
Abstract
Purpose
Preliminary studies have shown a significant decrease in severity of obstructive sleep apnea (OSA) with the use of a combination of atomoxetine and oxybutynin, with patients having moderate pharyngeal collapsibility during sleep more likely to respond. This study evaluated the efficacy and safety of AD036 (atomoxetine 80 mg and oxybutynin 5 mg) in the treatment of OSA.
Methods
This trial was a phase 2, randomized, placebo-controlled crossover study comparing AD036, atomoxetine 80 mg alone, and placebo during three home sleep studies, each separated by about 1 week. The trial included patients with OSA and moderate pharyngeal collapsibility as defined by a higher proportion of hypopneas to apneas and mild oxygen desaturation.
Results
Of 62 patients who were randomized, 60 were included in efficacy analyses. The apnea–hypopnea index (AHI) from a median (interquartile range) of 14.2 (5.4 to 22.3) events/h on placebo to 6.2 (2.8 to 13.6) with AD036 and 4.8 (1.4 to 11.6) with atomoxetine alone (p < .0001). Both drugs also decreased the oxygen desaturation index (ODI) and the hypoxic burden (p < .0001). AD036, but not atomoxetine alone, reduced the respiratory arousal index and improved ventilation at the respiratory arousal threshold (greater Vactive). There was a trend for total sleep time to be decreased more with atomoxetine alone than with AD036. The most common adverse event was insomnia (12% with AD036, 18% with atomoxetine).
Conclusion
AD036 significantly improved OSA severity in patients with moderate pharyngeal collapsibility. Atomoxetine may account for the majority of improvement in OSA severity, while the addition of oxybutynin may mitigate the disruptive effect of atomoxetine on sleep and further improve ventilation.
Trial registration
Clinical trial registered with www.clinicaltrials.gov (NCT04445688).
Similar content being viewed by others


Introduction
Obstructive sleep apnea (OSA) is a common sleep disorder, affecting over 930 million adults globally, and approximately 17% of women and 34% of men in the USA [1, 2]. Untreated OSA is associated with increased incidence of hypertension, coronary heart disease, arrhythmias, heart failure, and stroke [3]. Continuous positive airway pressure (CPAP), the primary therapy for OSA, is very effective, but its use is limited by poor tolerance and adherence in a substantial number of patients [4,5,6]. The principal alternative treatment options, which include mandibular advancement devices and upper airway surgery, are not effective in all patients, and prediction of efficacy is challenging [2]. There are currently no approved pharmacotherapies for OSA, and efforts to develop such therapies have been generally unsuccessful [7, 8]. However, recent advances in our understanding of the pathophysiological traits which contribute to OSA indicate that airway collapsibility and pharyngeal dilator muscle function are two of the most important traits contributing to OSA, suggesting that drugs that are capable of raising baseline upper airway muscle activity and its responsiveness to increased load have the potential to resolve OSA [9, 10]. Preliminary studies have shown a significant decrease in OSA severity using norepinephrine reuptake inhibitors such as atomoxetine and reboxetine in combination with antimuscarinics such as oxybutynin or hyoscine butylbromide [11,12,13]. In one study, the combined administration of atomoxetine and oxybutynin resulted in a reduction in the apnea–hypopnea index (AHI) by approximately 60% in a small group of patients with OSA [11]. The presumed mechanism for these findings is an increase in upper airway dilator muscle activity and responsiveness caused by noradrenergic and antimuscarinic stimulation [14, 15].
AD036 is a fixed-dose combination of atomoxetine and oxybutynin that is being developed for the treatment of OSA. Post hoc analyses of feasibility studies assessing several dose combinations of AD036 in patients with varying OSA severity and physiologic characteristics indicated that patients with clinical signs of moderate pharyngeal collapsibility during sleep (see below) were more likely to respond to the medication than those with more severe collapsibility [16]. This study was undertaken to assess the efficacy and safety of AD036 in this patient population with moderate pharyngeal collapsibility.
Methods
Study design
This trial was a phase 2, randomized, 3-period, single-dose, placebo-controlled crossover study to evaluate the efficacy and safety of AD036 versus placebo or atomoxetine alone in the treatment of OSA. The study was conducted at five clinical investigative sites in the USA and was approved by institutional review boards at each site and performed in accordance with the Declaration of Helsinki. All participants provided written informed consent (www.clinicaltrials.gov identifier NCT04445688).
Participants
Participants were adults with OSA who had low to moderate pharyngeal collapsibility during sleep as indicated by respiratory criteria based on either a baseline polysomnography (PSG) study from a prior AD036 study or a home sleep apnea test (HSAT) during screening. Respiratory criteria for moderate collapsibility were derived from previous analysis collected in the academic [16, 17] and sponsored research settings (unpublished observation) and included (1) an AHI = 10 to < 20, or (2) AHI ≥ 20 along with one of the following criteria: mean oxygen desaturation of obstructive events ≤ 4%, fraction of hypopneas > 90%, or fraction of hypopneas 50–90% with mean oxygen desaturation 4–8%. Previous studies (unpublished) found that these criteria indicated moderate pharyngeal collapsibility using the methods of Sands and Wellman [18]. Hypopneas were defined using a minimum oxygen desaturation criterion of 4% [19]. Demographic requirements included age 25–65 years (up to 67 for participants of the prior AD036 study) and body mass index (BMI) between 18.5 and 40 kg/m2. Key exclusion criteria were as follows: clinically important craniofacial malformation, cardiac disease, neurological disorder, cognitive dysfunction, constipation, gastric retention, urinary retention; history of narcolepsy, schizophrenia, schizoaffective disorder, bipolar disorder, suicide attempt or ideation in the past year; substance use disorder (within past 2 years) or positive screen for drugs of abuse; use of oxygen or medications with central nervous system effects.
Treatments and study protocol
After screening for eligibility, participants underwent three overnight HSATs after dosing of one of the following treatments on each study night: AD036 (comprised of commercially available formulations of atomoxetine 80 mg and oxybutynin 5 mg), atomoxetine 80 mg + placebo, placebo + placebo. Participants were centrally randomized to one of six treatment sequences in this three-period crossover design. There was no blinding, but the study treatments were packaged in a manner to reduce participants’ knowledge of the treatment assignment. A 1-week minimum washout period occurred between each of the crossover nights. Participants were instructed to take study drug orally immediately prior to bedtime. HSATs were utilized instead of in-lab PSGs to decrease the risk of transmission of coronavirus disease 2019 (COVID-19) as the study was initiated at the beginning of the COVID-19 pandemic. HSATs were conducted utilizing the Nox A1 system (Nox Medical, Reykjavık, Iceland) and included electroencephalogram (EEG), electrooculogram, respiratory airflow (using nasal pressure transducer), thoracic and abdominal respiratory effort, oximetry, heart rate, and body position. Because scalp EEG and submental electromyogram (EMG) leads are difficult to self-apply, we used only frontal leads for EEG recordings and excluded submental EMG leads. Site personnel contacted participants via video call during each HSAT night to assist with sensor and equipment placement, and to confirm study drug dosing. Adverse event and concomitant medication monitoring were conducted on HSAT nights and during each washout period. CPAP use was not permitted within 2 weeks of the first HSAT or anytime thereafter. Other treatments for OSA, including oral, nasal, and positional devices, were not permitted during study participation.
Data analysis
Sleep studies were scored by a blinded central reading center (Sleep Strategies Inc., Ontario, Canada) using American Academy of Sleep Medicine guidelines [19]. The 4% oxygen desaturation criterion was used for scoring of hypopneas and subsequent calculation of the AHI, and for calculation of the oxygen desaturation index (ODI). We also calculated AHI using the 3% desaturation or arousal criterion for hypopneas (AHI3A). Sleep staging was scored using the frontal EEG leads. In addition to standard sleep and respiratory variables, we also calculated hypoxic burden, a measure of the severity of oxygen desaturation over the entire night determined by quantifying the respiratory event–associated area under the desaturation curve from a pre-event baseline. This metric has been shown to predict adverse cardiovascular outcomes better than AHI [20]. We also calculated the fraction of hypopneas to total disordered breathing events (apneas plus hypopneas), arousals associated with respiratory events, and arousal burden [21], a validated measure which quantifies both the frequency and intensity of respiratory-related arousals expressed as the sum of arousal intensities per hour of sleep (arousal intensity ranging from 0 to 9). Finally, a validated algorithm was used to estimate endotypic traits, including passive airway collapsibility (Vmin; ventilation measured during obstructive events at the minimal ventilatory drive), pharyngeal muscle activation (Vactive; ventilation measured at maximum ventilatory drive during obstructive events prior to arousal), loop gain (stability of the respiratory control system), and arousal threshold (estimated ventilatory drive prior to arousal) [18].
Statistical analysis
The primary efficacy endpoint was the comparison of AHI between AD036 and placebo. Secondary endpoints were comparisons between AD036 and placebo and between AD036 and atomoxetine alone on hypoxic burden and ODI. Tertiary endpoints included total time with oxygen saturation (SaO2) < 90%, sleep stage distribution, and respiratory arousal index.
Approximately 54 participants (9 per sequence) were planned for enrollment. This sample size provided 90% power to detect a treatment difference in AHI of 11 events/h at a two-sided 0.05 significance level, using a within-subject standard deviation of 12 events/h.
Continuous variables were analyzed using a within-subject analysis of variance (ANOVA) model including treatment sequence, treatment, and visit as fixed effects. Robust variance estimates for the fixed effects were used for testing treatment differences. Within-subject variability was modeled using an unstructured covariance pattern. Endotypic traits and arousal burden were analyzed post hoc using a mixed model analysis with treatment as a fixed factor and subjects as a random factor. Data analyses were performed using SAS for Windows, version 9.4 or higher (SAS, Cary, NC). Data are presented as median and interquartile range or least square (LS) mean with 95% confidence interval.
Baseline (study entry criteria) data were not used in analyses because the time between the baseline sleep study and the initiation of this protocol was quiet variable (between 1 and 18 months earlier), and the type of study conducted was a full PSG for some and an HSAT for others. Thus, the placebo arm of the trial was used as the comparator for the medication arms.
Results
Participants
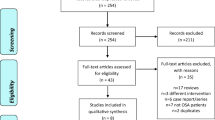
A total of 62 participants were randomized and 59 (95%) completed all three treatments (Fig. 1). Three participants discontinued study participation following the first study night (two because of treatment-emergent adverse events, one because of discomfort with the HSAT device). Safety analysis was conducted using all 62 participants (safety population). Efficacy analyses were conducted using the modified intent-to-treat population, which included all participants who took at least 1 dose of any study treatment and had at least one measurement on the primary endpoint (n = 60).

Consolidated Standards of Reporting Trials diagram of the clinical trial
Participants in the safety population were 50% male, primarily white (66%), and non-Hispanic (94%). Median age was 53.0 years (range: 28 to 66) and median BMI was 32.3 kg/m2 (range: 22.6 to 39.6).
Efficacy
Efficacy data are presented in Table 1. AD036 was superior to placebo for the primary and secondary endpoints of AHI, ODI, and hypoxic burden (Fig. 2). AD036 also resulted in a decrease in time spent with SaO2 < 90% and an increase in the fraction of hypopneas comprising the AHI vs. placebo. The respiratory arousal index was lower for AD036 compared to both placebo and atomoxetine. However, there was no difference between AD036 and atomoxetine in most other respiratory measures, with atomoxetine alone yielding a significant reduction in AHI, ODI, and hypoxic burden when compared to placebo. Post hoc analysis of arousal burden showed a decrease with AD036 compared to both placebo and atomoxetine (Table 2).

Box plots of apnea–hypopnea index (AHI), oxygen desaturation index (ODI), and hypoxic burden (HB) on placebo, AD036, and atomoxetine. Black lines indicate medians; boxes indicate 25th and 75th percentiles
Total sleep time was decreased by both drugs, with median reduction compared to placebo of 29.0 min for AD036 and 61.0 min for atomoxetine (Table 1). There was a trend for lower sleep time on atomoxetine vs. AD036 (p = 0.06). Both drugs reduced REM sleep.
Effects on endotypic traits
The post hoc analyses of endotypic traits are presented in Table 2. Compared to placebo, both AD036 and atomoxetine improved passive airway collapsibility (Vmin), reduced the arousal threshold, and reduced loop gain. AD036, but not atomoxetine, increased Vactive, compared to placebo.
Safety
No serious adverse events were reported during the study. A total of 78 treatment-emergent adverse events were reported in 26 (42%) participants in the safety population: 21 (36%) individuals with AD036; 18 (29%) with atomoxetine; and 5 (9%) with placebo. Two participants discontinued study participation because of adverse events considered related to study treatment with atomoxetine (anxiety in one person, hyperesthesia, nausea, and insomnia in one person.) A third participant discontinued participation because of discomfort with the HSAT device on the atomoxetine-treatment night. The most common adverse events for AD036 were insomnia (12%) and nausea (5%); for atomoxetine, insomnia (18%), nausea (5%), decreased appetite (5%), and feeling jittery (5%); and, for placebo, insomnia (7%). All adverse events were rated as mild to moderate except for one event (hyperesthesia) that was rated as severe.
Discussion
The main finding of this study is that AD036 significantly improved AHI and other measures of OSA severity during a single night of treatment. The magnitude of effect was clinically meaningful, with a 54% reduction in median AHI compared to placebo. On AD036 vs. placebo, residual respiratory events were milder, as indicated by an increase in the fraction of hypopneas comprising the AHI (from a median of 73% on placebo to 94% with AD036) and substantial reduction in the time spent with SaO2 < 90% (from a median 14.1 min to 5.1 min), as well as a decrease in hypoxic burden (from a median 30.5% min/h to 13.7% min/h). However, the effects of AD036 on AHI and most other respiratory endpoints were not significantly different from that of atomoxetine alone. A noted exception was a reduction in respiratory arousals with AD036 but not with atomoxetine alone, an effect seen with the respiratory arousal index (22% decrease) as well as with arousal burden (24% decrease), a related measure that quantifies both intensity and frequency of respiratory arousals. In addition, the post hoc analysis of endotypic traits indicated that only AD036 increased Vactive, suggesting that AD036 had a stronger effect than atomoxetine alone in improving ventilation by recruiting the upper airway dilator muscles, consistent with prior findings [11, 16]. AD036 was also less disruptive of sleep than atomoxetine alone, with a median decrease in total sleep time compared to placebo of 29.0 min for AD036 versus 61.0 min for atomoxetine alone.
Both AD036 and atomoxetine alone were safe and well tolerated in this single-night crossover exposure. The most common adverse events (insomnia and nausea with AD036; insomnia, nausea, decreased appetite, jitteriness with atomoxetine) were consistent with the expected profile of the individual approved drugs, atomoxetine and oxybutynin [22, 23]. Insomnia, the most common side effect, was reported more frequently with atomoxetine alone (18%) than with AD036 (12%), consistent with the objective sleep data.
These data extend the findings of a previous pilot study demonstrating improvement in AHI and other respiratory measures with the combination of atomoxetine and oxybutynin [11]. Unlike that study, we also found improvement in AHI with atomoxetine alone, with no difference between atomoxetine alone and the combination of atomoxetine-oxybutynin for most respiratory measures. Our data also differs from a study of atomoxetine 40–80 mg alone that showed no reduction in AHI or respiratory disturbance index after 4 weeks of use [24]. Differences in results may possibly be related to study design, sample size, or the specific focus on patients with milder collapsibility in the current study. The study by Sangal et al. [24] included 15 patients with very mild sleep apnea in a pre-post study design without placebo control. The evaluation of atomoxetine alone in the Taranto-Montemurro et al. study [11] occurred in a supplementary open-label investigation in a subsample (n = 9) of participants from the primary study and also found no effect of atomoxetine on AHI, although it was responsible for 38% improvement in ventilation during sleep, compared to the 18% improvement with oxybutynin alone [16]. Furthermore, in the previous trial, atomoxetine alone reduced the arousal threshold (patients woke up more easily due to obstructive events) and reduced loop gain (breathing control was more stable).
Results of endotypic trait analyses were similar to those of the prior atomoxetine-oxybutynin study [16]. Both AD036 and atomoxetine alone improved airway collapsibility (greater Vmin) and improved breathing stability (reduced loop gain), but only AD036 improved ventilation at the respiratory arousal threshold (greater Vactive), suggesting a stronger effect on upper airway dilator muscles. Both AD036 and atomoxetine alone reduced the arousal threshold by about 15%, an effect that may decrease sleep efficiency and increase the likelihood of arousal to small changes in ventilatory drive. The reduction in the arousal threshold may be a consequence of the adrenergic effect of atomoxetine or the result of spontaneous arousals during mild flow limitation and may be a limitation to its use.
One limitation of the current study was the use of a single night for each treatment condition. This is a potential problem as severity of OSA can vary from night to night, particularly in individuals with mild to moderate OSA [25, 26]. Our study population, a sample chosen to have moderate pharyngeal collapsibility, was comprised of individuals with mostly mild to moderate OSA as defined by AHI. Furthermore, 12 participants who met AHI entry criteria based on a prior study had AHI < 5 on the placebo night.
We do not believe that data quality issues with the use of HSATs in lieu of in-lab PSGs affected the study results. However, a validated [27], non-standard recording montage was used to facilitate the self-application of sensors by the patients. The use of this alternative montage may have affected the quantification of sleep staging, especially REM and N3, which were notably low even on the placebo night. REM was absent in 18%, 56%, and 70% of participants on the placebo, AD036, and atomoxetine nights, respectively. However, prior studies of atomoxetine [24] and atomoxetine combined with oxybutynin [16, 28] in patients with sleep apnea have shown reduced or absent REM while using standard in-lab PSG. It is unlikely that the reduction in REM sleep in both drug conditions in the current study accounts for the majority of decrease in the overall AHI, since sensitivity analysis of the AHI specifically during NREM sleep confirmed the main findings.
In conclusion, the current study demonstrated that AD036 improves AHI and other measures of OSA severity in a population of patients with moderate pharyngeal collapsibility as described by a higher proportion of hypopneas to apnea and mild degree of oxygen desaturation. The data suggest that atomoxetine is primarily responsible for the improvement in respiratory variables, while the addition of oxybutynin ameliorates the disruptive effect of atomoxetine on sleep and further improves ventilation. Longer-duration studies on a broader group of patients with more-diverse ethnicities are needed to determine if these findings persist over time and to further describe the characteristics of patients likely to respond to this type of pharmacologic treatment for OSA. The use of standard PSG recordings and quality-of-life metrics will be helpful to further elucidate the impact of these drug combinations on sleep architecture and daytime functioning.
Data availability
The datasets generated during and/or analyzed during the current study are available from the sponsor at ltaranto@apnimed.com on reasonable request.
References
Benjafield AV, Ayas NT, Eastwood PR, Heinzer R, Ip MSM, Morrell MJ, Nunez CM, Patel SR, Penzel T, Pépin JL, Peppard PE, Sinha S, Tufik S, Valentine K, Malhotra A (2019) Estimation of the global prevalence and burden of obstructive sleep apnoea: a literature-based analysis. Lancet Respir Med 8:687–698. https://doi.org/10.1016/S2213-2600(19)30198-5
Gottlieb DJ, Punjabi NM (2020) Diagnosis and management of obstructive sleep apnea: a Review. JAMA 14:1389–1400. https://doi.org/10.1001/jama.2020.3514
Javaheri S, Barbe F, Campos-Rodriguez F, Dempsey JA, Khayat R, Javaheri S, Malhotra A, Martinez-Garcia MA, Mehra R, Pack AI, Polotsky VY, Redline S, Somers VK (2017) Sleep apnea: types, mechanisms, and clinical cardiovascular consequences. J Am Coll Cardiol 69(7):841–858. https://doi.org/10.1016/j.jacc.2016.11.069
Patil SP, Ayappa IA, Caples SM, Kimoff RJ, Patel SR, Harrod CG (2019) Treatment of adult obstructive sleep apnea with positive airway pressure: an American Academy of Sleep Medicine systematic review meta-analysis and GRADE assessment. J Clin Sleep Med 15(2):301–334. https://doi.org/10.5664/jcsm.7638
Rotenberg BW, Murariu D, Pang KP (2016) Trends in CPAP adherence over twenty years of data collection: a flattened curve. J Otolaryngol Head Neck Surg 45:43. https://doi.org/10.1186/s40463-016-0156-0
Sawyer AM, Gooneratne NS, Marcus CL, Ofer D, Richards KC, Weaver TE (2011) A systematic review of CPAP adherence across age groups: clinical and empiric insights for developing CPAP adherence interventions. Sleep Med Rev 15:343–356. https://doi.org/10.1016/j.smrv.2011.01.003
Hedner J, Zou D (2018) Drug therapy in obstructive sleep apnea. Sleep Med Clin 13(2):203–217
Mason M, Welsh EJ, Smith I (2013) Drug therapy for obstructive sleep apnoea in adults. Cochrane Database Syst Rev (5):CD003002. https://doi.org/10.1002/14651858.CD003002.pub3.
Osman AM, Carter SG, Carberry JC and Eckert DJ (2018) Obstructive sleep apnea: current perspectives. Nature and science of sleep 21–34. https://doi.org/10.2147/NSS.S124657.
Taranto-Montemurro L, Messineo L, Wellman A (2019) Targeting endotypic traits with medications for the pharmacological treatment of obstructive sleep apnea A review of the current literature. J Clin Med 8(11):1846. https://doi.org/10.3390/jcm8111846
Taranto-Montemurro L, Messineo L, Sands SA, Azarbarzin A, Marques M, Edwards BA, Eckert DJ, White DP, Wellman A (2019) The combination of atomoxetine and oxybutynin greatly reduces obstructive sleep apnea severity A randomized, placebo-controlled double-blind crossover trial. Am J Respir Crit Care Med 199:1267–1276. https://doi.org/10.1164/rccm.201808-1493OC
Perger E, Montemurro LT, Rosa D, Vicini S, Marconi M, Zanotti L, Meriggi P, Azarbarzin A, Sands SA, Wellman A, Lombardi C, Parati G (2021) Reboxetine plus oxybutynin for OSA treatment: a 1-week randomized, placebo-controlled, double-blind crossover trial. Chest S0012–3692(21):03861–03867. https://doi.org/10.1016/j.chest.2021.08.080
Lim R, Messineo L, Grunstein RR, Carberry JC, Eckert DJ (2021) The noradrenergic agent reboxetine plus the antimuscarinic hyoscine butylbromide reduces sleep apnoea severity: a double-blind, placebo-controlled, randomised crossover trial. J Physiol 599(17):4183–4195. https://doi.org/10.1113/JP281912
Chan E, Steenland HW, Liu H, Horner RL (2006) Endogenous excitatory drive modulating respiratory muscle activity across sleep-wake states. Am J Respir Crit Care Med 174(11):1264–1273. https://doi.org/10.1164/rccm.200605-597OC
Grace KP, Hughes SW, Horner RL (2013) Identification of the mechanism mediating genioglossus muscle suppression in REM sleep. Am J Respir Crit Care Med 187(3):311–9. https://doi.org/10.1164/rccm.201209-1654OC
Taranto-Montemurro L, Messineo L, Azarbarzin A, Vena D, Hess LB, Calianese NA, White DP, Wellman A, Sands SA (2020) Effects of the combination of atomoxetine and oxybutynin on OSA endotypic traits. Chest 157(6):1626–1636. https://doi.org/10.1016/j.chest.2020.01.012
Sands SA, Edwards BA, Terrill PI, Taranto-Montemurro L, Azarbarzin A, Marques M, Hess LB, White DP, Wellman A (2018) Phenotyping pharyngeal pathophysiology using polysomnography in patients with obstructive sleep apnea. Am J Respir Crit Care Med 197:1187–1197. https://doi.org/10.1164/rccm.201707-1435OC
Vena D, Taranto-Montemurro L, Azarbarzin A, Op de Beeck S, Marques M, Vanderveken OM, Edwards BA, Gell L, Calianese N, Hess LB, Radmand R, Hamilton GS, Joosten SA, Verbraecken J, Braem M, White DP, Redline S, Sands SA, Wellman A (2022) Clinical polysomnographic methods for estimating pharyngeal collapsibility in obstructive sleep apnea. Sleep zsac050. https://doi.org/10.1093/sleep/zsac050
Berry RB, Quan SF, Abreu AR, et al; for the American Academy of Sleep Medicine (2020) The AASM manual for the scoring of sleep and associated events: rules, terminology and technical specifications. Version 2.6. Darien, IL: American Academy of Sleep Medicine
Azarbarzin A, Sands SA, Stone KL, Taranto-Montemurro L, Messineo L, Terrill PI et al (2019) The hypoxic burden of sleep apnoea predicts cardiovascular disease-related mortality: the Osteoporotic Fractures in Men Study and the Sleep Heart Health Study. Eur Heart J 40:1149–1157. https://doi.org/10.1093/eurheartj/ehy624
Amatoury J, Azarbarzin A, Younes M, Jordan AS, Wellman A, Eckert DJ (2016) Arousal intensity is a distinct pathophysiological trait in obstructive sleep apnea. Sleep 39(12):2091–2100. https://doi.org/10.5665/sleep.6304
Simpson D, Plosker GL (2004) Atomoxetine: a review of its use in adults with attention deficit hyperactivity disorder. Drugs 64(2):205–222. https://doi.org/10.2165/00003495-200464020-00005
Abramov Y, Sand PK (2004) Oxybutynin for treatment of urge urinary incontinence and overactive bladder: an updated review. Expert Opin Pharmacother 5(11):2351–2359. https://doi.org/10.1517/14656566.5.11.2351
Sangal RB, Sangal JM, Thorp K (2008) Atomoxetine improves sleepiness and global severity of illness but not the respiratory disturbance index in mild to moderate obstructive sleep apnea with sleepiness. Sleep Med 9(5):506–510. https://doi.org/10.1016/j.sleep.2007.07.013
Levendowski DJ, Zack N, Rao S, Wong K, Gendreau M, Kranzler J, Zavora T, Westbrook PR (2009) Assessment of the test-retest reliability of laboratory polysomnography. Sleep Breath 13:163–167. https://doi.org/10.1007/s11325-008-0214-6
Prasad B, Usmani S, Steffen AD, Van Dongen HP, Pack FM, Strakovsky I, Staley B, Dinges D, Maislin G, Pack AI, Weaver TE (2016) Short-term variability in apnea-hypopnea index during extended home portable monitoring. J Clin Sleep Med 12(6):855–863. https://doi.org/10.5664/jcsm.5886
Miettinen T, Myllymaa K, Westeren-Punnonen S et al (2018) Success rate and technical quality of home polysomnography with self-applicable electrode set in subjects with possible sleep bruxism. IEEE J Biomed Health Inform 22(4):1124–1132. https://doi.org/10.1109/JBHI.2017.2741522
Messineo L, Carter SG, Taranto-Montemurro L, Chiang A, Vakulin A, Adams RJ, Carberry JC, Eckert DJ (2021) Addition of zolpidem to combination therapy with atomoxetine-oxybutynin increases sleep efficiency and the respiratory arousal threshold in obstructive sleep apnoea: a randomized trial. Respirology 26(9):878–886. https://doi.org/10.1111/resp.14110
Funding
This study was funded by Apnimed, Inc. Dr. Sands is supported by the NIH NHLBI (R01HL146697) and American Academy of Sleep Medicine (228-SR-20).
Author information
Authors and Affiliations
Contributions
Concept: S. Sands; data acquisition: P. Schweitzer, J Maynard, P Wylie, H Emsellem; writing–original draft preparation: P. Schweitzer; writing–review and editing: all authors. All authors read and approved the final manuscript. The study was developed and funded by Apnimed, Inc.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by institutional review boards at each site and performed in accordance with the Declaration of Helsinki; all participants provided written informed consent (www.clinicaltrials.gov identifier NCT04445688, 24JUN2020).
Conflict of interest
P Schweitzer has received consultancy fees from Apnimed, Inc. and Jazz Pharmaceuticals; her institution has received research funding from Apnimed, Inc., Avadel, Inspire Medical Systems, Jazz Pharmaceuticals, and Suven Life Sciences.
J Maynard reports no conflicts.
P Wylie reports no conflicts.
H. Emsellem has received research funding from Apnimed, Inc., Jazz Pharmaceuticals, Avadel, NLS, Philips, Suven, Vanda Pharmaceuticals, and Takeda. She has participated in advisory boards for Avadel, Harmony Biosciences, and Takeda.
S Sands has received consulting fees from Apnimed, Nox Medical, Merck, and Inspire, and received grant support from Apnimed, Prosomnus, and Dynaflex.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schweitzer, P.K., Maynard, J.P., Wylie, P.E. et al. Efficacy of atomoxetine plus oxybutynin in the treatment of obstructive sleep apnea with moderate pharyngeal collapsibility. Sleep Breath 27, 495–503 (2023). https://doi.org/10.1007/s11325-022-02634-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11325-022-02634-x