
Abstract
Purpose
(S)-4-(3-[18F]Fluoropropyl)-L-glutamic acid ([18F]FSPG) is an L-glutamate derivative used as a PET biomarker to assess intracellular redox status in vivo through targeting of the cystine/glutamate antiporter protein, xc− transporter. In this report, we describe a radiosynthesis of [18F]FSPG for use in PET studies that address specific challenges in relation to the radiotracer purity, molar activity, and quality control testing methods.
Procedures
The radiosynthesis of [18F]FSPG was performed using a customised RNPlus Research automated radiosynthesis system (Synthra GmbH, Hamburg, Germany). [18F]FSPG was labelled in the 3-fluoropropylmoiety at the 4-position of the glutamic acid backbone with fluorine-18 via substitution of nucleophilic [18F]fluoride with a protected naphthylsulfonyloxy-propyl-L-glutamate derivative. Radiochemical purity of the final product was determined by radio HPLC using a new method of direct analysis using a Hypercarb C18 column.
Results
The average radioactivity yield of [18F]FSPG was 4.2 GBq (range, 3.4–4.8 GBq) at the end of synthesis, starting from 16 GBq of [18F]fluoride at the end of bombardment (n = 10) in a synthesis time of 50 min. The average molar activity and radioactivity volumetric concentration at the end of synthesis were 66 GBq µmol−1 (range, 48–73 GBq µmol−1) and 343–400 MBq mL−1, respectively.
Conclusion
Stability tests using a 4.6 GBq dose with a radioactivity volumetric concentration of 369 MBq mL−1 at the end of synthesis showed no observable radiolysis 3 h after production. The formulated product is of high radiochemical purity (> 95%) and higher molar activity compared to previous methods and is safe to inject into mice up to 3 h after production.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Within oncological PET imaging, 18F-labelled amino acids (AA) are important radiotracers for the detection of tumour proliferation [1]. Perhaps, the most notable is O-(2-[18F]fluoroethyl)-L-tyrosine [2], although, more recently, 18F-analogues of glutamine and glutamic acid have become radiotracers of considerable interest. Examples include [18F]-(2S,4R)4-fluoroglutamine [3,4,5,6,7], [18F](2S,4S)-4-(3-fluoropropyl)glutamine [8], 4-[18F]fluoroglutamic acid [9], and N‐(2‐[18F]fluoropropionyl)‐L‐glutamic acid [10,11,12,13]. (S)-4-(3-[18F]fluoropropyl)-L-glutamic acid ([18F]FSPG) is an L-glutamate derivative used with PET for tumour detection and visualisation through targeting of the cystine/glutamate antiporter protein (xCTor SLC7A11), which is a subunit of the transport system xc −[14,15,16]. In clinical imaging trials, [18F]FSPG has demonstrated utility in the detection of brain, lung, liver, and breast cancers [17, 18]. System xc− is important for maintaining intracellular redox homeostasis and shows increased activity in certain tumour cells compared to normal, healthy cells due to increased metabolic activity in tumour cells [19, 20]. [18F]FSPG has, therefore, gained interest as a PET biomarker to assess intracellular redox status in vivo. In the clinic, [18F]FSPG PET could prove valuable as a biomarker to assess xc− transporter activity in patients with cancer to identify those suitable for oxidant and antioxidant therapies (or assess treatment response to oxidant and antioxidant therapies).
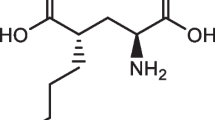
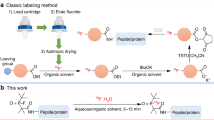
The radiosynthesis of [18F]FSPG (Fig. 1) is typically carried out as a one-pot, two-step synthesis followed by purification and formulation for i.v. injection. In the first step, fluorine-18 incorporation into a protected precursor is achieved via nucleophilic substitution. This is followed by deprotection via acid hydrolysis to yield [18F]FSPG. The first reported methods (Table 1) describe the use of a precursor with a 4-nitrophenylsulphonate, leaving the group as the starting material for [18F]nucleophilic displacement [21, 22]. Acid hydrolysis of O-tert-butyl and N-tert-butoxycarbonyl groups then furnished [18F]FSPG, with radiochemical yields (RCY) from [18F]fluoride varying from 30 to 63% (decay corrected). A more recent method has employed the 2-naphthylsulphonate substituent as the leaving group and O-tert-butyl and N-triphenylmethyl substituents as the protecting groups [17]. For this method, the reported yield of [18F]FSPG from [18F]fluoride was 35–49% (decay corrected). While the radiolabelling and hydrolysis steps are optimised, purification of the final product has proved challenging as several methods have been applied with varied success. One of the earliest methods used a combination of reverse-phase HPLC and solid phase extraction to isolate [18F]FSPG with 93% radiochemical purity (RCP) [15]. Later methods have switched to cartridge purification using a combination of reverse-phase and cation-exchange stationary phases [23, 24], sometimes as part of chemical reagent cassette-based methods [17, 25]. Radiochemical purities using this approach have been as low as 90% [17, 23], although more recently values above 95% have been achieved [25, 26] with molar activities (MA) in the range of 4–48 GBq µmol−1 at the end of synthesis (EOS). Here, we present a non-cassette based method for the radiosynthesis of [18F]FSPG followed by HPLC purification and formulation via solid phase extraction that delivers a high purity product in high molar activity that is suitable for preclinical and clinical PET research.

Synthesis routes to [18F]FSPG including a previously reported methods and b the method and conditions reported in this article.
Materials and Methods
Reagents and Materials
Kryptofix 222, potassium carbonate, 96% sulphuric acid, 37% hydrochloric acid, sodium hydroxide, sodium dihydrogen phosphate dihydrate, sodium chloride, potassium chloride, disodium hydrogen phosphate, potassium dihydrogen phosphate, and acetonitrile were purchased from Merck Life Science UK Ltd, di-tert-butyl-(2S,4S)-2-(3-((naphthalen-2-ylsulfonyl)oxy)propyl)-4-(tritylamino)pentanedioate (FSPG precursor) and (S)-4-(3-fluoropropyl)-L-glutamic acid (FSPG reference) were purchased from ABX-advanced biochemical compounds (Dresden, Germany); methanol, acetonitrile (HPLC grade), and ethanol were purchased from Rathburn Chemicals UK Ltd; QMA SepPakPlus Light cartridges were purchased from Waters UK Ltd; Chromafix PS-H+ (medium tube size) cartridges were purchased from Macherey–Nagel; o-phthalaldehyde (OPA) reagent solution reagent was purchased from Agilent Technologies UK Ltd; 0.9% saline was purchased from the Greater Glasgow & Clyde NHS Pharmacy Distribution Centre. Phosphate buffered saline (PBS), pH 7.4 was prepared as a 1-L stock solution by dissolving sodium chloride (8.00 g/137.0 mmol), potassium chloride (0.20 g/2.7 mmol), disodium hydrogen phosphate (1.44 g/10.0 mmol), and potassium dihydrogen phosphate (0.24 g/1.8 mmol) in 800 mL of water. The resultant solution was adjusted to pH 7.4 with 0.1 N HCl and then made up to a total volume of 1 L by adding water. No-carrier-added [18F]fluoride was obtained through the 18O(p, n)18F nuclear reaction by irradiation of 95–97 atom % oxygen-18-enriched water (purchased from Sercon UK Ltd) in a niobium target chamber (2.7-ml target volume) with a 16.4 MeV proton beam on the GE Healthcare PET trace cyclotron at the West of Scotland PET Centre. Typically, a 22 µA, 15-min target irradiation gave 15.4 GBq (16.0 GBq maximum) of [18F]fluoride for use at the start of a synthesis.
Radio-HPLC Analytical Chromatography
Radio-HPLC analysis was carried out on a DionexUltimate 3000 HPLC system, consisting of an IC-3000 solvent delivery system, a WPS-3000SL-Analyt autosampler, a VWD3100 absorbance detector, a TCC3000SD column compartment, and a BioscanFC-1000 Flow countrate counter coupled to a BioscanFC-3500 PIN diode detector. The system was operated using Chromeleon software (version 6.8).
Radiosynthesis of [18F]FSPG
The fully automated synthesis of [18F]FSPG was carried out on a customised RNPlusSynthra module (Fig. 2) as follows. Cyclotron produced, no-carrier-added [18F]fluoride was passed through a QMA SepPakPlus Light cartridge. The trapped [18F]fluoride was eluted from the cartridge into a reaction vessel using a 1.0-mL aliquot from a stock solution containing Kryptofix 2.2.2 (120 mg, 0.319 mmol)/K2CO3(22 mg, 0.159 mmol)/CH3CN (1 mL)/H2O (14 mL). The resultant mixture was azeotropically dried under a stream of nitrogen and vacuum at 110 °C for 8 min. A further 1 mL of CH3CN was added, and the mixture was dried again under a stream of nitrogen and vacuum at 110 °C for an additional 3 min. An FSPG precursor (3.0 mg) in anhydrous CH3CN (1.0 mL) was added to the dried [18F]fluoride residue, and the mixture was heated at 100 °C for 15 min. The resultant mixture was cooled to 40 °C; then, 1.0-M H2SO4 (1.0 ml) was added, and the reaction mixture was heated at 100 °C for 5 min under open vessel conditions. The resultant mixture was cooled to 40 °C; then, 4.0-M NaOH (0.75 mL) was added, and the reaction mixture was heated at 100 °C for a further 5 min under open-vessel conditions. After cooling to 40 °C, PBS (3.0 mL) was added, and the resultant mixture was injected onto an HPLC column (PhenomenexSynergi Hydro R, 4 micron, length = 150 mm, diameter = 10 mm) and eluted with 1.67% ethanol at a flow rate of 2.0 mL min−1. The eluent passing through the HPLC column was monitored continuously for radioactivity and absorbance at 218 nm. The fraction containing [18F]FSPG (retention time, Rt = 6.5–7.8 min) was collected and stirred in a round bottom flask containing 0.1-M H2SO4 (30 mL). After stirring, the resultant solution was passed through a Chromafix PS-H+ cartridge [conditioned before use by passing ethanol (10 mL) then deionised water (10 mL) through the cartridge]. The cartridge was washed with deionised water (12 mL), and the radioactive product was eluted with PBS (3.0 mL). Finally, the [18F]FSPG formulation was dispensed via a 0.2 micron filter into a sterile vial for preclinical application.

Automated system for the synthesis of [18F]FSPG. Reagent vials contained the following reagents: A1 = kryptofix/K2CO3 solution (1.0 mL), A2 = CH3CN (1.0 mL), B1 = FSPG precursor (3.0 mg) in CH3CN (1.0 mL), B2 = 1.0 M H2SO4 (1.0 mL), B3 = 4.0-M NaOH (0.75 mL), B4 = PBS (3.0 mL), C1 = H2O (6.0 mL), C2 = H2O (6.0 mL), C3 = PBS (3.0 mL).
Quality Control Analysis of [18F]FSPG
Chemical and Radiochemical Purity (Direct Analysis)
[18F]FSPG formulated in PBS (10-µL aliquot) was analysed by HPLC using a Thermo Fisher Hypercarb C18 column (150 × 4.6 mm) eluted at 0.9 mL min−1 with a mixture of 0.1% trifluoroacetic acid and acetonitrile (94:6). The eluate was monitored continuously for radioactivity and absorbance at 218 nm. The retention time for FSPG was 4.6 min. The formulated product gave one radioactive peak with the same retention time (4.6 min) as authentic FSPG. The radiochemical purity was 95–97%. For confirmation, [18F]FSPG formulated in PBS (50 µL) was mixed with a 50-µL sample of a 100-µM reference solution of authentic FSPG. A 10-µL aliquot of the resultant mixture was then analysed by HPLC as described above.
Molar Activity Determination (Analysis Using Pre-Column Derivatisation)
[18F]FSPG formulated in PBS (a 10-μL aliquot) was mixed with OPA reagent solution (10 μL) and stirred for 1 min at room temperature. The resultant sample was analysed by HPLC using a Waters Spherisorb C18, 3-µm column (250 × 4.6 mm) eluted at 1.0 mL min−1 with a mixture of 10-mM NaH2PO4 (pH adjusted to 7.0 using 5.0-M NaOH) and methanol (60:40). The eluate was monitored continuously for radioactivity and absorbance at 338 nm. A sample of authentic FSPG (100 µM, 50 µL) when incubated at room temperature for 1 min with 50 µL of OPA reagent gave a single peak with a retention time of 4.6 min. The derivatised18F-fluorinated product gave one radioactive peak and one stable peak both with the same retention time (4.6 min) as the OPA derivative of authentic FSPG. Typically, the radiochemical purity was 95–97%, and the average molar activity was 66 GBq µmol−1 (range, 44–73 GBq µmol−1) at the end of synthesis (EOS).
Results and Discussion
This work was undertaken to support a preclinical study of the PET imaging characteristics of the transport system xc− using [18F]FSPG in a mouse tumour model. The study requires that samples of [18F]FSPG be prepared on demand and transported off-site to an animal PET scanning facility prior to use. It was, therefore, necessary to have a reliable radiosynthesis method that was able to produce [18F]FSPG in high chemical and radiochemical stability at room temperature. Furthermore, the method had to yield [18F]FSPG in high radioactivity volumetric concentration and molar activity to provide the small volume doses needed for preclinical PET.
The substitution reaction of nucleophilic [18F]fluoride with a naphthylsulfonyloxy-propyl-L-glutamate derivative, containing tert-butyl and trityl-protecting groups at the carboxylic O-hydroxy and N-amino groups, respectively, was used to synthesise [18F]FSPG labelled in the fluoropropylmoiety with fluorine-18 (Fig. 1b). The radiolabelling step was performed in acetonitrile under sealed, thermal conditions. The resultant radiolabelled product was then heated under open-vessel conditions for cleavage of tert-butyl and trityl-protecting groups. While still under open-vessel conditions, the reaction mixture was heated at 100 °C with concentrated sodium hydroxide. This was to degrade or remove any potential volatile by-products of the radiolabelling and deprotection steps, such as tert-butyl alcohol (tert-butanol: boiling point = 83 °C) [27], which might hinder purification of the product. For purification, semi-preparative HPLC was used to isolate [18F]FSPG, while the lipophilic3-(2-naphthylsulfonyl)oxy)propyl precursor was strongly retained on the HPLC column and was later eluted using 50% ethanol (Fig. 3). The [18F]FSPG was collected in a dilute acid solution then trapped by solid phase extraction on a cation exchange cartridge, containing a sorbent with strong anionic functional groups [28]. This ion exchange trapping process requires that [18F]FSPG behaves in aqueous solution as a zwitterion in the same manner as its glutamic acid parent compound [29]. Thus, at low pH, under acidic and aqueous conditions, it exists in a predominantly protonated form with a net charge of + 1 due to a positive charge at the amino group. This allows it to be trapped on the cartridge due to ionic interaction with anionic groups on the cartridge sorbent. On elution of the cartridge with PBS (pH = 7.4), the [18F]FSPG is converted to a deprotonated form with a net charge of − 1 due to deprotonation at both carboxylic acid groups, while the amino group retains a positive charge. In this deprotonated form, it is released from the cartridge into the PBS solution (pH = 7.4) then filtered for use in clinical and preclinical PET imaging studies. Using this method, [18F]FSPG was produced typically in 39–43% radiochemical yield (decay corrected) from [18F]fluoride. After dispensing, the average radioactivity yield of [18F]FSPG as formulated for preclinical use was 4.2 GBq (range, 3.4–4.8 GBq), starting from 16 GBq of [18F]fluoride at the end of bombardment (EOB) (n = 10). The average molar activity was 66 GBq µmol−1 (range, 44–73 GBq µmol−1) at EOS, and the radioactivity volumetric concentration was 343–400 MBq mL−1 at EOS. No chemical impurities were observed in the final product at 218-nm absorbance (Fig. 4).

A typical HPLC chromatogram of [18F]FSPG purification.

Quality control direct analysis of [.18F]FSPG (UV absorbance peaks at 1.0–3.0 min are attributable to PBS, pH 7.4).
Values of [18F]FSPG molar activity at the end of synthesis for previously reported methods (Table 1) are typically between 4 and 48 GBq µmol−1 [16, 17, 24,25,26]. These methods rely on solid phase extraction (SPE) using multiple cation exchange cartridges in series for product purification and isolation. However, SPE in this way can be problematic in terms of poor recovery, reproducibility issues, and sample extracts being insufficiently clean. There is a potential for structurally similar side products to form and co-elute as part of the final formulated product. The authors of these reported methods discuss the challenges of SPE in relation to [18F]FSPG purification [22, 25]. Product purification is challenging because SPE does not involve real-time analysis of the recovered eluate. In the case of the synthesis of [18F]FSPG, which uses harsh acidic and basic reagents, the HPLC purification method employed here offers a more accurate and reproducible way to recover products in high purity and high molar activity. This is important from a PET imaging perspective since [18F]FSPG is used as a transporter radioligand for the transport system xc− [15, 16]. Glutamine and cystine are the endogenous AA targets for this transport system and so [18F]FSPG molar activity must be sufficiently high to avoid saturation of the transporter. Tumour cell uptake studies using L-[5-11C]glutamine with molar activity of 1.85 GBq µmol−1 at EOS have a maximum uptake of 22% in SF188 tumour cells and in dynamic small-animal PET studies using rats, bearing 9L tumor xenografts; it shows barely measurable tumour uptake and retention [30, 31]. By comparison, in the same xenograft model, [18F](2S,4S)-4-(3-Fluoropropyl)glutamine shows good tumour uptake compared to the muscle (background) regions [8]. PET studies using [18F]FSPG at the molar activity reported here could potentially allow improved tumour uptake characteristics.
Amino acids are typically difficult to detect by UV absorbance due to a lack of a suitable UV chromophore. Pre-column derivatisation using OPA Reagent is, therefore, used as a method for indirect HPLC analysis, whereby the amino acid analyte is converted to a UV detectable isoindoline derivative. The method is used for amino acid analysis in biological, industrial, and environmental applications [32,33,34] where a high detection sensitivity is necessary. However, the reagent is expensive, requires careful handling, has a limited shelf life, and, crucially, it may not react with product sample impurities to produce compounds identifiable by UV detection. Despite these drawbacks, the reagent is highly sensitive and reacts with trace amounts of a wide variety of compounds containing primary amino substituents. Using this indirect method, the radiochemical purity (RCP) and molar activity (MA) of the formulated radioactive product were measured at 95–97% and 44–73 GBq µmol−1, respectively, with [18F]fluoride as the only radioactive impurity (Fig. 5). Radiostability over time was tested using an18Fproduct dose of 4.6 GBq with a radioactivity volumetric concentration of 369 MBq mL−1 at EOS. Radioanalyses of the final product at EOS and at 1.5 and 3.0 h after EOS were performed, and, at all 3 time points, the RCP was measured at 95% with [18F]fluoride as the only radioactive impurity. In between radioanalyses, the product was stored under sealed conditions at room temperature. For confirmation, the radiostability analysis was repeated using an alternative method, which allowed direct analysis of [18F]FSPG. The method used a Thermo Fisher Hypercarb C18 column, which has a sufficiently high carbon loading to retain hydrophilic FSPG and allow it to be eluted with a reasonable retention time (Fig. 4). Using this method, the identity of the final product was first confirmed by co-elution with an authentic FSPG reference prior to radiostability testing. The results of the radiostability analysis were the same as those of the indirect method, confirming that the product is suitable for use up to 3-h post-radiosynthesis. The direct analysis method is simpler but has a lower limit of detection for FSPG of ca. 50 µM and, therefore, is not sufficiently sensitive for FSPG analysis at the no-carrier-added level. This simplified procedure could be used for routine radiochemical purity and chemical purity measurements as well as radiostability testing, whereas the indirect method involving analyte derivatisation is useful for determining molar activity.

Quality control indirect analysis of [18F]FSPG using pre-column OPA derivatisation.
Conclusions
FSPG was labelled in the fluoropropylmoiety at the 4-position of the glutamic acid backbone with 18F via the substitution reaction between nucleophilic [18F]fluoride and di-tert-butyl-(2S,4S)-2-(3-((naphthalen-2-ylsulfonyl)oxy)propyl)-4-(tritylamino)pentanedioate. Radiolabelling was achieved by a method utilising semi-preparative reverse-phase HPLC purification and solid-phase extraction. This is in contrast to previously reported methods, employing cartridge purification, which do not allow real-time analysis of the purification process and are, therefore, prone to issues with reproducibility and product separation efficiency. The method developed here delivers a product with improved molar activity at the end of synthesis compared to previously reported methods, which could potentially have better PET imaging characteristics with respect to imaging the amino acid transport system xc−. To simplify the product analysis, we have employed a direct method of radio-HPLC analysis, which avoids the need for pre-column derivatisation of the radiolabelled product using OPA Reagent, an expensive derivatising agent with limited chemical reactivity and a short shelf life. Stability tests using both direct and indirect methods of radioanalysis show that the formulated product is of high radiochemical purity (RCP > 95%), undergoes no observable radiolysis, has an acceptable range of molar activity (MA = 44–73 GBq µmol−1 EOS) and is suitable to inject into mice up to 3 h after production.
References
Qi Y, Liu X, Li J et al (2017) Fluorine-18 labeled amino acids for tumor PET/CT imaging. Oncotarget 8:60581–60588
Heinzel A, Dedic D, Galldiks N et al (2022) Two decades of brain tumour imaging with O-(2-[18F]fluoroethyl)-L-tyrosine PET: the Forschungszentrum Jülich experience. Cancers 14:3336
Qu W, Zha Z, Ploessl K et al (2011) Synthesis of optically pure 4-fluoro-glutamines as potential metabolic imaging agents for tumors. J Am ChemSoc 133:1122–1133
Lieberman BP, Ploessl K, Wang L et al (2011) PET imaging of glutaminolysis in tumors by 18F-(2S,4R)4-fluoroglutamine. J Nucl Med 52:1947–1955
Ploessl K, Wang L, Lieberman BP et al (2012) Comparative evaluation of 18F-labeled glutamic acid and glutamine as tumor metabolic imaging agents. J Nucl Med 53:1616–1624
Valtorta S, Toscani D, Martina Chiu M et al (2021) [18F](2S,4R)-4-fluoroglutamine as a new positron emission tomography tracer in myeloma. Front Oncol 11:760732
Seo Y, Craig MC, Murphy ST et al (2022) [18F]-(2S,4R)4-fluoroglutamine PET imaging of glutamine metabolism in murine models of hepatocellular carcinoma (HCC). Mol Imaging 2022:5185951
Wu Z, Zha Z, Li G, Lieberman BP et al (2014) [18F](2S,4S)-4-(3-fluoropropyl)glutamine as a tumor imaging agent. Mol Pharmaceutics 11:3852–3866
Krasikova RN, Kuznetsova OF, Fedorova OS et al (2011) 4-[18F]fluoroglutamic acid (BAY 85–8050), a new amino acid radiotracer for PET imaging of tumors: synthesis and in vitro characterization. J Med Chem 54:406–410
Hu K, Du K, Tang G et al (2014) Radiosynthesis and biological evaluation of N-[18F]labeled glutamic acid as a tumor metabolic imaging tracer. PLoS One 9:e93262
Sun A, Liu S, Tang X et al (2017) Simple and rapid radiosynthesis of N-18F-labeled glutamic acid as a hepatocellular carcinoma PET tracer. Nucl Med Biol 49:38–43
Liu S, Sun A, Zhang Z et al (2017) Automated synthesis of N-(2-[18F]fluoropropionyl)-L-glutamic acid as an amino acid tracer for tumor imaging on a modified [18F]FDG synthesis module. J Label CompdRadiopharm 60:331–336
Sun A, Liu S, Tang X et al (2021) N-(2–18F-fluoropropionyl)-L-glutamate as a potential oncology tracer for PET imaging of glioma. ApplRadiat Isotopes 168:109530
Smolarz K, Krause BJ, Graner FP et al (2013) (S)-4-(3–18F-fluoropropyl)-L-glutamic acid: an 18F-labeled tumor-specific probe for PET/CT imaging—dosimetry. J Nucl Med 54:861–866
Domercq M, Szczupak B, Gejo J et al (2016) PET imaging with [18F]FSPG evidences the role of system xc- on brain inflammation following cerebral ischemia in rats. Theranostics 6:1753–1767
Hoehne A, James ML, Alam IS et al (2018) [18F]FSPG-PET reveals increased cystine/glutamate antiporter (xc-) activity in a mouse model of multiple sclerosis. J Neuroinflammation 15:55
Kavanaugh G, Williams J, Morris AS et al (2016) Utility of [18F]FSPG PET to image hepatocellular carcinoma: first clinical evaluation in a US population. Mol Imaging Biol 18:924–934
Baek S, Mueller A, Lim YS et al (2013) (4S)-4-(3–18F-fluoropropyl)-L-glutamate for imaging of xC transporter activity in hepatocellular carcinoma using PET: preclinical and exploratory clinical studies. J Nucl Med 54:117–123
Muir A, Danai LV, Gui DY et al (2017) Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. eLife 6:e27713
Lyons SA, Chung WJ, Weaver AK et al (2007) Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res 67:9463–9471
Koglin N, Mueller A, Berndt M et al (2011) Specific PET imaging of xC- transporter activity using a 18F-labeled glutamate derivative reveals a dominant pathway in tumor metabolism. Clin Cancer Res 17:6000–6011
Berndt M, Schmitt-Willich H, Friebe M et al (2011) Method for production of F-18-labeled glutamic acid derivatives (Bayer Schering Pharma Aktiengesellschaft). WO 2011/060887.
Baek S, Choi CM, Ahn SH et al (2012) Exploratory clinical trial of (4S)-4-(3-[18F]fluoropropyl)-L-glutamate for imaging xC- transporter using positron emission tomography in patients with non-small cell lung or breast cancer. Clin Cancer Res 18:5427–5437
Shih KT, Huang YY, Yang CY et al (2020) Synthesis and analysis of 4-(3-fluoropropyl)- glutamic acid stereoisomers to determine the stereochemical purity of (4S)-4-(3-[18F] fluoropropyl)-L-glutamic acid ([18F]FSPG) for clinical use. PLoS One 15:e0243831
Edwards R, Greenwood HE, McRobbie G et al (2021) Robust and facile automated radiosynthesis of [18F]FSPG on the GE FASTlab. Mol Imaging Biol 23:854–864
McCormick PN, Greenwood HE, Glaser M et al (2019) Assessment of tumor redox status through (S)-4-(3-[18F]fluoropropyl)-L-glutamic acid positron emission tomography imaging of system xc- activity. Cancer Res 79:853–863
Haynes WM (ed.). (n.d) CRC handbook of chemistry and physics. 94th Edition. CRC Press LLC, Boca Raton: FL 2013–2014, p. 3–392
Fontanals N, Cormack PAG, Marcé RM, Borrull F (2010) Mixed-mode ion-exchange polymeric sorbents: dual-phase materials that improve selectivity and capacity. Trends Anal Chem 29:765–779
Dietzen DJ (2018) Amino acids, peptides, and proteins. In: Rifai N, Horvath AR, Wittwer CT (eds) Principles and applications of molecular diagnostics. Elsevier, 345–380.
Qu W, Oya S, Lieberman BP et al (2012) Preparation and characterization of l-[5-11C]-glutamine for metabolic imaging of tumors. J Nucl Med 53:98–105
Gleede T, Riehl B, Shea C et al (2015) Investigation of SN2 [11C]cyanation for base-sensitive substrates: an improved radiosynthesis of l-[5-11C]-glutamine. Amino Acids 47:525–533
Deyl Z, Hyanek J, Horakova M (1986) Profiling of amino acids in body fluids and tissues by means of liquid chromatography. J Chromatog 379:177–250
Schuster R (1988) Determination of amino acids in biological, pharmaceutical, plant and food samples by automated precolumn derivatization and high-performance liquid chromatography. J Chromatog 431:271–284
Qureshi GA, Qureshi AR (1989) Determination of free amino acids in biological samples: problems of quantitation. J Chromatog 491:281–289
Acknowledgements
We thank the team at the Radiopharmaceutical Unit of the Glasgow West of Scotland PET Centre for their generous support with cyclotron operations. We would like to thank the Advanced Technologies at the Cancer Research UK Beatson Institute, with particular thanks to Translational Molecular Imaging at the CRUK Beatson Institute.
Funding
We would like to acknowledge funding from Cancer Research UK for core funding to the CRUK Beatson Institute (A13827) and to David Lewis’ laboratory (A25006) and from the Beatson Cancer Charity and Beatson Endowment.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brown, G., Soloviev, D. & Lewis, D.Y. Radiosynthesis and Analysis of (S)-4-(3-[18F]Fluoropropyl)-L-Glutamic Acid. Mol Imaging Biol 25, 586–595 (2023). https://doi.org/10.1007/s11307-022-01793-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11307-022-01793-3