
Abstract
The possible application of BRAF-targeted therapy in brain tumors is growing continuously. We have analyzed clinical strategies that address BRAF activation in primary brain tumors and verified current recommendations regarding screening for BRAF mutations. There is preliminary evidence for a range of positive responses in certain brain tumor types harboring the BRAF V600E mutation. National Comprehensive Cancer Network Guidelines for central nervous system cancers recommend screening for the BRAF V600E mutation in pilocytic astrocytoma, pleomorphic xanthoastrocytoma, and ganglioglioma. We suggest additional testing in glioblastomas WHO grade IV below the age of 30 years, especially those with epithelioid features, papillary craniopharyngiomas, and pediatric low-grade astrocytomas. BRAF-targeted therapy should be limited to the setting of a clinical trial. If the patient harboring a V600E mutation does not qualify for a trial, multimodality treatment is recommended. Dual inhibition of both RAF and MEK is expected to provide more potent and durable effects than anti-BRAF monotherapy. First-generation RAF inhibitors should be avoided. Gain-of-function mutations of EGFR and KIAA fusions may compromise BRAF-targeted therapy. BRAF alterations that result in MAPK pathway activation are common events in several types of brain tumors. BRAF V600E mutation emerges as a promising molecular target. The proposed algorithm was designed to help oncologists to provide the best therapeutic options for brain tumor patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Patients with certain brain tumors require screening for the BRAF V600E mutation. |
BRAF V600E-mutant tumors need to be considered in the context with other genetic alterations (e.g., coexisting gain-of-function mutation of EGFR or KIAA1549-BRAF fusion). |
Dual inhibition of both RAF and MEK is expected to provide more potent and durable effects than anti-BRAF monotherapy. |
BRAF-targeted therapy in brain tumors should be limited to the setting of a clinical trial. |
1 Introduction
Primary brain tumors remain the leading cause of mortality from malignant neoplasms in children and young adults. Glioblastoma (GBM), the most common brain tumor, is characterized by a median survival of < 21 months, despite surgical resection, radiation therapy, high-dose chemotherapy, and alternative approaches such as Tumor Treating Fields (TTFields) [1,2,3,4]. Within the central nervous system (CNS), immune cells follow different principles. The blood–brain barrier (BBB) not only restricts the movement of soluble mediators and leukocytes from the periphery [5], but also prevents the brain uptake of most neurotherapeutics [6]. Finally, brain neoplasms are exceptionally heterogeneous, further hindering the development of successful treatment modalities [7].
The mitogen-activated protein kinase (MAPK) is an essential signaling pathway in a number of malignancies. Alterations in various components of the MAPK pathway, especially the BRAF gene, have been thoroughly described in melanoma, colorectal cancer, thyroid cancer, non-small-cell lung cancer (NSCLC), and hairy cell leukemia [8,9,10,11]. BRAF encodes the B-Raf kinase that activates MAPK signaling through phosphorylation of MAPK kinase (MEK) and subsequently MAPK. Activating mutations of BRAF lead to constitutive downstream activation of RAF-MEK-MAPK signaling cascade, promoting cell proliferation and survival while inhibiting apoptosis, and eventually driving tumor growth [12, 13].
Currently available combinations of RAF and MEK inhibitors approved by the FDA include vemurafenib/cobimetinib, dabrafenib/trametinib, and encorafenib/binimetinib in melanoma and dabrafenib/trametinib in NSCLC [14,15,16,17]. Given the remarkable responses seen in these patients, BRAF-targeted approaches have attracted significant attention in the field of neuro-oncology.
The possible application of BRAF-targeted therapy in CNS tumors grows continuously. While clinical trials are still ongoing, there is preliminary evidence for a range of positive responses in certain brain tumor types harboring BRAF V600E mutation. Herein, we propose a management algorithm for brain tumor patients who could benefit from BRAF-targeted therapy.
2 Relevance of V600E Among BRAF Mutations
In vivo xenograft studies confirm the previously described role of BRAF in MAPK signaling regulation within CNS tumors [18, 19]. To date, over 30 BRAF alterations have been associated with human cancers. They have been grouped into three classes according to their kinase activity, rat sarcoma protein (RAS) dependency, and dimerization status. Although they all lead to MAPK activation, only class I mutations are sensitive to currently available BRAF inhibitors.
Class I mutations are independent of both upstream RAS activation and the need for dimerization. This class is represented by four V600 subtypes (V600E, V600D, V600K, and V600R). V600E is a single nucleotide mutation at codon 600 resulting in substitution of glutamic acid (E) for valine (V). The glutamate residue interacts with glycine-rich loop that ordinarily suppresses the activity of BRAF. The loss of this inhibitory effect results in an increase in BRAF basal activity and contributes to oncogenesis [8, 20].
Compared with V600E, the remaining class I mutations occur far less frequently and, therefore, their clinical relevance is harder to assess [21]. V600E is present in a significant subset of CNS tumors (Table 1). Its highest incidence is observed in papillary craniopharyngioma and pleomorphic xanthoastrocytoma (PXA), in 95% and up to 78% of cases, respectively [26, 50]. It also frequently occurs in pilocytic astrocytoma (PA), ganglioglioma (GG), and pediatric low-grade astrocytoma. While this mutation is rare in GBM in general, it is relatively common among young adults and when diagnosed as an epithelioid type [34].
Class II mutations include several point mutations and fusions that activate MEK through RAS-independent dimerization. The most common point mutations include K601E/N/T, L597Q/V, and G469A/V/R; however, their relative frequency in brain tumors remains unknown [21]. Among fusions, the most common is KIAA1549-BRAF, which is predominantly identified in low-grade gliomas.
Class III mutations are kinase impaired and enhance MAPK signaling through RAS and subsequent CRAF activation. This class is represented by G466E, D594G, and G596D; however, their incidence in brain tumors remains unclear [21, 54].
3 Strategies for the Management of BRAF-Mutant Brain Tumors
Table 1 summarizes the outcomes gathered from experiences with BRAF-targeted therapy in primary brain tumors; a PubMed database search for valid records was conducted until April 3, 2020; keywords used were BRAF mutation, V600, V600E, CNS tumors, brain tumors, MAPK, ERK, MEK1/2, RAF, glioma, glioblastoma, ependymoma, medulloblastoma, oligodendroglioma, xanthoastrocytoma, ganglioglioma, craniopharyngioma, spindle cell oncocytoma, and astrocytoma. The search results were narrowed by selecting studies in humans published in English. Twenty-six studies mentioning BRAF-targeted therapy in primary brain tumors were included [23,24,25, 27,28,29,30, 32, 33, 35,36,37,38,39, 41,42,43,44,45,46,47,48,49, 51,52,53]. The PubMed database was chosen as it remains the most widely used resource of medical journals and indexes only peer-reviewed literature [55]. Experience with BRAF-targeting therapies in brain tumors is generally limited to case reports and series that are likely biased toward unusually positive responses. In the majority of cases, the targeted therapy was initiated after progression on standard therapy. The assessment of its efficacy is further complicated by concurrent treatments including TTF, chloroquine, or vinblastine. Also, there is mixed use of second-generation RAF inhibitors (vemurafenib, dabrafenib) and MEK inhibitors (trametinib, cobimetinib). In cerebral metastatic melanoma, the response rates are better for dabrafenib (31%) compared with vemurafenib (16%) [56,57,58,59]. This difference is probably due to smaller size and molecular structure of dabrafenib that enables better penetration of the BBB [60]. In primary brain tumors, however, there are no clinical trials that support any combination of the aforementioned agents. Authors generally agree that first-generation RAF inhibitors, such as sorafenib, have poor blood–brain penetrance and efficient response is only to be expected in tumors with significant BBB breakdown [61]. In fact, sorafenib was tested in pediatric patients with low-grade gliomas and contributed to unexpected and unprecedented acceleration of tumor growth, irrespective of the BRAF status [62].
Finally, tumor heterogeneity often affects responses in ways not yet fully understood. Whether BRAF mutations are present in all or in only a subset of tumor cells seems fundamental for the selection of appropriate treatment.
3.1 BRAF Inhibition May Be Insufficient
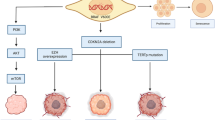
While MAPK signaling is essential in glioma formation, BRAF activation alone is not sufficient for the development of high-grade tumors [63]. In fact, following the positive response to V600E-targeted monotherapy, patients often succumb to disease progression [25, 29, 30, 33]. The resistance mechanisms have been described in detail in other malignancies, especially in melanoma [64]. Their common denominator is the maintenance of MAPK upregulation through alternative activators, either upstream or downstream of mutant BRAF (Fig. 1) [65,66,67].

Mechanisms of resistance to BRAF-targeted therapy. V600E and KIAA1549-BRAF fusion lead to constitutive activation of MAPK signaling in tumor cells. MAPK phosphorylates downstream nuclear effectors that ultimately enhance cell survival and proliferation. One recognized mechanism of resistance to BRAF inhibition is the upregulation of CRAF, which activates MAPK through MEK. Concurrent inhibition of MEK overcomes this resistance. Both RAF and MEK inhibition, as well as EGFR gain-of-function mutation contribute to amplification of the PI3K/AKT/mTOR pathway that represents another mechanism of resistance to BRAF-targeted therapy. EGF epidermal growth factor, EGFR epidermal growth factor receptor, MAPK mitogen-activated protein kinase, MEK MAPK kinase, mTOR mammalian target of rapamycin, PI3K phosphoinositide 3-kinase, RAS rat sarcoma protein
MEK inhibitors effectively prevent reactivation of MAPK signaling in various BRAF-mutated malignancies [14,15,16,17]. Hence, dual BRAF and MEK inhibition is associated with both prolonged clinical endpoints and improved side effect profiles compared with the BRAF inhibition alone. To date, multiple combinations of BRAF and MEK inhibitors have been approved by the FDA for the treatment of patients with melanoma and NSCLC [14,15,16,17]. In primary brain tumors, adequate brain distribution of both compounds is essential. While BRAF inhibitors show some level of BBB penetrance in melanoma patients with intracranial metastasis, brain distribution of MEK inhibitors may be significantly limited. A possible reason for that is the P-glycoprotein (P-gp) that was reported to efficiently restrict brain distribution of trametinib [68]. Therefore, inhibiting the efflux transporters represents an attractive approach to further increasing the therapeutic efficacy of BRAF-targeted therapy.
In V600E-mutated glioma cell lines, epidermal growth factor receptor (EGFR) amplification causes resistance to BRAF inhibition and blocking EGFR overcomes this resistance [69]. Presumably, this resistance emanates from adaptive feedback reactivation of MAPK signaling, in which EGFR activates other kinases, such as CRAF, which are resistant to BRAF inhibitors. One case report supports this observation, where a patient with GBM and concurrent gain of function EGFR mutation experienced no clinical benefit, despite RAF and MEK inhibition [38].
Also, the KIAA1549-BRAF fusion may hinder the effect of targeted treatment [30]. Although this fusion occurs in a mutually exclusive pattern with other activating mutations in the MAPK signaling pathway [70, 71], a few cases were reported with both the fusion and V600E mutations [30, 72]. In such patients, KIAA1549-BRAF fusion is believed to function as a homodimer that not only renders cells resistant to BRAF inhibition, but also displays paradoxical activation of MAPK signaling [73]. It might be an explanation for rapid progression of low-grade astrocytoma in pediatric patients with both BRAF alterations after treatment with selumetinib [30].
Mutations in receptor tyrosine kinase (RTK) growth factor receptors resulting in constitutive downstream signaling of the canonical MAPK and PI3K/Akt pathways are found in almost all astrocytic tumors [63]. While MEK/MAPK signaling represents one arm of the tumor growth and survival network in gliomas, the MAPK and PI3K pathways regulate a common set of downstream apoptotic regulators, such as Bad, Mcl-1, Bcl-xL and Bcl2. In such cases, the combination of a BH3 mimetic with MEK/RAF and PI3K inhibitors may prove necessary.
3.2 BRAF and the Oncogene-Induced Senescence (OIS)
BRAF activation in neural stem and progenitor cells not only promotes tumor growth, but also mediates oncogene-induced senescence in low-grade brain tumors [74]. This explains the relatively high frequency of BRAF-mutant brain tumors associated with favorable prognosis [71]. Up to 78% of PXAs WHO grade II and only 12% of anaplastic astrocytoma WHO grade III harbor the V600E mutation [26, 40]. This phenomenon suggests that BRAF activation entails the oncogene-induced senescence (OIS) in PXA [74]. The major role of OIS is to abolish the increased mitogenic signaling and protect from tumor progression [74]. The potential protective role of BRAF in brain tumors might put under question its suitability as a molecular target. Nonetheless, recent results from an open-label, nonrandomized, multicohort study for BRAFV600-mutant nonmelanoma cancers supports the detrimental effect of BRAF activation in PXA. From 24 patients treated with vemurafenib, seven with grade II PXA achieved the highest response rate [25]. Another three case reports support the suitability of BRAF inhibitors against grade III PXA (Table 1) [38, 39, 41]. On the other hand, OIS may potentially promote carcinogenesis through alternative combinations of downstream effectors [75], and whether it occurs in brain tumors remains to be seen.
3.3 Current Recommendations Regarding BRAF Mutations in CNS Tumors
Only a few molecular markers to determine prognosis and guide treatment have been integrated into the clinical care of brain tumor patients. This applies especially for MGMT promoter methylation and TERT promoter mutation in IDH-wild type gliomas. To date, however, the validity of this testing is not supported by strong evidence and no large consensus exists on the optimal method of assessment of these alterations [76, 77]. National Comprehensive Cancer Network (NCCN) Guidelines for central nervous system cancers are the only ones to mention gain of function BRAF mutation in brain tumors. The guidelines recommend treatment with BRAF and MEK inhibitors in V600E-mutated PA, PXA and GG in the adjuvant setting [78].
3.4 Screening for V600E in CNS Tumors
Anti-BRAF V600E (clone VE1), a mouse monoclonal antibody, is used in the immunohistochemical identification of mutant BRAF V600E protein and recently has been successfully validated in CNS tumors [79,80,81]. Because of its unsuitability for detecting V600E in particular neoplasms, such as pituitary adenomas [82], it is generally recommended to perform sequencing validation in positive cases prior to application for targeted treatment [34].
Behling et al. suggest routine screening for V600E for glioblastomas WHO grade IV below the age of 30 years, especially those with epithelioid features, PXAs, and GGs [34]. We propose additional testing in papillary craniopharyngiomas, pediatric low-grade astrocytomas, and PAs (Fig. 2) due to significant subsets of V600E positive cases within these groups (Table 1).

Management algorithm for brain tumor patients—the candidates for BRAF-targeted therapy. The initial step in primary brain tumor treatment is for the neurosurgeon to remove as much of the tumor as possible while minimally disturbing the surrounding brain tissue. Patients with GBM WHO grade IV below the age of 30 years (especially those with epithelioid features), PLGA, PA, PXA, GG, and PCP should be screened for BRAF V600E mutation using the anti-BRAF V600E. The positive IHC result needs to be confirmed by sequencing. BRAF-targeted therapy should be limited to the setting of a clinical trial. If the patient harboring a V600E mutation does not qualify for a trial, multidirectional treatment is recommended. Dual inhibition of both RAF and MEK is expected to provide more potent and durable effect than anti-BRAF monotherapy. First-generation RAF inhibitors, such as sorafenib, are not recommended. The oncologist that is directly involved in treatment of the patient should decide whether and when to test for gain-of-function mutation of EGFR and KIAA fusion, since both of these alterations may compromise BRAF-targeted therapy. BBB blood–brain barrier, eGBM epithelioid glioblastoma, GBM glioblastoma, GG ganglioglioma, IHC immunohistochemical, PA pilocytic astrocytoma, PCP papillary craniopharyngioma, PLGA pediatric low grade astrocytoma, PXA pleomorphic xanthoastrocytoma
3.5 Selection of Patients with V600E for Targeted Therapy
There is no strong evidence of benefit from BRAF-targeted therapy as a first-line treatment in CNS tumors. Regardless of tumor histology, maximal safe resection is recommended whenever possible. BRAF-targeted therapy may improve survival in some patients, however its initiation requires a multidisciplinary consultation.
Schreck et al. suggest limiting the BRAF-targeted therapy to the setting of a clinical trial and considering combination therapy only in patients who are not eligible for a trial [21]. The NCCN Guidelines recommend single-modality therapies only for patients with poor performance status (Karnofsky Performance Score [KPS] < 60), since they frequently do not tolerate the toxicity associated with combination regimens [78].
Since the therapy must be tailored to the specific mutational context and distinct mechanisms of action of the mutant kinase, we recommend considering additional testing for the gain of function EGFR mutation and KIAA1549-BRAF fusion beforehand (Fig. 2). KIAA1549-BRAF fusion can be detected with RNA-Seq. FISH is a less reliable method of identifying BRAF fusions because the KIAA1549 and BRAF genes lie in close proximity and it is difficult to distinguish fusion signal from a normal signal [83]. If, however, the RNA extraction was unsuccessful, FISH may represent a useful adjunct.
Ongoing trials will further clarify targeted treatment options in the presence of BRAF-mutant brain tumors. Three phase II clinical trials are evaluating the biological activity of trametinib (NCT03363217), MEK 162 (NCT02285439), and a combination of dabrafenib with trametinib (NCT02684058) in pediatric gliomas. In adults, two phase II clinical trials using binimetinib with encorafenib in HGG (NCT03973918) and vemurafenib with cobimetinib in craniopharyngioma (NCT03224767) are currently underway.
4 Conclusions
BRAF mutations that result in MAPK pathway activation are common events in several types of brain tumors. The efficacy of targeted therapy for BRAF-mutant brain tumors is currently being investigated. Given the encouraging early results, in the not-too-distant future, such approaches may shift the treatment paradigms and increase survival of patients with CNS tumors. The proposed algorithm was designed to help oncologists to provide the best therapeutic options for brain tumor patients.
References
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96.
Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370:699–708.
Stupp M, Idbaih A, Steinberg DM, Read W, Toms S, Barnett G, et al. LTBK-01: prospective, multi-center phase III trial of tumor treating fields together with temozolomide compared to temozolomide alone in patients with newly diagnosed glioblastoma. Neuro Oncol. 2016;18:i1–i1.
Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma. JAMA. 2017;318:2306–16.
Larochelle C, Alvarez JI, Prat A. How do immune cells overcome the blood-brain barrier in multiple sclerosis? FEBS Lett. 2011;585:3770–80.
Pardridge WM. The blood-brain barrier: Bottleneck in brain drug development. NeuroRX. 2005;2:3–14.
Sang L, Coller HA, Roberts JM. Control of the reversibility of cellular quiescence by the transcriptional repressor HES1. Science. 2008;321:1095–100.
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54.
Tan YH, Liu Y, Eu KW, Ang PW, Li WQ, Salto-Tellez M, et al. Detection of BRAF V600E mutation by pyrosequencing. Pathology. 2008;40:295–8.
Puxeddu E, Moretti S, Elisei R, Romei C, Pascucci R, Martinelli M, et al. BRAF V599E mutation is the leading genetic event in adult sporadic papillary thyroid carcinomas. J Clin Endocrinol Metab. 2004;89:2414–20.
Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011;364:2305–15.
Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–67.
Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell. 2012;22:668–82.
Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877–88.
Larkin J, Ascierto PA, Dréno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371:1867–76.
Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19:1315–27.
Planchard D, Besse B, Groen HJM, Souquet PJ, Quoix E, Baik CS, et al. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol. 2016;17:984–93.
Ahn J-H, Lee YW, Ahn SK, Lee M. Oncogenic BRAF inhibitor UAI-201 induces cell cycle arrest and autophagy in BRAF mutant glioma cells. Life Sci. 2014;104:38–46.
Lyustikman Y, Momota H, Pao W, Holland EC. Constitutive activation of Raf-1 induces glioma formation in Mice. Neoplasia. 2008;10:501–10.
Ritterhouse LL, Barletta JA. BRAF V600E mutation-specific antibody: a review. Semin Diagn Pathol. 2015;32:400–8.
Schreck KC, Grossman SA, Pratilas CA. BRAF mutations and the utility of RAF and MEK inhibitors in primary brain tumors. Cancers (Basel). 2019;11:1262.
Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011;121:397–405.
Drobysheva A, Klesse LJ, Bowers DC, Rajaram V, Rakheja D, Timmons CF, et al. Targeted MAPK pathway inhibitors in patients with disseminated pilocytic astrocytomas. J Natl Compr Cancer Netw. 2017;15:978–82.
Bernstein A, Mrowczynski OD, Greene A, Ryan S, Chung C, Zacharia BE, et al. Dual BRAF/MEK therapy in BRAF V600E-mutated primary brain tumors: a case series showing dramatic clinical and radiographic responses and a reduction in cutaneous toxicity. J. Neurosurg. 2019. https://doi.org/10.3171/2019.8.JNS19643.
Kaley T, Touat M, Subbiah V, Hollebecque A, Rodon J, Lockhart AC, et al. BRAF inhibition in BRAFV600-mutant gliomas: results from the VE-BASKET study. J Clin Oncol. 2018;36:3477–84.
Lassaletta A, Zapotocky M, Mistry M, Ramaswamy V, Honnorat M, Krishnatry R, et al. Therapeutic and prognostic implications of BRAF V600E in pediatric low-grade gliomas. J Clin Oncol. 2017;35:2934–41.
Kieran MW, Bouffet E, Tabori U, Broniscer A, Cohen K, Hansford J, et al. CNS tumours the first study of dabrafenib in pediatric patients with BRAF V600–mutant relapsed or refractory low-grade gliomas. Ann Oncol. 2016;27:1–36.
Hargrave DR, Bouffet E, Tabori U, Broniscer A, Cohen KJ, Hansford JR, et al. Efficacy and safety of dabrafenib in pediatric patients with BRAF V600 mutation-positive relapsed or refractory low-grade glioma: results from a phase I/IIa study. Clin Cancer Res. 2019;25:7303–11.
Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, et al. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol. 2019;20:1011–22.
Banerjee A, Jakacki RI, Onar-Thomas A, Wu S, Nicolaides T, Young Poussaint T, et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a Pediatric Brain Tumor Consortium (PBTC) study. Neuro Oncol. 2017;19:1135–44.
Dahiya S, Emnett RJ, Haydon DH, Leonard JR, Phillips JJ, Perry A, et al. BRAF-V600E mutation in pediatric and adult glioblastoma. Neuro Oncol. 2014;16:318–9.
Toll SA, Tran HN, Cotter J, Judkins AR, Tamrazi B, Biegel JA, et al. Sustained response of three pediatric BRAFV600E mutated high-grade gliomas to combined BRAF and MEK inhibitor therapy. Oncotarget. 2019;10:551–7.
Bautista F, Paci A, Minard-Colin V, Dufour C, Grill J, Lacroix L, et al. Vemurafenib in pediatric patients with BRAFV 600E mutated high-grade gliomas. Pediatr Blood Cancer. 2014;61:1101–3.
Behling F, Barrantes-Freer A, Skardelly M, Nieser M, Christians A, Stockhammer F, et al. Frequency of BRAF V600E mutations in 969 central nervous system neoplasms. Diagn Pathol. 2016;11:55.
Meletath SK, Pavlick D, Brennan T, Hamilton R, Chmielecki J, Elvin JA, et al. Personalized treatment for a patient with a BRAF V600E mutation using dabrafenib and a tumor treatment fields device in a high-grade glioma arising from ganglioglioma. J Natl Compr Cancer Netw. 2016;14:1345–50.
Wen P, Alexander S, Yung-Jue B, van den Bent M, Gazzah A, Dietrich S, et al. RARE-09. Efficacy and safety of dabrafenib + trametinib in patients with recurrent/refractory BRAF V600E–mutated high-grade glioma (HGG). Neuro Oncol. 2018;20:vi238.
Johanns TM, Ferguson CJ, Grierson PM, Dahiya S, Ansstas G. Rapid clinical and radiographic response with combined dabrafenib and trametinib in adults with BRAF-mutated high-grade glioma. J Natl Compr Cancer Netw. 2018;16:4–10.
Smith-Cohn M, Davidson C, Colman H, Cohen AL. Challenges of targeting BRAF V600E mutations in adult primary brain tumor patients: a report of two cases. CNS Oncol. 2019;8:CNS48.
Schreck KC, Guajardo A, Lin DDM, Eberhart CG, Grossman SA. Concurrent BRAF/MEK inhibitors in BRAF V600–mutant high-grade primary brain tumors. J Natl Compr Cancer Netw. 2018;16:343–7.
Dias-Santagata D, Lam Q, Vernovsky K, Vena N, Lennerz JK, Borger DR, et al. BRAF V600E mutations are common in pleomorphic xanthoastrocytoma: diagnostic and therapeutic implications. PLoS ONE. 2011;6:e17948.
Piña Y, Fusco MJ, Macaulay RJ, Walko CM, Peguero E, Evernden BR, et al. Using personalized medicine in gliomas: a genomic approach to diagnosis and overcoming treatment resistance in a case with pleomorphic xanthoastrocytoma. J Neurol. 2020;267:783–90.
Chamberlain MC. Salvage therapy with BRAF inhibitors for recurrent pleomorphic xanthoastrocytoma: a retrospective case series. J Neurooncol. 2013;114:237–40.
Marks AM, Bindra RS, DiLuna ML, Huttner A, Jairam V, Kahle KT, et al. Response to the BRAF/MEK inhibitors dabrafenib/trametinib in an adolescent with a BRAF V600E mutated anaplastic ganglioglioma intolerant to vemurafenib. Pediatr Blood Cancer. 2018;65:e26969.
Beland B, Tsang RY, Sutherland G. Unprecedented response to combination BRAF and MEK inhibitors in adult anaplastic ganglioglioma. J Neurooncol. 2018;137:667–9.
del Bufalo F, Carai A, Figà-Talamanca L, Pettorini B, Mallucci C, Giangaspero F, et al. Response of recurrent BRAFV600E mutated ganglioglioma to Vemurafenib as single agent. J Transl Med. 2014;12:356.
Garnier L, Ducray F, Verlut C, Mihai MI, Cattin F, Petit A, et al. Prolonged response induced by single agent vemurafenib in a BRAF V600E spinal ganglioglioma: a case report and review of the literature. Front Oncol. 2019;9:177.
Aguilera D, Janss A, Mazewski C, Castellino RC, Schniederjan M, Hayes L, et al. Successful retreatment of a child with a refractory brainstem ganglioglioma with vemurafenib. Pediatr Blood Cancer. 2016;63:541–3.
Touat M, Gratieux J, Auliac SC, Sejean K, Aldea S, Savatovsky J, et al. Vemurafenib and cobimetinib overcome resistance to vemurafenib in BRAF-mutant ganglioglioma. Neurology. 2018;91:523–5.
Rush S, Foreman N, Liu A. Brainstem ganglioglioma successfully treated with vemurafenib. J Clin Oncol. 2013;31:e159–e160160.
Brastianos PK, Taylor-Weiner A, Manley PE, Jones RT, Dias-Santagata D, Thorner AR, et al. Exome sequencing identifies BRAF mutations in papillary craniopharyngiomas. Nat Genet. 2014;46:161–5.
Himes BT, Ruff MW, Van Gompel JJ, Park SS, Galanis E, Kaufmann TJ, et al. Recurrent papillary craniopharyngioma with BRAF V600E mutation treated with dabrafenib: case report. J Neurosurg. 2019;130:1299–303.
Roque A, Odia Y. BRAF -V600E mutant papillary craniopharyngioma dramatically responds to combination BRAF and MEK inhibitors. CNS Oncol. 2017;6:95–9.
Sollfrank L, Lettmaier S, Erdmann M, Uslu U. Panniculitis under successful targeted inhibition of the MAPK/ERK signaling pathway in a patient with BRAF V600E-mutated spindle cell oncocytoma of the pituitary gland. Anticancer Res. 2019;39:3955–9.
Dankner M, Rose AAN, Rajkumar S, Siegel PM, Watson IR. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene. 2018;37:3183–99.
Key MEDLINE® Indicators.
Maraka S, Janku F. BRAF alterations in primary brain tumors. Discov Med. 2018;26:51–60.
Arozarena I, Wellbrock C. Overcoming resistance to BRAF inhibitors. Ann Transl Med. 2017;5:387.
Long GV, Trefzer U, Davies MA, Kefford RF, Ascierto PA, Chapman PB, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13:1087–95.
McArthur GA, Maio M, Arance A, Nathan P, Blank C, Avril M-F, et al. Vemurafenib in metastatic melanoma patients with brain metastases: an open-label, single-arm, phase 2, multicentre study. Ann Oncol. 2017;28:634–41.
Mittapalli RK, Vaidhyanathan S, Dudek AZ, Elmquist WF. Mechanisms limiting distribution of the threonine-protein kinase B-RaF(V600E) inhibitor dabrafenib to the brain: implications for the treatment of melanoma brain metastases. J Pharmacol Exp Ther. 2013;344:655–64.
Sun Y, Alberta JA, Pilarz C, Calligaris D, Chadwick EJ, Ramkissoon SH, et al. A brain-penetrant RAF dimer antagonist for the noncanonical BRAF oncoprotein of pediatric low-grade astrocytomas. Neuro Oncol. 2017;19:774–85.
Karajannis MA, Legault G, Fisher MJ, Milla SS, Cohen KJ, Wisoff JH, et al. Phase II study of sorafenib in children with recurrent or progressive low-grade astrocytomas. Neuro Oncol. 2014;16:1408–16.
Robinson JP, VanBrocklin MW, Guilbeault AR, Signorelli DL, Brandner S, Holmen SL. Activated BRAF induces gliomas in mice when combined with Ink4a/Arf loss or Akt activation. Oncogene. 2010;29:335–44.
Lim SY, Menzies AM, Rizos H. Mechanisms and strategies to overcome resistance to molecularly targeted therapy for melanoma. Cancer. 2017;123:2118–299.
Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011;480:387–90.
Nissan MH, Pratilas CA, Jones AM, Ramirez R, Won H, Liu C, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014;74:2340–50.
Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–7.
Vaidhyanathan S, Mittapalli RK, Sarkaria JN, Elmquist WF. Factors Influencing the CNS distribution of a Novel MEK-1/2 inhibitor: implications for combination therapy for melanoma brain metastases. Drug Metab Dispos. 2014;42:1292–300.
Yao TW, Zhang J, Prados M, Weiss WA, James CD, Nicolaides T. Acquired resistance to BRAF inhibition in BRAFV600E mutant gliomas. Oncotarget. 2017;8:583–95.
Ross JS, Wang K, Chmielecki J, Gay L, Johnson A, Chudnovsky J, et al. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int J Cancer. 2016;138:881–90.
Behling F, Schittenhelm J. Oncogenic BRAF alterations and their role in brain tumors. Cancers (Basel). 2019;11:794.
Cin H, Meyer C, Herr R, Janzarik WG, Lambert S, Jones DTW, et al. Oncogenic FAM131B–BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol. 2011;121:763–74.
Sievert AJ, Lang SS, Boucher KL, Madsen PJ, Slaunwhite E, Choudhari N, et al. Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc Natl Acad Sci. 2013;110:5957–62.
Raabe EH, Lim KS, Kim JM, Meeker A, Mao XG, Nikkhah G, et al. BRAF activation induces transformation and then senescence in human neural stem cells: a pilocytic astrocytoma model. Clin Cancer Res. 2011;17:3590–9.
Liu X, Ding J, Meng L. Oncogene-induced senescence: a double edged sword in cancer. Acta Pharmacol Sin. 2018;39:1553–8.
NICE. Recommendations | Brain tumours (primary) and brain metastases in adults | Guidance | NICE 2019. (2018).
Mansouri A, Hachem LD, Mansouri S, Nassiri F, Laperriere NJ, Xia D, et al. MGMT promoter methylation status testing to guide therapy for glioblastoma: refining the approach based on emerging evidence and current challenges. Neuro Oncol. 2019;21:167–78.
Nabors, L. et al. National Comprehensive Cancer Network. Central Nervous System Cancers. (Version 1.2020). 2020. https://www.nccn.org/professionals/physician_gls/pdf/cns_blocks.pdf. Accessed 17th May 2020.
Ida CM, Vrana JA, Rodriguez FJ, Jentoft ME, Caron AA, Jenkins SM, et al. Immunohistochemistry is highly sensitive and specific for detection of BRAF V600E mutation in pleomorphic xanthoastrocytoma. Acta Neuropathol Commun. 2013;1:20.
Tosuner Z, Geçer MÖ, Hatiboğlu MA, Abdallah A, Turna S. BRAF V600E mutation and BRAF VE1 immunoexpression profiles in different types of glioblastoma. Oncol Lett. 2018;16:2402–8.
Breton Q, Plouhinec H, Prunier-Mirebeau D, Boisselier B, Michalak S, Menei P, et al. BRAF-V600E immunohistochemistry in a large series of glial and glial-neuronal tumors. Brain Behav. 2017;7:e00641.
Mordes DA, Lynch K, Campbell S, Dias-Santagata D, Nose V, Louis DN, et al. VE1 Antibody immunoreactivity in normal anterior pituitary and adrenal cortex without detectable BRAF V600E mutations. Am J Clin Pathol. 2014;141:811–5.
Tian Y, Rich BE, Vena N, Craig JM, Macconaill LE, Rajaram V, et al. Detection of KIAA1549-BRAF fusion transcripts in formalin-fixed paraffin-embedded pediatric low-grade gliomas. J Mol Diagn. 2011;13:669–77.
Author information
Authors and Affiliations
Contributions
A.K. and Ł.S. conceived of the presented idea. A.K., J.D., and M.Z. wrote the manuscript. D.G. and Ł.S. were in charge of overall direction and planning.
Corresponding author
Ethics declarations
Funding
No external funding was used in the preparation of this manuscript.
Conflict of interest
A.K., J.D., M.Z., D.G., and Ł.S. declare that they have no conflicts of interest that might be relevant to the contents of this manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Kowalewski, A., Durślewicz, J., Zdrenka, M. et al. Clinical Relevance of BRAF V600E Mutation Status in Brain Tumors with a Focus on a Novel Management Algorithm. Targ Oncol 15, 531–540 (2020). https://doi.org/10.1007/s11523-020-00735-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-020-00735-9