
Abstract
Introduction
ABP 798 is being developed as a biosimilar to rituximab reference product (RP), a CD20-directed cytolytic antibody that is approved in the US and EU for the treatment of non-Hodgkin lymphoma (NHL).
Methods
This randomized, double-blind, comparative clinical study (JASMINE) evaluated the efficacy and safety of ABP 798 compared with rituximab RP. Adult, anti-CD20 treatment naïve patients diagnosed with grade 1, 2, or 3a follicular B-cell NHL expressing CD20 were randomized 1:1 to receive a 375 mg/m2 infusion of either ABP 798 or rituximab RP once weekly for 4 weeks and at weeks 12 and 20. Tumor assessments were performed at baseline and weeks 12 and 28. Primary endpoint was the risk difference (RD) of overall response rate (ORR) of complete response, unconfirmed complete response, or partial response by week 28 based on data from central, independent, and blinded assessments of disease.
Results
Of the 256 randomized patients, 254 were treated with ABP 798 (n = 128; 100%) or rituximab RP (n = 126; 98.4%); 96 (78.0%) patients in the ABP 798 group and 87 (70.2%) in the rituximab RP group had a best ORR by week 28. The point estimate of RD in ORR between ABP 798 and rituximab RP from the adjusted generalized linear model for stratification factors was 7.7%. Clinical equivalence was based on sequential testing of the one-sided 95% lower confidence limits and one-sided 95% upper confidence limits of RD in ORR (− 1.4% and 16.8%, respectively) which was within the prespecified non-inferiority margin (− 15%) and non-superiority margin (35.5%), respectively. Results of sensitivity analyses were consistent with the primary efficacy analysis. ABP 798 was also comparable to rituximab RP across additional secondary endpoints, further supporting the conclusion of similarity, and including: RD of ORR at week 12; trough serum concentrations; percent of patients with complete depletion of CD19+ cell count at day 8; safety; and immunogenicity.
Conclusions
These results support a conclusion of similar clinical efficacy between ABP 798 and rituximab RP in patients with follicular lymphoma.
NCT Number
NCT02747043; first posted April 21, 2016.
EudraCT Number
2013-005,542-11; submitted 14 October, 2014.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
ABP 798 is being developed as a biosimilar to rituximab reference product (RP). |
Similarity in efficacy and safety of ABP 798 compared with rituximab RP was evaluated in patients with CD20+ follicular B-cell non-Hodgkin lymphoma in a comparative clinical study. |
Clinical equivalence was confirmed based on similarity of efficacy, safety, and other secondary endpoints. |
1 Introduction
ABP 798Footnote 1 is being developed as a biosimilar to rituximab reference product (RP) (licensed as Rituxan® in the United States [US] and MabThera® in the European Union [EU] and Australia). Rituximab RP binds specifically to the antigen cluster of differentiation 20 (CD20) expressed on B-cells (also known as B lymphocytes) and mediates B-cell depletion. Rituximab RP is approved for the treatment of non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis (Wegener’s granulomatosis), microscopic polyangiitis, and pemphigus vulgaris [1, 2]. A biosimilar is a biological drug that is highly similar to an approved, branded biological RP [3, 4].
For the development of biosimilar ABP 798, a stepwise approach was used, according to the Food and Drug Administration (FDA) and European Medicines Agency (EMA) guidance and program-specific scientific advice, starting with the similarity assessment of analytical (structural, functional, and physiochemical) characteristics, including critical quality attributes, which are product characteristics that can impact safety, potency, and pharmacokinetics (PK), as well as assessments of non-clinical and functional similarity related specifically to the mechanisms of action [5,6,7,8,9,10]. ABP 798 is a genetically engineered chimeric mouse/human immunoglobulin (Ig) isotype class G subclass 1 monoclonal antibody with an amino acid sequence identical to rituximab RP. ABP 798 has the same formulation and pharmaceutical form as intravenous (IV) rituximab RP. Comprehensive analytical and functional characterization has demonstrated that ABP 798 is similar to rituximab RP in terms of structure, purity, and potency as well as chemotherapeutic synergy, primary and secondary mechanisms of action including the mediation of B-cell lysis both in vitro and in vivo, and toxicokinetics [11, 12].
The final steps in the development of a biosimilar include comparisons of PK, safety, immunogenicity, and efficacy between the biosimilar and the RP in human clinical trials. The PK/pharmacodynamic (PD) similarity of ABP 798 with rituximab RP has been demonstrated in a three-arm study in patients with rheumatoid arthritis [13]. The current study (JASMINE) was designed to demonstrate the clinical similarity between ABP 798 and rituximab RP in terms of efficacy, PK, PD, safety, and immunogenicity in patients with previously untreated CD20-positive (CD20+), low tumor-burden follicular lymphoma, as an appropriately sensitive population for the demonstration of clinical similarity. The dose of 375 mg/m2 administered as an IV infusion was chosen because it is the therapeutic dose of the RP for the NHL indication [1]. The weekly 375 mg/m2 dose given for 4 weeks and followed by a maintenance schedule at 2-monthly intervals is a treatment option for patients with asymptomatic, advanced-stage, low tumor-burden follicular lymphoma [14].
2 Methods
2.1 Study Design
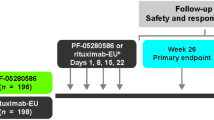
This was a randomized, double-blind, active-controlled, two-arm study in adult patients with grade 1, 2, or 3a follicular B-cell NHL and low tumor burden (asymptomatic). The study schema is provided in Fig. 1. Patients received a 375 mg/m2 IV infusion of either ABP 798 or rituximab RP once weekly (QW) for 4 weeks, followed by dosing at weeks 12 and 20, and remained in the study until week 28. Tumor assessments were performed at screening (i.e., baseline) and weeks 12 and 28.

Study design in patients with NHL grade 1–3a follicular lymphoma stage II–IV CD20 +. IV intravenous, NHL non-Hodgkin lymphoma, RP reference product. ↓ indicates IV infusion. *Post-treatment tumor assessments
An interactive voice/web response system (IXRS) was used to randomize the subjects centrally to receive either ABP 798 or rituximab in a 1:1 manner. Randomization data were kept strictly confidential, filed securely by the sponsor (or designee), and were accessible only to authorized persons as per the sponsor’s (or designee’s) standard operating procedures (SOPs) until the time of unblinding. Because the investigational product (IP) containers are different for ABP 798 and rituximab RP, IP (ABP 798 or rituximab RP) was prepared by an unblinded pharmacist, or designee, to make it into a common IV preparation for administration to the patient. Patients, sponsor, contract research organization (CRO) designees, and other clinical site staff were blinded to the IP allocation for each patient. This study was conducted in accordance with International Conference on Harmonisation (ICH) Good Clinical Practice (GCP) regulations/guidelines. All participants provided written informed consent prior to entering the study and before initiation of any study-related procedure (including administration of IP). Patients who discontinued IP before week 20 were followed for 8 weeks after the last dose of IP and then completed the end-of-study (EOS) visit.
2.2 Study Population
Eligible patients were adults ≥ 18 years of age with histologically confirmed grade 1, 2, or 3a follicular B-cell lymphoma expressing CD20 within 12 months before randomization. Disease was classified as stage II, III, or IV based on Cotswold’s modification of the Ann Arbor Staging System, with measurable disease and low tumor burden (based on Groupe d'Etudes des Lymphomes Folliculaires criteria and an Eastern Cooperative Oncology Group performance status score of 0 or 1) [15, 16]. Patients were excluded from participation if they had a history or known presence of central nervous system metastases or if they had used either commercially available or investigational chemotherapy, biological therapy, or immunological therapy for NHL (including rituximab RP or biosimilar rituximab, or other anti-CD20 treatments).
2.3 Investigational and Reference Products
ABP 798 was manufactured and packaged by Amgen, Inc. (Thousand Oaks, CA) and was supplied as a sterile, preservative-free liquid concentrate for IV infusion at a concentration of 10 mg/mL in either 100 mg/10 mL or 500 mg/50 mL single-dose vials. Rituximab RP (Rituxan®, Roche Genentech) was procured from commercial supplies. Patients received either ABP 798 or rituximab RP at a dose of 375 mg/m2 administered as an IV infusion QW for 4 weeks, followed by dosing at weeks 12 and 20. Concomitant medications were given before each infusion according to the local practice for administration of rituximab RP therapy. The following were prohibited at any time during the study: any non-study anti-cancer treatment (other than palliative radiotherapy to non-index lesions), commercial rituximab (other than as specified in this study design), any experimental (biological or non-biological) therapy (within or outside a clinical study), and live and attenuated vaccinations.
2.4 Efficacy Assessments
Disease was assessed by the investigator according to the International Working Group (IWG) response criteria for NHL at baseline and at weeks 12 and 28 [17]. Assessment included both a physical examination and a radiographic examination by computed tomography (CT) scan. Copies of all scheduled and unscheduled screening and on-study CT scans performed to monitor or diagnose NHL were submitted for a central, blinded, independent radiological review (Perceptive Informatics, LLC). Clinical disease assessments, including physical examinations, were performed by the investigator or sub-investigator and were submitted to the central imaging vendor, if applicable. Responses were categorized according to RECIST V.1.1 as complete response (CR), unconfirmed CR (CRu), partial response (PR), stable disease, relapsed disease, and progressive disease, and a response category of not evaluable was provided for situations in which there was inadequate information to otherwise categorize response status [17]. Overall response rate (ORR) was defined as the percentage of patients with a CR, CRu, or PR, and patients without post-baseline disease assessments were counted as non-responders in calculating the ORR.
2.5 Pharmacokinetic Assessments
Serum samples for PK analysis were collected at baseline; predose at weeks 2, 3, 4, 12, and 20; immediately after the end of infusion at week 12; and at week 28/EOS. Patients who agreed to optional additional PK sampling also had PK samples collected at 2 h (± 1 h) post-dose at weeks 1, 4, and 5.
2.6 Pharmacodynamic Assessments
CD19 + cell count, IgG, and IgM levels for PD analyses were collected at baseline and at weeks 2, 3, 4, and 28. Complete depletion of CD19 + cell count at any post-dose time was defined as CD19 + cell counts < 20 cell/μL (0.02 × 109 cell/L). Patients with missing CD19 + cell count at baseline or patients with CD19 + cell count < 20 cell/μL at baseline were excluded from the derivation of complete depletion of CD19 + cell count.
2.7 Safety Assessments
Safety endpoints were summarized descriptively. The adverse events of interest (AEOIs) prespecified for this study were infusion reactions including hypersensitivity, cardiac disorders, serious infections, progressive multifocal leukoencephalopathy, hematological reactions, hepatitis B reactivation, opportunistic infections, severe mucocutaneous reactions, tumor lysis syndrome, gastrointestinal perforation, and reversible posterior leukoencephalopathy syndrome. The AEOIs were retrieved utilizing Standardized Medical Dictionary for Regulatory Activities (MedDRA) Queries (SMQs), MedDRA system organ classes (SOCs) or a customized search strategy as best applicable to the AEOI medical concept.
2.8 Antidrug Antibody Assessments
Blood samples for antidrug antibody (ADA) assessments were collected at baseline/week 1, week 12, 20, and 28/EOS. Samples testing positive for binding antibodies were also tested for neutralizing antibodies. If patients tested positive for neutralizing antibodies to ABP 798 or rituximab RP at the final scheduled study visit, they were asked to return for additional follow-up testing. The number and percentages of patients who developed binding ADAs and those who developed neutralizing ADAs were tabulated separately.
2.9 Statistical Methods
The primary endpoint was risk difference (RD) of ORR from the central, independent, blinded tumor assessments by week 28. Secondary endpoints included RD of ORR at week 12, PK, PD, safety, and immunogenicity. A sample size of 250 patients was chosen to provide approximately 95% power at a 0.05 significance level to demonstrate equivalence between ABP 798 and rituximab RP on the primary efficacy endpoint (RD of ORR by week 28) assuming an ORR of 85% for both groups and a non-inferiority margin of − 15% and a non-superiority margin of + 35.5% [18]. The primary analysis was based on the modified full analysis set, which included all randomized patients with evidence of disease at baseline (ABP 798, n = 123 and rituximab RP, n = 124). Clinical equivalence of the primary endpoint was first to be demonstrated by comparing the one-sided 95% lower confidence limit of the RD of ORR by week 28 for ABP 798 and rituximab RP, with a non-inferiority margin of − 15%. If this was successful, the one-sided 95% upper confidence limit of the RD of ORR by week 28 was compared with the non-superiority margin of + 35.5%. The point estimate and confidence limits of the RD of ORR were estimated using a generalized linear model adjusted for the stratification factors (geographic region and age group). To assess the robustness of the primary ORR results, sensitivity analyses were performed using the central, independent, blinded assessments on the per-protocol (PP) analysis set (n = 120 for each ABP 798 and rituximab RP) according to actual treatment received and using the investigator’s assessment of disease on the full analysis set. The PP analysis set included subjects who completed all four weekly doses of IP or who permanently discontinued IP prior to completing four weekly doses due to reasons allowed per protocol (i.e., disease progression, adverse events [AEs], and death), who had at least one post-baseline tumor assessment prior to the EOS from the central, independent, blinded assessments, and who did not experience a protocol deviation that would affect their evaluation for the primary objective of the study.
Serum ABP 798 and rituximab RP concentrations were summarized by treatment for each sampling visit using the safety analysis set. The area under the curve (AUC) from week 4 to week 12 was calculated for the subset of patients with results at all optional PK visits within the analysis visit windows and results at predose week 4 and week 12, regardless of whether a partial or full dose was received at the dose prior, and the geometric least squares means of the AUC for each treatment and corresponding ratio and 90% confidence interval (CI) were calculated. All PD analyses were performed using the full analysis set. All safety analyses were performed on the safety analysis set.
3 Results
3.1 Enrollment and Disposition of Patients
A total of 380 patients were screened, and 256 patients were randomized (128 in the ABP 798 group and 128 in the rituximab RP group) at 91 centers across 20 countries. All 256 randomized patients were included in the full analysis set. Demographic and baseline physical characteristics were generally comparable between ABP 798 and rituximab RP (see Table 1). Overall, 50.8% of patients were female, 79.3% were white, and the median (range) age was 58.5 (24–84) years. Baseline disease characteristics were also generally comparable between ABP 798 and rituximab RP groups (see Table 1). Overall, the median (range) interval from diagnosis was 2.3 (0.3–142.6) months. Over half of the patients had histological grade 2 disease at screening (147 [57.4%]), and most had no bone marrow involvement (181 [70.7%]). Disease stage at screening was stage IV in 74 patients (28.9%) (ABP 798: 34 [26.6%]; rituximab RP: 40 [31.3%]), stage III in 119 patients (46.6%) (ABP 798: 60 [46.9%]; rituximab RP: 59 [46.1%]), stage II in 63 patients (24.7%) (ABP 798: 34 [26.6%]; rituximab RP: 29 [22.7%]), and stage I in no patients (0.0%). Median (range) number of nodal sites was 2.0 (1–6). Follicular Lymphoma International Prognostic Index (FLIPI) risk group classification was low in 113 patients (44.1%), intermediate in 104 patients (40.6%), and high in 39 patients (15.2%). Six patients (2.3%) had received previous local radiation treatment at a mean time of 18.22 (range of 7.0–37.3) months before study start. There were no patients who had had prior chemotherapy, biological therapy, or immunological therapy.
A summary of the study and IP disposition for patients in the full analysis set is provided in Fig. 2. Of the 256 randomized patients, 254 (99.2%) were treated (ABP 798: 128 patients; rituximab RP: 126 patients). Overall, 242 patients (94.5%) (ABP 798: 119 [93.0%]; rituximab RP: 123 [96.1%]) completed treatment and 14 patients (5.5%) (ABP 798: nine [7.0%]; rituximab RP: five [3.9%]) discontinued treatment. For the ABP 798 group, the most common reason for discontinuing IP was disease progression (four patients [3.1%]), followed by AE (three patients [2.3%]) and physician decision and other reasons (one patient each [0.8%]). In the rituximab RP group, the reasons for discontinuing IP were AE, patient request, patient dissatisfaction with treatment efficacy, physician decision, and protocol violation (n = 1 each).

Study disposition. RP reference product
3.2 Primary Efficacy Endpoint
Results of the analysis of the primary efficacy endpoint of RD of ORR by week 28, based on data from the central, independent, blinded assessments of disease for the modified full analysis set (ABP 798, n = 123 and rituximab RP, n = 124), are provided in Table 2. Overall, 96 patients (78.0%) in the ABP 798 group and 87 patients (70.2%) in the rituximab RP group had a best ORR by week 28; the point estimate of RD of ORR between treatment groups (ABP 798 − rituximab RP) from the generalized linear model adjusting for stratification factors was 7.7%. The one-sided 95% lower confidence limit of RD of ORR by week 28 was − 1.4% and was within the prespecified non-inferiority margin of − 15%. Subsequently, the one-sided 95% upper confidence limit was determined to be 16.8% and was within the pre-specified non-superiority margin of 35.5%. The results of the sequential testing thus established equivalence in clinical efficacy between ABP 798 and rituximab RP. Additionally, the two-sided 95% CI of RD of ORR by week 28 (− 3.2% to 18.6%) was also consistent with similarity of clinical efficacy (see Fig. 3). The results of the sensitivity analyses (central, independent, blinded assessment of disease on the PP analysis set; investigator’s assessment of disease on the full analysis set; and impact of baseline covariates using the central, independent, blinded assessment of disease on the modified full analysis set) support the conclusion of primary efficacy analysis (data not shown). Results of the analysis of the primary efficacy endpoint of ORR by week 28 by subgroup of geographic region showed that, in general, for subgroups with larger sample sizes, results were consistent with results from the primary analysis.

Primary efficacy endpoint of RD of ORR by week 28, based on data from the central, independent, blinded assessments of disease for the modified full analysis set. CI confidence interval, ORR overall response rate, RD risk difference. *The CI limits correspond to the one-sided 95% CI lower limit and the one-sided 95% CI upper limit
3.3 Secondary Efficacy Endpoint: Difference of Overall Response Rate at Week 12
Results of the analysis of the secondary efficacy endpoint of RD of ORR at week 12 support a conclusion of comparable efficacy between the treatment groups. Overall, 73 patients (59.3%) in the ABP 798 group and 72 patients (58.1%) in the rituximab RP group had best overall response at week 12; the point estimate of RD of ORR between treatment groups (ABP 798 − rituximab RP) from the generalized linear model adjusting for stratification factors was 0.9%. At week 12, the one-sided 95% lower confidence limit of RD of ORR was − 9.3%, and the one-sided 95% upper confidence limit of RD of ORR at week 12 was 11.2%. Additionally, the two-sided 95% CI of RD of ORR at week 12 was − 11.3% to 13.2%, and was also consistent with similarity of clinical efficacy.
3.4 Pharmacodynamics
The percentage of patients with complete depletion of CD19 + cell count at day 8 was similar between the treatment groups (113/115 patients [98.3%] in the ABP 798 group and 114/116 patients [98.3%] in the rituximab RP group), indicating that the extent of B-cell depletion in patients with NHL was similar between ABP 798 and rituximab RP. In general, only minor changes from baseline were noted in IgG and IgM levels for both treatment groups. Mean baseline IgG (SD) was 9.740 (2.5988) g/L and 10.570 g/L (2.5970) for ABP 798 and rituximab RP, respectively. At week 4, the mean change from baseline (SD) was − 0.018 (1.2459) and − 0.407 (1.4065) for ABP 798 and rituximab RP, respectively. Mean baseline IgM (SD) was 1.021 (1.0072) g/L and 1.247 g/L (3.4018) for ABP 798 and rituximab RP, respectively. At week 4, the mean change from baseline (SD) was − 0.033 (0.3158) and 0.137 (1.9442) for ABP 798 and rituximab RP, respectively.
3.5 Pharmacokinetics
The resulting mean serum concentrations in the safety analysis set at predose (trough) at each of the planned PK visits (except baseline and EOS) and immediately after the end of infusion at week 12 support similarity of ABP 798 and rituximab RP and were consistent with similarity of ABP 798 relative to rituximab RP. Mean (± SD) serum ABP 798 and rituximab RP concentration–time profiles from week 4 through week 12 for the patients who agreed to the optional additional PK sampling are presented in Fig. 4. The geometric mean ratio (GMR) (90% CI) for the AUC from week 4 to week 12 was 0.9240 (0.7958–1.0729).

Serum concentrations over time by treatment (week 4–12) in the subset of patients who agreed to the optional PK testing. Note: This graph represents PK results from the 45 patients treated with ABP 798 and the 41 patients treated with rituximab RP who agreed to the optional PK sampling. The sample sizes for each visit (ABP 798 vs rituximab) were as follows: week 4 predose (44 vs 39), week 4 post-dose (36 vs 32), week 5 (38 vs 37), and week 12 predose (44 vs 41). PK pharmacokinetic, RP reference product
3.6 Antidrug Antibodies
A summary of ADA results is provided in Table 3. All patients in the safety analysis set (100%) had at least one on-study ADA result. One patient (0.8%) in the ABP 798 group and three patients (2.4%) in the rituximab RP group tested positive for pre-existing binding ADAs at baseline, but no patients tested positive for pre-existing neutralizing ADAs at baseline. Post-baseline, from day 1 through week 28/EOS, three patients (2.4%) in the ABP 798 group and one patient (0.8%) in the rituximab RP group tested positive for the development of binding ADAs; the results were transient (i.e., negative results at the patient’s last time point tested within the study period) for two patients (1.6%) and zero patients (0.0%), respectively. The incidence of developing ADAs was consistent across the treatment groups over the entire study and consistent with similarity of ABP 798 relative to rituximab RP.
3.7 Safety
Of the 256 randomized patients, 254 (99.2%) were treated with IP (128 patients [100.0%] in the ABP 798 group and 126 [98.4%] in the rituximab RP group) and were included in the safety analysis set. The number of doses, the cumulative dose of IP received, and the total IP exposure duration were similar between the treatment groups across both time periods summarized. In general, exposure to IP was similar between the treatment groups, thereby facilitating comparisons of safety and immunogenicity across groups. Overall, 202 patients (79.5%) reported at least one AE (107 patients [83.6%] in the ABP 798 group and 95 patients [75.4%] in the rituximab RP group).
An overall summary of AEs, including treatment-emergent events in ≥ 5% of patients as well as AEOIs, is provided in Table 4. Overall, 63 patients (49.2%) in the ABP 798 treatment group and 57 patients (45.2%) in the rituximab RP treatment group experienced an AEOI. The RD for experiencing any AEOI was 4.0 (95% CI − 8.3 to 16.3) for the difference between the ABP 798 and rituximab RP treatment groups. Overall survival was defined as the time from randomization to the date of death. All patients (100.0%) in the safety analysis set remained alive at the EOS.
At least one grade ≥ 3 AE was reported in 27 patients (10.6%) (14 patients [10.9%] in the ABP 798 group and 13 patients [10.3%] in the rituximab RP group). Grade ≥ 3 AEs by preferred term occurring in more than one patient overall were neutropenia (three patients [2.3%] in the ABP 798 treatment group and three patients [2.4%] in the rituximab RP group), pyrexia (two [1.6%] and zero patients [0.0%], respectively), hypertension (one [0.8%] and two patients [1.6%], respectively), urticaria (one [0.8%] and two patients [1.6%], respectively), and abdominal pain (one [0.8%] and one patient [0.8%], respectively).
The subject incidence of serious adverse events (SAEs) was balanced between the treatment groups (five patients [3.9%] in the ABP 798 group and five patients [4.0%] in the rituximab RP group). In the ABP 798 group, SAEs were most frequently reported in the SOCs of infections and infestations, and injury, poisoning, and procedural complications (two patients [1.6%] each in the ABP 798 group and no patient [0.0%] each in the rituximab RP group); in the rituximab RP group, SAEs were most frequently reported in the SOC of gastrointestinal disorders (one patient [0.8%] in the ABP 798 group and three patients [2.4%] in the rituximab RP group). Overall, the severities of all AEs observed were within the grades of severity expected with rituximab RP, and no new safety signals were identified.
Discontinuation of IP or study due to one or more AEs occurred in four patients (3.1%) in the ABP 798 group and one patient (0.8%) in the rituximab RP group. Several AEs leading to discontinuation of IP or study were infusion-related reactions (i.e., anaphylactic reaction, dyspnea, erythema, and stomatitis), which are expected events for rituximab RP [19]. There were no AEs leading to discontinuation of IP or discontinuation of the study that occurred in more than a single patient. Only one SAE, stomatitis in one patient (0.8%) in the ABP 798 group, resulted in IP or study discontinuation.
4 Discussion
This comparative clinical trial was designed to establish the similarity of efficacy and safety of ABP 798 compared to rituximab RP in patients with CD20-positive B-cell follicular lymphoma. A population of follicular lymphoma patients with low tumor burden was selected for this study to evaluate the use of ABP 798 as monotherapy in a homogeneous patient population. Patients with advanced-stage, low tumor-burden follicular lymphoma conventionally undergo watchful waiting until disease progression. Rituximab 375 mg/m2 weekly for 4 weeks followed by a maintenance schedule at 2-monthly intervals up to 2 years is a treatment option in this patient population [14]. This ABP 798 comparative trial used two maintenance cycles only as was required for biosimilar development; however, patients could continue maintenance treatment using commercially available rituximab RP according to standard of care.
Equivalence in clinical efficacy was established by the primary endpoint analysis of RD of ORR by week 28 with central independent, blinded assessment. The one-sided 95% lower confidence limit of RD of ORR was − 1.4% and was contained within the prespecified non-inferiority margin of − 15%. The one-sided 95% upper confidence limit of RD of ORR was 16.8% and was contained within the prespecified non-superiority margin of 35.5%. This demonstration of both non-inferiority and non-superiority is supportive of equivalence in the clinical efficacy of ABP 798 relative to rituximab RP. The results of sensitivity analyses that were performed to ensure that the results are robust with different imputations were consistent with the results from the primary efficacy analysis, thereby supporting the conclusion of efficacy similarity.
The IWG response criteria for NHL described in Cheson et al. were selected for use in this study, to align with the assessment method used in the phase 3 rituximab RP study upon which this study design was based and upon which the margin for demonstrating clinical equivalence was calculated [14, 17, 18]. The sample size of 250 patients was chosen to provide approximately 95% power at a 0.05 significance level to demonstrate equivalence between ABP 798 and the RP on the primary efficacy endpoint (RD of ORR by week 28), assuming an ORR of 85% for both groups and a non-inferiority margin of − 15% and a non-superiority margin of + 35.5%. The statistically derived lower margin (− 35.5%) following FDA non-inferiority guidance preserves 50% of the rituximab RP effect from the 80% lower CI of the difference in ORR of rituximab RP monotherapy induction plus maintenance compared with watchful waiting as reported in a study in patients with asymptomatic stage 2, 3, or 4 follicular lymphomas [18]. A more conservative lower margin (− 15%) preserving 79% of the rituximab RP treatment effect from the 80% lower CI was selected to rule out that ABP 798 is inferior to rituximab RP; the upper margin (+ 35.5%) was chosen to be symmetric to the statistically derived lower margin to demonstrate that ABP 798 is non-superior to rituximab RP. Clinical equivalence of the primary efficacy endpoint was first demonstrated by comparing the one-sided 95% lower confidence limit of the RD of ORR by week 28 between ABP 798 and the RP with a non-inferiority margin of − 15%. Since this was successful, the one-sided 95% upper confidence limit of the RD of ORR by week 28 was then compared with a non-superiority margin of + 35.5%. The point estimate and confidence limits of the RD of ORR by week 28, which were estimated using a generalized linear model adjusted for the stratification factors (geographic region and age group), were within the prespecified margin.
Equivalence in clinical efficacy of ABP 798 with respect to rituximab RP in patients with CD20-positive B-cell NHL was further supported by the results of efficacy assessment at week 12, with a two-sided 90% CI (− 9.3% to 11.2%). In addition to similarity of efficacy by week 28, this similarity of efficacy at week 12 was also determined based on RD of ORR for the central, independent, blinded assessment of disease using the modified full analysis set and the investigator’s assessment of disease using the full analysis set.
Similarity of clinical PK was supported by trough serum concentrations and, in a subset of patients who agreed to the optional PK sampling, AUC values that were similar across ABP 798 and rituximab RP groups. Similarity of PD between ABP 798 and rituximab RP was demonstrated, with a similar percentage of patients with complete depletion of CD19 + cell count at day 8, indicating similarity in the extent of B-cell depletion. The levels of IgG and IgM levels were similar for treatment groups, with only minor changes from baseline noted. Similarity in clinical safety and immunogenicity was established by demonstrating that the incidence, type, and severity of AEs and the subject incidence of developing ADAs were similar between treatment groups, with no clinically meaningful differences, and were consistent with the safety profile of rituximab RP. These additional measures beyond the primary efficacy endpoint all support a conclusion of clinical similarity of ABP 798 relative to rituximab RP.
NHL is the most common hematological malignancy and comprises a diverse group of malignancies, most of B-cell origin [20]. Follicular lymphoma, a subtype of B-cell NHL characterized as indolent in nature with a slowly progressive natural history [21], is the second most common lymphoma diagnosed in the US and Western Europe. An overwhelming majority of patients have advanced-stage disease at diagnosis. Patients with asymptomatic follicular lymphoma do not require immediate treatment unless they have symptomatic nodal disease, compromised end-organ function B symptoms, symptomatic extranodal disease, or cytopenias [22]. Rituximab RP monotherapy presents an opportunity to significantly delay disease progression and death, with limited side effects, and has changed the paradigm for treating follicular lymphoma, both as monotherapy and in combination with chemotherapy (usually in the symptomatic setting) [23], resulting in an overall increase in survival of patients with follicular lymphoma [24, 25].
An additional two biosimilars of rituximab RP are available in the US for the treatment of NHL—Truxima® and Ruxience® [26, 27]. Similar to that of ABP 798, the development of other biosimilars to rituximab RP also began with non-clinical and preclinical investigations and clinical trials in rheumatoid arthritis prior to the comparative clinical trials to demonstrate similarity with rituximab RP in follicular lymphoma. CT-P10 (rituximab-abbs; Truxima® [Teva Pharmaceuticals USA, Inc, North Wales, PA]) was evaluated in a comparative clinical study to assess non-inferior efficacy and PK equivalence of CT-P10 compared with rituximab when used in combination with cyclophosphamide, vincristine, and prednisone in patients with newly diagnosed, advanced-staged (Ann Arbor stage III–IV) follicular lymphoma. The primary efficacy endpoint was the proportion of patients who had an overall response over eight cycles and was assessed in the efficacy population (all randomized patients). CT-P10 exhibited non-inferior efficacy and PK equivalence to rituximab RP, and the safety profile of CT-P10 was comparable to that of rituximab RP [28]. GP2013 (rituximab-pvvr; Ruxience® [Pfizer Inc., NY, NY USA]) was evaluated in a comparative clinical study of efficacy, safety, tolerability, PK, and PD of GP2013 in combination with cyclophosphamide, vincristine, and prednisone compared with rituximab in the same combination dosing paradigm in patients with previously untreated, advanced-stage (Ann Arbor stage III or IV) follicular lymphoma. The primary endpoint of comparability in overall response to eight cycles of GP2013 in the combination phase was met [29]. Though not currently approved in the US, HLX01 (Shanghai Henlius Biotech Inc, Shanghai, China) has been evaluated in a comparative clinical study to assess the efficacy, safety, and immunogenicity of HLX01 plus cyclophosphamide, doxorubicin, vincristine, and prednisone compared with rituximab RP in the same combination dosing paradigm in patients with histologically confirmed CD20 + diffuse large B-cell lymphoma (Ann Arbor stages I–IV) every 21 days for up to six cycles. The primary efficacy endpoint of comparability of best ORR within six cycles of treatment was met [30].
While rituximab RP has changed the paradigm for treating follicular lymphoma, the cost of treatment may represent a barrier to patient access. Approval of biosimilars offers the hope of increased access and improved patient outcomes. Comparative clinical studies, including the current ABP 798 study, have demonstrated similarity of efficacy of proposed biosimilars of rituximab RP with respect to the RP in follicular lymphoma. Overall, the results of this study support a conclusion of similarity in efficacy and safety/tolerability with regard to ABP 798 and rituximab RP in patients with CD20 + follicular B-cell NHL.
Change history
12 November 2020
Page 608, 4 Discussion, right column, second paragraph.
Notes
At the time of publication, ABP 798 has not been approved by the FDA or another relevant regulatory agency and the indications are yet undetermined. Please consult ABP 798′s later approved label in the relevant country for information regarding the approved uses for ABP 798 in your country.
References
Rituxan® (rituximab) prescribing information. Genentech Inc.; 2019.
MabThera. Summary of product characteristics. Grenzach-Wyhlen, Germany: Roche Pharma AG; 2019.
US Food and Drug Administration. Guidance for industry—questions and answers on biosimilar development and the BPCI Act (revision 1). 2018. https://www.fda.gov/media/119258/download. Accessed May 11, 2020.
European Medicines Agency. Guideline on similar biological medicinal products. 2014. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-rev1_en.pdf. Accessed 11 May 2020.
U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research. Guidance for industry. Scientific considerations in demonstrating biosimilarity to a reference product. 2015. https://www.fda.gov/media/82647/download. Accessed May 11, 2020.
US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research. Guidance for industry. Quality considerations in demonstrating biosimilarity of a therapeutic protein product to a reference product. 2015. https://www.fda.gov/media/135612/download. Accessed May 11, 2020.
US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research. Guidance for industry. Clinical pharmacology data to support a demonstration of biosimilarity to a reference product. Guidance for industry. 2016. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM397017.pdf. Accessed May 11, 2020.
European Medicines Agency, Committee for Medicinal Products for Human Use. Guideline on similar biological medicinal products containing monoclonal antibodies—non-clinical and clinical issues. 2012. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-monoclonal-antibodies-non-clinical_en.pdf. Accessed 11 May 2020.
European Medicines Agency. Committee for Medicinal Products for Human Use. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues (revision 1). 2014.https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-0.pdf. Accessed May 11, 2020.
European Medicines Agency. Committee for Medicinal Products for Human Use. Guideline on similar biological medicinal products (revision 1). 2014. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-rev1_en.pdf. Accessed May 11, 2020.
Seo N, Huang A, Kuhns S, Sweet H, Cao S, Wikström M, Liu J. Analytical and functional similarity of biosimilar ABP 798 with rituximab reference product. Biologicals. 2020. https://doi.org/10.1016/j.biologicals.2020.08.002.
McBride H, Maher G, Sweet H, Foltz I, Kuhns S. Functional similarity of proposed biosimilar ABP 798 with rituximab. Blood. 2017;130(1):5001.
Burmester G, Chien D, Chow V, Gessner M, Pan J, Cohen S. A randomized, double-blind study comparing pharmacokinetics and pharmacodynamics of proposed biosimilar ABP 798 with rituximab reference product in subjects with moderate to severe rheumatoid arthritis. Clin Pharmacol Drug Dev. 2020. https://doi.org/10.1002/cpdd.845.
Ardeshna KM, Qian W, Smith P, Braganca N, Lowry L, Patrick P, et al. Rituximab versus a watch-and-wait approach in patients with advanced-stage, asymptomatic, non-bulky follicular lymphoma: an open-label randomised phase 3 trial. Lancet Oncol. 2014;15:424–35.
Armitage JO. Staging non-Hodgkin lymphoma. CA Cancer J Clin. 2005;55:368–76.
Solal-Céligny P, Lepage E, Brousse N, Tendler CL, Brice P, Haïoun C, et al. Doxorubicin-containing regimen with or without interferon alfa-2b for advanced follicular lymphomas: final analysis of survival and toxicity in the Groupe d'Etude des lymphomes folliculaires 86 trial. J Clin Oncol. 1998;16(7):2332–8.
Cheson BD, Horning SJ, Coiffier B, Shipp MA, Fisher RI, Connors JM, et al. Report of an international workshop to standardize response criteria for non-Hodgkins lymphomas. NCI Sponsored International Working Group. J Clin Oncol. 1999;17(4):1244.
Ardeshna K, Qian W, Smith P, Warden J, Stevens L, Pocock CFE, et al. An intergroup randomised trial of rituximab versus a watch and wait strategy in patients with stage II, III, IV, asymptomatic, non-bulky follicular lymphoma (grades 1, 2 and 3a). A preliminary analysis. Blood. 2010;116:6.
Plosker GL, Figgitt DP. Rituximab: a review of its use in non-Hodgkin's lymphoma and chronic lymphocytic leukaemia. Drugs. 2003;63(8):803–43.
American Cancer Society. Cancer Facts and Figures 2020. Atlanta: American Cancer Society; 2020. Available at https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2020/cancer-facts-and-figures-2020.pdf. Accessed August 17, 2020.
Pileri SA, Zinzani PL, Went P, Pileri A Jr, Bendandi M. Indolent lymphoma: the pathologist's viewpoint. Ann Oncol. 2004;15(1):12–8.
Ardeshna KM, Smith P, Norton A, Hancock BW, Hoskin PJ, MacLennan KA, et al. Long-term effect of a watch and wait policy versus immediate systemic treatment for asymptomatic advanced-stage non-Hodgkin lymphoma: a randomised controlled trial. Lancet. 2003;362(9383):516–22. https://doi.org/10.1016/s0140-6736(03)14110-4.
Herold M, Haas A, Srock S, Neser S, Al-Ali KH, Neubauer A, et al. Rituximab added to first-line mitoxantrone, chlorambucil, and prednisolone chemotherapy followed by interferon maintenance prolongs survival in patients with advanced follicular lymphoma: an East German Study Group Hematology and Oncology Study. J Clin Oncol. 2007;25(15):1986–92. https://doi.org/10.1200/JCO.2006.06.4618.
Chen Q, Ayer T, Nastoupil LJ, Seward M, Zhang H, Sinha R, et al. Initial management strategies for follicular lymphoma. Int J Hematol Oncol. 2012;1(1):35–45. https://doi.org/10.2217/ijh.12.7.
Freedman A. Follicular lymphoma: 2018 update on diagnosis and management. Am J Hematol. 2018;93(2):296–305. https://doi.org/10.1002/ajh.24937.
Truxima® (rituximab-abbs) prescribing information. CELLTRION, Inc.; 2020.
Ruxience® (rituximab-pvvr) prescribing information. Pfizer, Inc.; 2020.
Kim WS, Buske C, Ogura M, Jurczak W, Sancho JM, Zhavrid E, et al. Efficacy, pharmacokinetics, and safety of the biosimilar CT-P10 compared with rituximab in patients with previously untreated advanced-stage follicular lymphoma: a randomised, double-blind, parallel-group, non-inferiority phase 3 trial. Lancet Haematol. 2017;4(8):e362–e373373. https://doi.org/10.1016/S2352-3026(17)30120-5.
Jurczak W, Moreira I, Kanakasetty GB, Munhoz E, Echeveste MA, Giri P, et al. Rituximab biosimilar and reference rituximab in patients with previously untreated advanced follicular lymphoma (ASSIST-FL): primary results from a confirmatory phase 3, double-blind, randomised, controlled study. Lancet Haematol. 2017;4(8):e350–e361361. https://doi.org/10.1016/S2352-3026(17)30106-0.
Shi Y, Song Y, Qin Y, Zhang Q, Han X, Hong X, et al. A phase 3 study of rituximab biosimilar HLX01 in patients with diffuse large B-cell lymphoma. J Hematol Oncol. 2020;13(1):38. https://doi.org/10.1186/s13045-020-00871-9.
Acknowledgements
Medical writing support was provided by Sonya Lehto, PhD, Amgen Inc., under the guidance of Monica Ramchandani, PhD, Amgen Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was supported by Amgen Inc., Thousand Oaks, CA, USA. Open Access funding provided by Projekt DEAL.
Conflicts of Interest
Dietger Niederwieser reports personal fees from Cellectis, Daiichi, and Novartis. David Chien, Vladimir Hanes, and Mindy Mo are employees and stockholders of Amgen Inc. Patrick Cobb, Vincent Delwail, Cecily Forsyth, Roman Hajek, Caroline Hamm, and Alessandra Tucci have nothing to disclose.
Ethics Approval
This study was conducted in accordance with International Conference on Harmonisation (ICH) Good Clinical Practice (GCP) regulations/guidelines.
Consent to Participate
All subjects provided written informed consent prior to entering the study and before initiation of any study-related procedure (including administration of investigational product).
Availability of Data and Material
There is a plan to share data. This may include de-identified individual patient data for variables necessary to address the specific research question in an approved data sharing request, and also related data dictionaries, study protocol, statistical analysis plan, informed consent form, and/or clinical study report. Data sharing requests relating to data in this manuscript will be considered after the publication date and (1) this product and indication (or other new use) have been granted marketing authorization in both the US and Europe or (2) clinical development discontinues and the data will not be submitted to regulatory authorities. There is no end date for eligibility to submit a data sharing request for these data. Qualified researchers may submit a request containing the research objectives, the Amgen product(s), and Amgen study/studies in scope, endpoints/outcomes of interest, statistical analysis plan, data requirements, publication plan, and qualifications of the researcher(s). In general, Amgen does not grant external requests for individual patient data for the purpose of re-evaluating safety and efficacy issues already addressed in the product labeling. A committee of internal advisors reviews requests. If not approved, a Data Sharing Independent Review Panel may arbitrate and make the final decision. Requests that pose a potential conflict of interest or an actual or potential competitive risk may be declined at Amgen’s sole discretion and without further arbitration. Upon approval, information necessary to address the research question will be provided under the terms of a data sharing agreement. This may include anonymized individual patient data and/or available supporting documents, containing fragments of analysis code where provided in analysis specifications. Further details are available at https://www.amgen.com/datasharing.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Niederwieser, D., Hamm, C., Cobb, P. et al. Efficacy and Safety of ABP 798: Results from the JASMINE Trial in Patients with Follicular Lymphoma in Comparison with Rituximab Reference Product. Targ Oncol 15, 599–611 (2020). https://doi.org/10.1007/s11523-020-00748-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-020-00748-4