib dose should be adjusted, if necessary [14].
As mentioned previously, hypertension is commonly associated with bevacizumab. In PAOLA-1, the incidence of hypertension was numerically lower in patients receiving olaparib plus bevacizumab than in those receiving bevacizumab alone (46% vs. 60% of patients) [34]. In patients receiving a PARPi in combination with bevacizumab, BP should be monitored every 2‒3 weeks and appropriate antihypertensive therapy should be initiated in patients who develop hypertension. For severe hypertension, bevacizumab should be withheld until BP is controlled, and bevacizumab should be discontinued in patients who develop hypertensive crisis or hypertensive encephalopathy [3].
5.3 Dose Modifications and Discontinuations
AEs associated with PARPis can usually be managed with dose modifications, including dose interruption and dose reduction. The rates of dose modification and treatment discontinuation and the durations of treatment in key trials of PARPis approved in the US are shown in Table 6.
In patients receiving olaparib, most AEs were managed with dose interruptions or reductions, with ≤21% of patients requiring treatment discontinuation across trials (Table 6) [22, 34, 46, 63]. Rates of dose modification or treatment discontinuation in patients receiving olaparib were generally consistent across treatment settings; the higher rate of olaparib discontinuation in PAOLA-1 [31] versus SOLO1 [16] may partly reflect differences between the studies (e.g. combination therapy) and differences between the populations (patients with a BRCAm vs. all comers) [89]. In SOLO1, of the 162 patients still receiving olaparib at month 24, the majority (64%) were receiving the recommended starting dose of olaparib 300 mg twice daily without requiring a dose reduction, with 17% receiving a reduced olaparib dose of 250 mg twice daily [19].
In patients receiving niraparib, although most AEs were managed by niraparib dose modification, with ≤21% of patients requiring treatment discontinuation, a high proportion of patients required niraparib dose interruptions (≤80% of patients) or dose reductions (≤71%; Table 6) [17, 38, 69]. The rate of niraparib dose modification appeared higher in the first-line maintenance setting [17] than in the later-line maintenance [38] or treatment [69] settings, although comparisons across trials should be made with caution given the protocol amendment permitting use of an ISD in PRIMA [17].
AEs occurring in patients receiving rucaparib as maintenance or treatment were also usually managed with dose reductions or interruptions, and few (≤15%; Table 6) patients required treatment discontinuation of rucaparib [18, 59]. Rates of dose modification or treatment discontinuation in patients receiving rucaparib were generally consistent across treatment settings.
5.4 Impact on Health-Related Quality of Life
The PFS benefit seen with PARPi maintenance monotherapy and combination therapy in patients with newly diagnosed advanced OC was achieved with no detrimental effect on health-related quality of life (HRQoL) and was supported by patient-centered outcomes such as quality-adjusted PFS (QA-PFS) and time without significant symptoms of toxicity (TWiST) or quality-adjusted TWiST (Q-TWiST); these outcomes take into account the adverse effects of PARPis.
In SOLO1, there was no clinically meaningful difference in the mean change from baseline in the Functional Assessment of Cancer Therapy–Ovarian Cancer (FACT-O) Trial Outcome Index (TOI) score over 24 months between maintenance olaparib and placebo (Table 2) [16, 90]. In PRIMA, mean Functional Assessment of Cancer Therapy–Ovarian Symptom Index (FOSI), European Organization for the Research and Treatment of Cancer Quality of Life Questionnaire (EORTC-QLQ-C30), and EORTC-QLQ-ovarian cancer module (EORTC-QLQ-OV28) scores did not indicate a difference in HRQoL between maintenance niraparib and placebo [17, 91, 92]. In ATHENA-MONO, changes from baseline in the FACT-O TOI score were similar in the rucaparib and placebo groups [18]. In SOLO1 and PRIMA, PARPi maintenance monotherapy was associated with significant gains in QA-PFS [17, 90,91,92] and TWiST [90] or Q-TWiST [17, 91, 92] (Table 2). In PAOLA-1, no clinically meaningful difference in the global health status quality of life (GHS-QOL) score was observed between maintenance olaparib plus bevacizumab and placebo plus bevacizumab [31]; olaparib plus bevacizumab was associated with significant gains in TWiST over placebo plus bevacizumab [93] (Table 3).
Similarly, maintenance monotherapy had no detrimental effect on HRQoL in the relapsed disease setting. In SOLO2, the mean change from baseline in FACT-O TOI score did not significantly differ between maintenance olaparib and placebo [37, 94], in NOVA, the adjusted mean FOSI and 5-level EQ-5D (EQ-5D-5L) scores were generally similar between maintenance niraparib and placebo [38, 95], and in ARIEL3, there was no significant difference between maintenance rucaparib and placebo in the time to worsening in the FOSI-18 disease-related symptoms–physical (DRS-P) subscale score [39] (Table 4). Patient-centered benefits were seen in QA-PFS [94, 96] and TWiST [94, 97] or Q-TWIST [96] (Table 4).
Later-line olaparib treatment did not adversely affect HRQoL in SOLO3, with no clinically or statistically significant difference in the mean change from baseline in the FACT-O TOI score with olaparib treatment versus non-platinum chemotherapy [63] (Table 5).
6 Evidence From Real-World Studies
Real-world data support the use of PARPis in OC, although differences between olaparib, niraparib, and rucaparib are apparent in real-world settings.
In a US real-world evidence study, differences between olaparib, niraparib, and rucaparib were seen in the maintenance and treatment settings [98]. For example, the risk of a clinical event of interest was significantly higher with niraparib than with either olaparib (odds ratio [OR] 3.36; 95% CI 2.00–5.65) or rucaparib (OR 2.09; 95% CI 1.10–3.95); dose reductions were seen in significantly (P < 0.05) fewer olaparib patients (21%) than in rucaparib (30%) or niraparib (35%) patients; persistence and adherence were significantly (P < 0.05) higher with olaparib than with niraparib or rucaparib; and healthcare resource utilization was higher with niraparib and rucaparib than with olaparib [98].
In support of clinical trial findings, a PFS benefit was seen in newly diagnosed OC patients who did, compared with those who did not, receive PARPi maintenance therapy in a real-world setting [99]. Prolonging the time to disease progression also has the potential to delay the high costs associated with progression in newly diagnosed patients [100]. Despite this, utilization of PARPi maintenance therapy in the first-line setting is currently suboptimal; this is especially noticeable in those patients who will benefit the most, with PARPi maintenance therapy administered to 56% of patients with BRCAm tumors and 57% of patients whose tumors tested HRD positive [99].
The low utilization raises the question: if those patients whose tumors are found to have a sBRCAm or gBRCAm are not strongly encouraged to receive PARPi maintenance therapy, are they not being recommended the standard of care given the overwhelming evidence supports PARPi maintenance therapy in this population? If patients with BRCAm tumors are not encouraged to be treated with PARPi maintenance therapy, then the potential to markedly increase the curative intent is lost [22]. A similar question could be asked of those patients whose tumors are found to be HRD-positive.
Although utilization of PARPi maintenance therapy in the relapsed disease setting has improved over time, real-world data show that a proportion of eligible patients with OC are still not receiving PARPi maintenance therapy [101, 102].
Optimizing adherence to PARPi therapy is critical. Real-world data found that as many as one-quarter of OC patients may have suboptimal adherence to PARPi therapy, with non-adherent patients more likely to receive niraparib and have a longer duration of therapy [103]. Appropriate management of AEs such as nausea and vomiting is also key to maintaining adherence to the recommended PARPi dosage [86, 87]. Patient preference data indicate that patients with OC would be willing to accept a shorter PFS to avoid severe AEs, particularly nausea and vomiting [104].
Although rates of BRCAm and HRD testing in OC patients are improving [102], a proportion of patients are still not being tested [105]. Universal biomarker testing of all patients with newly diagnosed OC remains the goal [102].
7 Challenges and Future Directions
The optimal sequencing of therapies in OC (including the potential impact of PARPi therapy on the efficacy of subsequent platinum-based chemotherapy [106]), mechanism of PARPi resistance, and use of novel combination therapies remain areas of interest.
In terms of maintenance combination therapy in the newly diagnosed setting, the Phase III ATHENA-COMBO (GOG-3020/ENGOT-ov45) trial (NCT03522246) is comparing rucaparib plus nivolumab with rucaparib alone. A non-analytical arm (nivolumab alone) will be analyzed as an exploratory endpoint to assess the relative contribution of nivolumab alone [107].
A number of other studies are investigating PARPi (with or without anti-angiogenic agents) in combination with immune checkpoint inhibitors and novel targeted agents, including inhibitors of WEE-1, ATR, MEK, AKT, and mTORC1/2, in OC. Key Phase III trials currently underway investigating triplet therapy in patients with newly diagnosed advanced OC include: DUO-O/ENGOT-ov46/GOG-3025, evaluating combinations of platinum-based chemotherapy, bevacizumab, and the anti-programmed death-ligand 1 (PD-L1) antibody durvalumab, followed by maintenance bevacizumab, durvalumab, and olaparib in patients without BRCAm [108]; KEYLYNK-001/MK-7339-001/ENGOT-ov43/GOG-3036, evaluating the anti-programmed cell death protein 1 (PD-1) agent pembrolizumab combined with carboplatin/paclitaxel followed by maintenance olaparib in non-BRCAm patients (concurrent, maintenance bevacizumab is optional) [109]; and FIRST/ENGOT-ov44, evaluating the anti-PD-L1 agent dostarlimab in combination with first-line paclitaxel/carboplatin, followed by maintenance dostarlimab plus niraparib (concurrent, maintenance bevacizumab is optional) [110].
Although some patients exhibit primary resistance to PARPi, various acquired resistance mechanisms (e.g. BRCA reversion mutations, restoration of HRR function, replication fork stabilization, epigenetic changes) can lead to disease progression during PARPi therapy [111,112,113]. Combining PARPis with novel targeted agents (e.g. WEE-1 or ATR inhibitors) may help achieve PARPi resensitization in patients who develop PARPi resistance and progress during PARPi therapy [114,115,116]. The potential benefit and limitations of PARPi rechallenge were shown in OReO/ENGOT Ov-38 (NCT03106987), the first Phase IIIb study to provide data on PARPi maintenance rechallenge [117]. Patients in OReO had PSROC and were heavily pretreated; maintenance olaparib rechallenge provided a statistically significant improvement in PFS over placebo in patients with a BRCAm (HR 0.57; 95% CI 0.37–0.87; P = 0.022) or without a BRCAm (HR 0.43; 95% CI 0.26–0.71; P = 0.002) [117]. Although OReO selected patients who had previously demonstrated sensitivity to a PARPi, some olaparib patients most likely progressed during OReO because PARPi resistance had developed during their prior PARPi therapy. Currently, there are limited clinical data on rechallenging patients with a single-agent PARPi as maintenance therapy; rechallenge may be an option for some patients, such as those patients who have not progressed on a prior PARPi followed by response to platinum-based chemotherapy.
8 Conclusions
To our knowledge, this narrative review provides the most comprehensive and up-to-date review of PARPi as maintenance therapy and treatment in OC.
Strong evidence supports the use of PARPis in OC. Over time, the treatment landscape has shifted from use of PARPis in the relapsed disease setting to the first-line maintenance therapy setting. Indeed, data support the early introduction of PARPis as they appear to provide the most benefit in the newly diagnosed setting, rather than reserving their use for the relapsed setting. Our proposed treatment algorithms for both newly diagnosed and PSROC are shown in Figs. 2 and 3.

Adapted from DiSilvestro et al. Maintenance treatment of newly diagnosed advanced ovarian cancer: time for a paradigm shift? Cancers 2021;13(22):5756 [118] (https://doi.org/10.3390/cancers13225756), an open access article distributed under the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). *Germline BRCAm testing should be offered to all patients with epithelial ovarian cancer at diagnosis, with tumor testing for somatic BRCAm for patients in whom a germline BRCAm is not detected; †Olaparib and niraparib approved in the US; ‡Niraparib approved in the US; §Olaparib + bev approved in the US; ǁBev approved in the US. bev bevacizumab, BRCAm BRCA1 and/or BRCA2 mutation, CR complete response, HRD homologous recombination deficiency, IV intravenous, NACT neoadjuvant therapy, NED no evidence of disease, PARPi poly(ADP-ribose) polymerase inhibitor, PBC platinum-based chemotherapy, PDS primary debulking surgery, PR partial response, q3w every 3 weeks
Proposed treatment algorithm for newly diagnosed ovarian cancer.

Proposed treatment algorithm for platinum-sensitive relapsed ovarian cancer. *Olaparib, niraparib, and rucaparib approved in the US (niraparib approved in patients with a germline BRCAm and rucaparib approved in patients with a BRCAm); †Bev approved in the US. bev bevacizumab, BRCAm BRCA1 and/or BRCA2 mutation, IV intravenous, PARPi poly(ADP-ribose) polymerase inhibitor, PBC platinum-based chemotherapy, q3w every 3 weeks
Within the newly diagnosed setting, PARPis provide the greatest clinical benefit in patients with BRCAm or who test positive for HRD, meaning biomarker testing is critical to identify patients most likely to benefit from PARPi maintenance therapy and guide treatment decisions. For this reason, biomarker testing, including evaluation of HRD and genomic instability, should be conducted in all newly diagnosed OC patients.
It is becoming clear that curative intent is an achievable outcome in advanced OC. In the first-line setting, 7-year results from SOLO1 showed a clinically meaningful improvement in OS in patients with a BRCAm who received maintenance olaparib, and 5-year results from PAOLA-1 showed a clinically meaningful improvement in OS in patients whose tumors tested positive for HRD who received maintenance olaparib plus bevacizumab. Identifying which factors predict the patients who will experience long-term remission with PARPi maintenance therapy remains critical.
Although similarities are evident in the tolerability profiles of the different PARPis, distinct differences are also observed, requiring customization of monitoring recommendations and/or dosing regimens (e.g. use of a lower starting dose of niraparib in the first-line setting in patients with a lower bodyweight or baseline platelet count to help ameliorate thrombocytopenia). Importantly, new safety signals have not been observed with longer-term follow-up. The risk of MDS/AML with PARPis remained low in the newly diagnosed setting with longer-term follow-up. The risk of MDS/AML appears higher when PARPis are utilized in the recurrent setting, although multiple factors may potentially impact the imbalance in MDS/AML seen with PARPi usage versus placebo in the relapsed disease setting. Long-term follow-up of patients receiving PARPis for MDS/AML remains important.
Data from trials investigating novel combination strategies including PARPis are awaited with interest; the optimal sequencing of novel therapies in OC remains to be established.
References
National Comprehensive Cancer Network (NCCN). NCCN Clinical Practice Guidelines in Oncology: Ovarian cancer including fallopian tube cancer and primary peritoneal cancer. Version 1. 2022. Available at: https://www.nccn.org/professionals/physician_gls/pdf/ovarian.pdf. Accessed 2 Feb 2022.
Ledermann JA, Raja FA, Fotopoulou C, Gonzalez-Martin A, Colombo N, Sessa C, et al. Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24:24–32.
Genentech Inc. AVASTIN® (bevacizumab) injection, for intravenous use: prescribing information. 2022. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/125085s340lbl.pdf. Accessed 5 Apr 2023.
Burger RA, Brady MF, Bookman MA, Fleming GF, Monk BJ, Huang H, et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365(26):2473–83.
Tewari KS, Burger RA, Enserro D, Norquist BM, Swisher EM, Brady MF, et al. Final overall survival of a randomized trial of bevacizumab for primary treatment of ovarian cancer. J Clin Oncol. 2019;37(26):2317–28.
Perren TJ, Swart AM, Pfisterer J, Ledermann JA, Pujade-Lauraine E, Kristensen G, et al. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365(26):2484–96.
Oza AM, Cook AD, Pfisterer J, Embleton A, Ledermann JA, Pujade-Lauraine E, et al. Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): overall survival results of a phase 3 randomised trial. Lancet Oncol. 2015;16(8):928–36.
Aghajanian C, Blank SV, Goff BA, Judson PL, Teneriello MG, Husain A, et al. OCEANS: a randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol. 2012;30(17):2039–45.
Aghajanian C, Goff B, Nycum LR, Wang YV, Husain A, Blank SV. Final overall survival and safety analysis of OCEANS, a phase 3 trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent ovarian cancer. Gynecol Oncol. 2015;139(1):10–6.
Coleman RL, Brady MF, Herzog TJ, Sabbatini P, Armstrong DK, Walker JL, et al. Bevacizumab and paclitaxel-carboplatin chemotherapy and secondary cytoreduction in recurrent, platinum-sensitive ovarian cancer (NRG Oncology/Gynecologic Oncology Group study GOG-0213): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017;18(6):779–91.
Pujade-Lauraine E, Hilpert F, Weber B, Reuss A, Poveda A, Kristensen G, et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: The AURELIA open-label randomized phase III trial. J Clin Oncol. 2014;32(13):1302–8.
Colombo N, Sessa C, du Bois A, Ledermann J, McCluggage WG, McNeish I, et al. ESMO-ESGO consensus conference recommendations on ovarian cancer: pathology and molecular biology, early and advanced stages, borderline tumours and recurrent diseasedagger. Ann Oncol. 2019;30(5):672–705.
AstraZeneca. LYNPARZA® (olaparib) tablets, for oral use: prescribing information. 2022. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/208558s024lbl.pdf. Accessed 5 Apr 2023.
GlaxoSmithKline. ZEJULA® (niraparib) capsules, for oral use: prescribing information. 2022. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/208447s025lbl.pdf. Accessed 5 Apr 2023.
Clovis Oncology. RUBRACA® (rucaparib) tablets, for oral use: prescribing information. 2022. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/209115s013lbl.pdf. Accessed 5 Apr 2023.
Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2018;379(26):2495–505.
González-Martín A, Pothuri B, Vergote I, DePont CR, Graybill W, Mirza MR, et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2019;381(25):2391–402.
Monk BJ, Parkinson C, Lim MC, O’Malley DM, Oaknin A, Wilson MK, et al. A randomized, phase III trial to evaluate rucaparib monotherapy as maintenance treatment in patients with newly diagnosed ovarian cancer (ATHENA-MONO/GOG-3020/ENGOT-ov45). J Clin Oncol. 2022;40(34):3952–64.
Colombo N, Moore K, Scambia G, Oaknin A, Friedlander M, Lisyanskaya A, et al. Tolerability of maintenance olaparib in newly diagnosed patients with advanced ovarian cancer and a BRCA mutation in the randomized phase III SOLO1 trial. Gynecol Oncol. 2021;163(1):41–9.
Colombo N, Bradley W, Moore K, Gonzalez Martin A, Scambia G, Oaknin A, et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer and a BRCA mutation: subgroup analysis by risk in the phase III SOLO1 study. Int J Gynecol Cancer. 2020;30(Suppl 4):A76.
Banerjee S, Moore KN, Colombo N, Scambia G, Kim B-G, Oaknin A, et al. Maintenance olaparib for patients with newly diagnosed advanced ovarian cancer and a BRCA mutation (SOLO1/GOG 3004): 5-year follow-up of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2021;22(12):1721–31.
DiSilvestro P, Banerjee S, Colombo N, Scambia G, Kim BG, Oaknin A, et al. Overall survival with maintenance olaparib at 7 years of follow-up in patients with newly diagnosed advanced ovarian cancer and a BRCA mutation: the SOLO1/GOG 3004 trial. J Clin Oncol. 2023;41(3):609–17.
Braicu E, Pothuri B, Pérez-Fidalgo JA, et al. Efficacy of niraparib therapy in patients with newly diagnosed advanced ovarian cancer by brcawt status: prima/ENGOT-OV26/GOG-3012 study. Int J Gynecol Cancer. 2020;30(4):70–1.
Mirza MR, Gonzalez Martin A, Graybill W, O’Malley DM, Gaba L, Yap OWS, et al. Evaluation of an individualized starting-dose of niraparib in the PRIMA/ENGOT-OV26/GOG-3012 study. J Clin Oncol. 2020;38(15):6050.
Graybill W, Mirza M, González-Martin A, O’Malley D, Gaba L, Yap O, et al. Efficacy on individualized starting dose (ISD) and fixed starting dose (FSD) of niraparib per investigator-assessment (IA) in newly diagnosed advanced ovarian cancer (OC). Int J Gynecol Cancer. 2020;30(Suppl 3):10.
European Medicines Agency. Zejula: assessment report. 2020. Available at: https://www.ema.europa.eu/en/documents/variation-report/zejula-h-c-003943-ii-0019-epar-assessment-report-variation_en.pdf. Accessed 5 Apr 2023.
González MA. PRIMA/ENGOT-OV26/GOG-3012 study: updated long-term PFS and safety. Ann Oncol. 2022;33(7):S789.
Li N, Zhu J, Yin R, Wang J, Pan L, Kong B, et al. Efficacy and safety of niraparib as maintenance treatment in patients with newly diagnosed advanced ovarian cancer using an individualized starting dose (PRIME study): a randomized, double-blind, placebo-controlled, phase 3 trial. Gynecol Oncol. 2022;166(Suppl. 1):5.
Kristeleit R. Rucaparib maintenance treatment in patients (pts) with newly diagnosed ovarian cancer (OC): defining benefit according to disease risk subgroups within the phase 3 ATHENA–MONO study. Ann Oncol. 2022;33(Suppl):S786.
Banerjee S, Gonzalez-Martin A, Harter P, Lorusso D, Moore KN, Oaknin A, et al. First-line PARP inhibitors in ovarian cancer: summary of an ESMO Open—Cancer Horizons round-table discussion. ESMO Open. 2020;5(6): e001110.
Ray-Coquard I, Pautier P, Pignata S, Pérol D, González-Martín A, Berger R, et al. Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. N Engl J Med. 2019;381(25):2416–28.
Hardesty MM, Krivak TC, Wright GS, Hamilton E, Fleming EL, Belotte J, et al. OVARIO phase II trial of combination niraparib plus bevacizumab maintenance therapy in advanced ovarian cancer following first-line platinum-based chemotherapy with bevacizumab. Gynecol Oncol. 2022;166(2):219–29.
Harter P, Mouret-Reynier MA, Pignata S, Cropet C, Gonzalez-Martin A, Bogner G, et al. Efficacy of maintenance olaparib plus bevacizumab according to clinical risk in patients with newly diagnosed, advanced ovarian cancer in the phase III PAOLA-1/ENGOT-ov25 trial. Gynecol Oncol. 2022;164(2):254–64.
González Martín A, Desauw C, Heitz F, Cropet C, Gargiulo P, Berger R, et al. Maintenance olaparib plus bevacizumab in patients with newly diagnosed advanced high-grade ovarian cancer: main analysis of second progression-free survival in the phase III PAOLA-1/ENGOT-ov25 trial. Eur J Cancer. 2022;174:221–31.
Ray-Coquard I, Leary A, Pignata S, Cropet C, González-Martín A, Bogner G, et al. Final overall survival (OS) results from the Phase III PAOLA-1/ENGOT-ov25 trial evaluating maintenance olaparib (ola) plus bevacizumab (bev) in patients (pts) with newly diagnosed advanced ovarian cancer (AOC). Ann Oncol. 2022;33(Suppl):S1396.
Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366(15):1382–92.
Pujade-Lauraine E, Ledermann JA, Selle F, Gebski V, Penson RT, Oza AM, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(9):1274–84.
Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154–64.
Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10106):1949–61.
Poveda A, Lheureux S, Colombo N, Cibula D, Lindemann K, Weberpals J, et al. Olaparib maintenance monotherapy in platinum-sensitive relapsed ovarian cancer patients without a germline BRCA1/BRCA2 mutation: OPINION primary analysis. Gynecol Oncol. 2022;164(3):498–504.
Pignata S, Oza A, Hall G, Pardo B, Madry R, Cibula D, et al. ORZORA: maintenance olaparib in patients with platinum-sensitive relapsed ovarian cancer: outcomes by somatic and germline BRCA and other homologous recombination repair gene mutation status. Gynecol Oncol. 2021;162:S29.
Wu XH, Zhu JQ, Yin RT, Yang JX, Liu JH, Wang J, et al. Niraparib maintenance therapy in patients with platinum-sensitive recurrent ovarian cancer using an individualized starting dose (NORA): a randomized, double-blind, placebo-controlled phase III trial. Ann Oncol. 2021;32(4):512–21.
Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15(8):852–61.
Friedlander M, Matulonis U, Gourley C, du Bois A, Vergote I, Rustin G, et al. Long-term efficacy, tolerability and overall survival in patients with platinum-sensitive, recurrent high-grade serous ovarian cancer treated with maintenance olaparib capsules following response to chemotherapy. Br J Cancer. 2018;119(9):1075–85.
Matulonis UA, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in patients with platinum-sensitive, relapsed serous ovarian cancer and a BRCA mutation: overall survival adjusted for postprogression poly(adenosine diphosphate ribose) polymerase inhibitor therapy. Cancer. 2016;122(12):1844–52.
Poveda A, Floquet A, Ledermann JA, Asher R, Penson RT, Oza AM, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a final analysis of a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2021;22(5):620–31.
Poveda A, Lheureux S, Colombo N, Cibula D, Elstrand M, Weberpals J, et al. Maintenance olaparib monotherapy in patients (pts) with platinum-sensitive relapsed ovarian cancer (PSR OC) without a germline BRCA1/BRCA2 mutation (non-gBRCAm): final overall survival (OS) results from the OPINION trial. Ann Oncol. 2022;33(Suppl. 7):S790.
Pignata S, Oza AM, Hall G, Pardo B, Madry R, Cibula D, et al. Maintenance olaparib in patients (pts) with platinum-sensitive relapsed ovarian cancer (PSROC) by somatic (s) or germline (g) BRCA and other homologous recombination repair (HRR) gene mutation status: overall survival (OS) results from the ORZORA study. J Clin Oncol. 2022;40(Suppl 16):5519.
Mirza MR, Feng B, Shan M, Sun K, Yalcin I, Coleman K, et al. Elucidation of PARP inhibitor activity in BRCAwt recurrent ovarian cancer by hrr mutational gene profile analysis. J Clin Oncol. 2019;37(15):5568.
Matulonis U, Herrstedt J, Oza A, Mahner S, Redondo A, Berton D, et al. Long-term safety and secondary efficacy endpoints in the ENGOT-OV16/NOVA phase III trial of niraparib in recurrent ovarian cancer. Gynecol Oncol. 2021;162:S24–5.
GlaxoSmithKline. Important drug warning: Subject: Zejula (niraparib) important drug warning for the maintenance treatment in recurrent ovarian cancer (2L+). 2022. Available at: https://www.zejulahcp.com/content/dam/cf-pharma/hcp-zejulahcp-v2/en_US/pdf/ZEJULA%20(niraparib)%20Dear%20HCP%20Letter.pdf. Accessed 5 Apr 2023.
GlaxoSmithKline. Important prescribing information: Subject: Zejula (niraparib) Important Prescribing Information for the maintenance treatment of adult patients with non-gBRCAmut recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in a complete or partial response to platinum-based chemotherapy in second or later line setting. 2022. Available at: https://www.zejulahcp.com/content/dam/cf-pharma/hcp-zejulahcp-v2/en_US/pdf/ZEJULA%20(niraparib)%20Dear%20HCP%20Letter%20November%202022.pdf. Accessed 5 Apr 2023.
O’Malley DM, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, et al. Clinical and molecular characteristics of ARIEL3 patients who derived exceptional benefit from rucaparib maintenance treatment for high-grade ovarian carcinoma. Gynecol Oncol. 2022;167(3):404–13.
Coleman RL, Oza A, Lorusso D, Aghajanian C, Oaknin A, Dean A, et al., editors. Overall survival results from ARIEL3: a Phase 3 randomized, double-blind study of rucaparib vs. placebo following response to platinum-based chemotherapy for recurrent ovarian carcinoma. IGCS 2022 Annual Global Meeting; 29 September–1 October 2022; New York; abstract O003.
Drew Y, Ledermann J, Hall G, Rea D, Glasspool R, Highley M, et al. Phase 2 multicentre trial investigating intermittent and continuous dosing schedules of the poly(ADP-ribose) polymerase inhibitor rucaparib in germline BRCA mutation carriers with advanced ovarian and breast cancer. Br J Cancer. 2016;114(7):723–30.
Kristeleit R, Shapiro GI, Burris HA, Oza AM, LoRusso P, Patel MR, et al. A phase I-II study of the oral PARP inhibitor rucaparib in patients with germline BRCA1/2-mutated ovarian carcinoma or other solid tumors. Clin Cancer Res. 2017;23(15):4095–106.
Oza AM, Tinker AV, Oaknin A, Shapira-Frommer R, McNeish IA, Swisher EM, et al. Antitumor activity and safety of the PARP inhibitor rucaparib in patients with high-grade ovarian carcinoma and a germline or somatic BRCA1 or BRCA2 mutation: integrated analysis of data from Study 10 and ARIEL2. Gynecol Oncol. 2017;147(2):267–75.
Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18(1):75–87.
Kristeleit R, Lisyanskaya A, Fedenko A, Dvorkin M, de Melo AC, Shparyk Y, et al. Rucaparib versus standard-of-care chemotherapy in patients with relapsed ovarian cancer and a deleterious BRCA1 or BRCA2 mutation (ARIEL4): an international, open-label, randomised, phase 3 trial. Lancet Oncol. 2022;23(4):465–78.
Oza AM, Lisyanskaya AS, Fedenko AA, de Melo AC, Shparik Y, Bondarenko I, et al. Overall survival results from ARIEL4: a phase III study assessing rucaparib vs chemotherapy in patients with advanced, relapsed ovarian carcinoma and a deleterious BRCA1/2 mutation. Ann Oncol. 2022;33(Suppl. 7):S780.
Clovis Oncology. Important prescribing information: Subject: Rubraca (rucaparib) for treatment of BRCA-mutated ovarian cancer after 2 or more chemotherapies is voluntarily withdrawn in the U.S. 2022. Available at: https://clovisoncology.com/pdfs/US_DHCPL_final_signed.pdf. Accessed 5 Apr 2023.
Domchek SM, Aghajanian C, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, et al. Efficacy and safety of olaparib monotherapy in germline BRCA1/2 mutation carriers with advanced ovarian cancer and three or more lines of prior therapy. Gynecol Oncol. 2016;140(2):199–203.
Penson RT, Valencia RV, Cibula D, Colombo N, Leath CA 3rd, Bidzinski M, et al. Olaparib versus nonplatinum chemotherapy in patients with platinum-sensitive relapsed ovarian cancer and a germline BRCA1/2 mutation (SOLO3): a randomized phase III trial. J Clin Oncol. 2020;38(11):1164–74.
Penson RT, Valencia RV, Colombo N, Leath CA 3rd, Bidzinski M, Kim BG, et al. Final overall survival results from SOLO3: Phase III trial assessing olaparib monotherapy versus non-platinum chemotherapy in heavily pretreated patients with germline BRCA1 and/or BRCA2-mutated platinum-sensitive relapsed ovarian cancer. Gynecol Oncol. 2022;166(Suppl 1):S19-20.
Leath CA, Scambia G, Valencia RV, Colombo N, Cibula D, Bidzinski M, et al., editors. Overall survival by number of prior lines of chemotherapy in patients with BRCA-mutated platinum-sensitive relapsed ovarian cancer receiving olaparib treatment or non-platinum chemotherapy in SOLO3. IGCS 2022 Annual Global Meeting; 29 September-1 October 2022; New York; abstract.
AstraZeneca. Important prescribing information: Lynparza (Olaparib) for treatment of adult patients with deleterious or suspected deleterious germline BRCA-mutated (gBRCAm) advanced ovarian cancer who have been treated with three or more prior lines of chemotherapy is voluntarily withdrawn in the U.S. 2022. Available at: https://www.lynparzahcp.com/content/dam/physician-services/us/590-lynparza-hcp-branded/hcp-global/pdf/solo3-dhcp-final-signed.pdf. Accessed 5 Apr 2023.
Cadoo K, Simpkins F, Mathews C, Liu YL, Provencher D, McCormick C, et al. Olaparib treatment for platinum-sensitive relapsed ovarian cancer by BRCA mutation and homologous recombination deficiency status: Phase II LIGHT study primary analysis. Gynecol Oncol. 2022;166(3):P425–31.
Mathews CA, Simpkins F, Cadoo KA, Liu YL, Provencher DM, McCormick C, et al. Olaparib treatment (Tx) in patients (pts) with platinum-sensitive relapsed ovarian cancer (PSR OC) by BRCA mutation (BRCAm) and homologous recombination deficiency (HRD) status: overall survival (OS) results from the phase II LIGHT study. J Clin Oncol. 2021;39(15):5515.
Moore KN, Secord AA, Geller MA, Miller DS, Cloven N, Fleming GF, et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019;20(5):636–48.
GlaxoSmithKline. Subject: ZEJULA® (niraparib) for the treatment of adult patients with advanced ovarian, fallopian tube, or primary peritoneal cancer who have been treated with 3 or more prior chemotherapy regimens is voluntarily withdrawn in the U.S. 2022. Available at: https://medinfo.gsk.com/5f95dbd7-245e-4e65-9f36-1a99e28e5bba/57e2a3fa-7b9b-432f-a220-5976a509b534/57e2a3fa-7b9b-432f-a220-5976a509b534_viewable_rendition__v.pdf. Accessed 5 Apr 2023.
Miller RE, Leary A, Scott CL, Serra V, Lord CJ, Bowtell D, et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann Oncol. 2020;31(12):1606–22.
Christinat Y, Ho L, Clément S, Genestie C, Sehouli J, Cinieri S, et al. The Geneva HRD test: clinical validation on 469 samples from the PAOLA-1 trial. Virchows Arch. 2022;481(Suppl 1):S49.
Konstantinopoulos PA, Norquist B, Lacchetti C, Armstrong D, Grisham RN, Goodfellow PJ, et al. Germline and somatic tumor testing in epithelial ovarian cancer: ASCO guideline. J Clin Oncol. 2020;38(11):1222–45.
Pujol P, Barberis M, Beer P, Friedman E, Piulats JM, Capoluongo ED, et al. Clinical practice guidelines for BRCA1 and BRCA2 genetic testing. Eur J Cancer. 2021;146:30–47.
Callens C, Vaur D, Soubeyran I, Rouleau E, Just PA, Guillerm E, et al. Concordance between tumor and germline BRCA status in high-grade ovarian carcinoma patients in the Phase III PAOLA-1/ENGOT-ov25 trial. J Natl Cancer Inst. 2021;113(7):917–23.
Hodgson DR, Brown JS, Dearden SP, Lai Z, Elks CE, Milenkova T, et al. Concordance of BRCA mutation detection in tumor versus blood, and frequency of bi-allelic loss of BRCA in tumors from patients in the phase III SOLO2 trial. Gynecol Oncol. 2021;163(3):563–8.
Pujade-Lauraine E, Brown J, Barnicle A, Wessen J, Lao-Sirieix P, Criscione SW, et al. Homologous recombination repair gene mutations to predict olaparib plus bevacizumab efficacy in the first-line ovarian cancer PAOLA-1/ENGOT-ov25 Trial. JCO Precis Oncol. 2023;7: e2200258.
Swisher EM, Kwan TT, Oza AM, Tinker AV, Ray-Coquard I, Oaknin A, et al. Molecular and clinical determinants of response and resistance to rucaparib for recurrent ovarian cancer treatment in ARIEL2 (Parts 1 and 2). Nature Commun. 2021;12(1):2487.
Cadoo K, Simpkins F, Mathews C, Altman A, Gilbert L, Liu Y, et al. Olaparib treatment in patients with platinum-sensitive relapsed ovarian cancer by BRCA mutation and homologous recombination deficiency status: secondary safety results from the phase II LIGHT study. Gynecol Oncol. 2021;162:S67–8.
Mirza MR, Benigno B, Dorum A, Mahner S, Bessette P, Barcelo IB, et al. Long-term safety in patients with recurrent ovarian cancer treated with niraparib versus placebo: results from the phase III ENGOT-OV16/NOVA trial. Gynecol Oncol. 2020;159(2):442–8.
Kristeleit RS, Oza AM, Oaknin A, Aghajanian C, Tinker AV, Tredan O, et al. Integrated safety analysis of the poly (ADP-ribose) polymerase (PARP) inhibitor rucaparib in patients (pts) with ovarian cancer in the treatment and maintenance settings. Ann Oncol. 2019;30:409–10.
Kristeleit RS, Oaknin A, Ray-Coquard I, Leary A, Balmaña J, Drew Y, et al. Antitumor activity of the poly(ADP-ribose) polymerase inhibitor rucaparib as monotherapy in patients with platinum-sensitive, relapsed, BRCA-mutated, high-grade ovarian cancer, and an update on safety. Int J Gynecol Cancer. 2019;29(9):1396–404.
Ledermann JA, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, et al. Rucaparib for patients with platinum-sensitive, recurrent ovarian carcinoma (ARIEL3): post-progression outcomes and updated safety results from a randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21(5):710–22.
Berek JS, Matulonis UA, Peen U, Ghatage P, Mahner S, Redondo A, et al. Safety and dose modification for patients receiving niraparib. Ann Oncol. 2018;29(8):1784–92.
Tookman L, Krell J, Nkolobe B, Burley L, McNeish IA. Practical guidance for the management of side effects during rucaparib therapy in a multidisciplinary UK setting. Ther Adv Med Oncol. 2020;12:1758835920921980.
Moore KN, Monk BJ. Patient counseling and management of symptoms during olaparib therapy for recurrent ovarian cancer. Oncologist. 2016;21(8):954–63.
Friedlander M, Banerjee S, Mileshkin L, Scott C, Shannon C, Goh J. Practical guidance on the use of olaparib capsules as maintenance therapy for women with BRCA mutations and platinum-sensitive recurrent ovarian cancer. Asia Pac J Clin Oncol. 2016;12(4):323–31.
McLaren A, Cartwright D, Ross E, Roxburgh P. First-line PARP inhibitors—emerging side effects require caution: a case of PARPi-induced pneumonitis. J Immunother Precis Oncol. 2021;4:171–5.
Vergote I, Ray-Coquard I, Anderson DM, Cantuaria G, Colombo N, Garnier-Tixidre C, et al. Population-adjusted indirect treatment comparison of the SOLO1 and PAOLA-1/ENGOT-ov25 trials evaluating maintenance olaparib or bevacizumab or the combination of both in newly diagnosed, advanced BRCA-mutated ovarian cancer. Eur J Cancer. 2021;157:415–23.
Friedlander M, Moore KN, Colombo N, Scambia G, Kim BG, Oaknin A, et al. Patient-centred outcomes and effect of disease progression on health status in patients with newly diagnosed advanced ovarian cancer and a BRCA mutation receiving maintenance olaparib or placebo (SOLO1): a randomised, phase 3 trial. Lancet Oncol. 2021;22(5):632–42.
Mäenpää J, Pothuri B, Han S, Chase D, Bender D, Follana P, et al. 294 Patient-reported outcomes (PROS) in patients (PTS) receiving niraparib in the prima/engot-ov26/gog-3012 trial. Int J Gynecol Cancer. 2020;30(Suppl 4):A63–4.
Barretina-Ginesta MP, Monk BJ, Han S, Pothuri B, Auranen A, Chase D, et al. 738P Quality-adjusted time without symptom or toxicity (QA-TWiST) and quality-adjusted progression-free survival (QA-PFS) of first-line (1L) maintenance niraparib in patients with advanced ovarian cancer (OC): results from the PRIMA trial. Ann Oncol. 2021;32:S736–7.
Joly F, Chabaud S, Cropet C, Anota A, Demarchi M, Atasevan B, et al. Time without symptoms or toxicity (TWiST) in patients with newly diagnosed advanced ovarian cancer receiving maintenance olaparib or placebo plus bevacizumab: Analysis of PAOLA-1/ENGOT-ov25 phase III trial. J Clin Oncol. 2022;40(Suppl 16):5562.
Friedlander M, Gebski V, Gibbs E, Davies L, Bloomfield R, Hilpert F, et al. Health-related quality of life and patient-centred outcomes with olaparib maintenance after chemotherapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT Ov-21): a placebo-controlled, phase 3 randomised trial. Lancet Oncol. 2018;19(8):1126–34.
Oza AM, Matulonis UA, Malander S, Hudgens S, Sehouli J, Del Campo JM, et al. Quality of life in patients with recurrent ovarian cancer treated with niraparib versus placebo (ENGOT-OV16/NOVA): results from a double-blind, phase 3, randomised controlled trial. Lancet Oncol. 2018;19(8):1117–25.
Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, Colombo N, et al. Patient-centered outcomes in ARIEL3, a phase III, randomized, placebo-controlled trial of rucaparib maintenance treatment in patients with recurrent ovarian carcinoma. J Clin Oncol. 2020;38(30):3494–505.
Matulonis UA, Walder L, Nøttrup TJ, Bessette P, Mahner S, Gil-Martin M, et al. Niraparib maintenance treatment improves time without symptoms or toxicity (TWiST) versus routine surveillance in recurrent ovarian cancer: a TWiST analysis of the ENGOT-OV16/NOVA trial. J Clin Oncol. 2019;37(34):3183–91.
Arend RC, O’Malley DM, Banerjee S, McLaurin K, Davidson R, Long GH. Utilization of poly(ADP-Ribose) polymerase inhibitors in ovarian cancer: a retrospective cohort study of US healthcare claims data. Adv Ther. 2022;39(1):328–45.
Chan JK, Liu J, Song J, Xiang C, Wu EQ, Kalilani L, et al. Real-world trends of PARPi maintenance treatment uptake and progression-free survival (PFS) in patients (pts) with newly diagnosed advanced ovarian cancer (AOC) in the United States. J Clin Oncol. 2022;40(Suppl 16):6580.
Simmons D, Blank SV, ElNaggar AC, Chastek B, Bunner SH, McLaurin K. Health care resource utilization and costs associated with disease progression in ovarian cancer. Adv Ther. 2022;39(6):2544–61.
Garofalo D, Aydin E, Labrador M, Webster J, Brown G, Donaldson J, et al. Real-world data analysis of ovarian cancer (OC) maintenance utilization among maintenance eligible patients. J Clin Oncol. 2019;37(15):5579.
Moss H, Secord AA, Perhanidis J, Hawkes C. Real-world treatment patterns of maintenance therapy in platinum-sensitive recurrent epithelial ovarian cancer: are some patients missing out? J Clin Oncol. 2020;38(15): e18042.
Moss HA, Chen L, Hershman DL, Davidson B, Wright JD. Adherence to PARP inhibitor therapy among women with ovarian cancer. Gynecol Oncol. 2021;163(2):262–8.
Havrilesky LJ, Alvarez Secord A, Ehrisman JA, Berchuck A, Valea FA, Lee PS, et al. Patient preferences in advanced or recurrent ovarian cancer. Cancer. 2014;120(23):3651–9.
Moore K, Mirza MR, Gourley C, Pignata S, Ali T, Lechpammer S, et al. Evolution of the ovarian cancer treatment paradigm, including maintenance treatment, in the US and Europe: a real-world chart review analysis (2017–2020). Gynecol Oncol. 2022;166(1):26.
Frenel JS, Kim JW, Berton-Rigaud D, Asher R, Vidal L, Pautier P, et al. 813MO Efficacy of subsequent chemotherapy for patients with BRCA1/2 mutated platinum-sensitive recurrent epithelial ovarian cancer (EOC) progressing on olaparib vs placebo: the SOLO2/ENGOT Ov-21 trial. Ann Oncol. 2020;31:S615.
Monk BJ, Coleman RL, Fujiwara K, Wilson MK, Oza AM, Oaknin A, et al. ATHENA (GOG-3020/ENGOT-ov45): a randomized, phase III trial to evaluate rucaparib as monotherapy (ATHENA-MONO) and rucaparib in combination with nivolumab (ATHENA-COMBO) as maintenance treatment following frontline platinum-based chemotherapy in ovarian cancer. Int J Gynecol Cancer. 2021;31(12):1589–94.
Okamoto A, Kim J-W, Yin R, Trillsch F, Reuss A, Aghajanian C, et al. A randomized Phase III trial of durvalumab with chemotherapy and bevacizumab, followed by maintenance durvalumab, bevacizumab and olaparib in newly diagnosed advanced ovarian cancer (DUO-O): updated trial endpoint and inclusion of China cohort. Gynecol Oncol. 2022;166(Suppl. 1):329.
Vergote I, Sehouli J, Salutari V, Zola P, Madry R, Wenham RM, et al. ENGOT-OV43/KEYLYNK-001: a phase III, randomized, double-blind, active- and placebo-controlled study of pembrolizumab plus chemotherapy with olaparib maintenance for first-line treatment of BRCA-nonmutated advanced epithelial ovarian cancer. J Clin Oncol. 2019;37(15 Suppl):25.
Hardy-Bessard A-C, Moore KN, Mirza MR, Asselain B, Redondo A, Pfisterer J, et al. ENGOT-OV44/FIRST study: a randomized, double-blind, adaptive, phase III study of standard of care (SOC) platinum-based therapy ± dostarlimab followed by niraparib ± dostarlimab maintenance as first-line (1L) treatment of stage 3 or 4 ovarian cancer (OC). J Clin Oncol. 2020;38(15 Suppl):25.
Tobalina L, Armenia J, Irving E, O’Connor MJ, Forment JV. A meta-analysis of reversion mutations in BRCA genes identifies signatures of DNA end-joining repair mechanisms driving therapy resistance. Ann Oncol. 2021;32(1):103–12.
Pham MM, Hinchcliff E, Avila M, Westin SN. The Clinical challenges, trials, and errors of combatting poly(ADP-Ribose) polymerase inhibitors resistance. Cancer J. 2021;27(6):491–500.
Lin KK, Harrell MI, Oza AM, Oaknin A, Ray-Coquard I, Tinker AV, et al. BRCA reversion mutations in circulating tumor DNA predict primary and acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2019;9(2):210–9.
Mahdi H, Hafez N, Doroshow D, Sohal D, Keedy V, Do KT, et al. Ceralasertib-mediated ATR inhibition combined with olaparib in advanced cancers harboring DNA damage response and repair alterations (olaparib combinations). JCO Precis Oncol. 2021;5:439.
Kim H, Xu H, George E, Hallberg D, Kumar S, Jagannathan V, et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat Commun. 2020;11(1):3726.
Gomez MK, Illuzzi G, Colomer C, Churchman M, Hollis RL, O’Connor MJ, et al. Identifying and overcoming mechanisms of PARP inhibitor resistance in homologous recombination repair-deficient and repair-proficient high grade serous ovarian cancer cells. Cancers (Basel). 2020;12(6):1503.
Pujade-Lauraine E, Selle F, Scambia G, Asselain B, Marmé F, Lindemann K, et al. LBA33 Maintenance olaparib rechallenge in patients (pts) with ovarian carcinoma (OC) previously treated with a PARP inhibitor (PARPi): Phase IIIb OReO/ENGOT Ov-38 trial. Ann Oncol. 2021;32:1308.
DiSilvestro P, Colombo N, Harter P, Gonzalez-Martin A, Ray-Coquard I, Coleman RL. Maintenance treatment of newly diagnosed advanced ovarian cancer: time for a paradigm shift? Cancers (Basel). 2021;13(22):5756.
Monk BJ, Parkinson C, Lim MC, O’Malley DM, Oaknin A, Wilson MK, et al. ATHENA–MONO (GOG-3020/ENGOT-ov45): a randomized, double-blind, phase 3 trial evaluating rucaparib monotherapy versus placebo as maintenance treatment following response to first-line platinum-based chemotherapy in ovarian cancer. J Clin Oncol. 2022;40(Suppl 17):5500.
Matulonis UA, Monk BJ, Secord AA, Geller MA, Miller DS, Cloven NG, et al. Baseline platelet count and body weight as predictors of early dose modification in the quadra trial of niraparib monotherapy for the treatment of heavily pretreated (≥4th line), advanced, recurrent high-grade serous ovarian cancer. Gynecol Oncol. 2019;154:3.
Swisher EM, Kristeleit RS, Oza AM, Tinker AV, Ray-Coquard I, Oaknin A, et al. Characterization of patients with long-term responses to rucaparib treatment in recurrent ovarian cancer. Gynecol Oncol. 2021;163(3):490–7.
Acknowledgements
Medical writing assistance was provided by Gillian Keating MBChB, of Cence, funded by AstraZeneca and Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, US.
Funding
This review was funded by AstraZeneca and Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, US.
Conflict of Interest
David M. O’Malley: Personal fees and/or advisory board/consulting fees from AbbVie, AdaptImmune, Agenus Inc., Arquer Diagnostics, Arcus Biosciences Inc., AstraZeneca, Atossa Therapeutics, Boston Biomedical, Cardiff Oncology, Celcuity, Clovis Oncology, Corcept Therapeutics, Duality Bio, Eisai, Elevar, Exelixis, Genentech Inc., Genelux, GlaxoSmithKline, GOG Foundation, Hoffmann-La Roche Inc., ImmunoGen Inc., Imvax, InterVenn, INXMED, IOVANCE Biotherapeutics, Janssen, Jazz Pharmaceuticals, Laekna, Leap Therapeutics, Inc., Luzsana Biotechology, Merck & Co, Merck Sharp & Dohme Corp., Mersana Therapeutics Inc., Myriad, Novartis, NovoCure, OncoC4 Inc., Onconova, Regeneron Pharmaceuticals Inc., RepImmune, R Pharm, Roche Diagnostics, Seattle Genetics (SeaGen), Sorrento, Sutro Biopharma, Tarveda Therapeutics, Toray, Trillium, Umoja, Verastem Inc., VBL Therapeutics, Vincerx Pharma, Xencor and Zentalis and research funding to his institution from AbbVie, Advaxis, Agenus Inc., Alkermes, Aravive Inc., Arcus Biosciences Inc., AstraZeneca, BeiGene USA Inc., Boston Biomedical, Bristol Myers Squibb, Clovis Oncology, Deciphera Pharma, Eisai, EMD Serono Inc., Exelixis, Genentech Inc., Genmab, GlaxoSmithKline, GOG Foundation, Hoffmann-La Roche Inc., ImmunoGen Inc., Incyte Corporation, IOVANCE Biotherapeutics, Karyopharm, Leap Therapeutics Inc., Ludwig Institute for Cancer Research, Merck & Co, Merck Sharp & Dohme Corp., Mersana Therapeutics Inc., NCI, Novartis, NovoCure, NRG Oncology, OncoC4 Inc., OncoQuest Inc., Pfizer Inc., Precision Therapeutics Inc., Prelude Therapeutics, Regeneron Pharmaceuticals Inc., RTOG, Rubius Therapeutics, Seattle Genetics (SeaGen), Sutro Biopharma, SWOG, TESARO, Verastem Inc. Thomas C. Krivak: Consulting fees, speaking fees and research support from AstraZeneca and GlaxoSmithKline; consulting fees and speaking fees from Myriad and Seagen/Genmab; and speaking fees and research support from Merck. Nashwa Kabil: Employment and stock ownership with AstraZeneca. Jiefen Munley: Employment and stock ownership with AstraZeneca. Kathleen N. Moore: Contracts from Genentech/Roche, Lilly Pharmaceuticals and PTC Therapeutics, for ovarian cancer investigator-initiated trials; consulting fees from IMab; payment to her institution for educational content in gynecological cancers from Onc Live, Physician Education Resource (PER), PRIME Oncology and Research to Practice; payment to her institution for advisory boards for use of assets in gynecological cancers from Alkemeres, Aravive, Blueprint Pharmaceuticals, Eisai, Genentech/Roche, Immunogen, Mersana, Mereo and VBL Therapeutics; participation on a data safety monitoring board for Incyte; payments to her institution for being an Associate Director of GOG partners and committee chair for NRG Ovarian Cancer.
Author Contributions
DMOM, TCK, NK, JM, and KNM contributed to data acquisition, analysis, and interpretation and to writing, review, and/or revision of the manuscript.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Availability of Data and Material
No datasets were generated or analyzed during the current study.
Code Availability
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
O’Malley, D.M., Krivak, T.C., Kabil, N. et al. PARP Inhibitors in Ovarian Cancer: A Review. Targ Oncol 18, 471–503 (2023). https://doi.org/10.1007/s11523-023-00970-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-023-00970-w