
Abstract
The human equilibrative nucleoside transporter 1 (SLC29A1, hENT1) is a solute carrier that modulates the passive transport of nucleosides and nucleobases, such as adenosine. This nucleoside regulates various physiological processes, such as vasodilation and -constriction, neurotransmission and immune defense. Marketed drugs such as dilazep and dipyridamole have proven useful in cardiovascular afflictions, but the application of hENT1 inhibitors can be beneficial in a number of other diseases. In this study, 39 derivatives of dilazep’s close analogue ST7092 were designed, synthesized and subsequently assessed using [3H]NBTI displacement assays and molecular docking. Different substitution patterns of the trimethoxy benzoates of ST7092 reduced interactions within the binding pocket, resulting in diminished hENT1 affinity. Conversely, [3H]NBTI displacement by potentially covalent compounds 14b, 14c, and 14d resulted in high affinities (Ki values between 1.1 and 17.5 nM) for the transporter, primarily by the ability of accommodating the inhibitors in various ways in the binding pocket. However, any indication of covalent binding with amino acid residue C439 remained absent, conceivably as a result of decreased nucleophilic residue reactivity. In conclusion, this research introduces novel dilazep derivatives that are active as hENT1 inhibitors, along with the first high affinity dilazep derivatives equipped with an electrophilic warhead. These findings will aid the rational and structure-based development of novel hENT1 inhibitors and pharmacological tools to study hENT1's function, binding mechanisms, and its relevance in (patho)physiological conditions.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The in- and efflux of nucleosides, nucleobases and nucleoside-derived xenobiotics in mammalian cells is primarily regulated by the membrane-bound nucleoside transporters [1, 2]. As part of the protein superfamily of solute carriers (SLCs), these transporters can be further characterized in the sodium-dependent concentrative nucleoside transporters (SLC28) and the sodium-independent equilibrative transporters (ENTs, SLC29) [3]. Of the four defined isoforms belonging to the subfamily of ENTs (ENT1, ENT2, ENT3 and ENT4), ENT1 is most prevalent and widely distributed amongst different cell types such as cardiovascular cells and various neuronal tissues in the central nervous system [4,5,6]. Exemplary of the physiological relevance of ENT1 is the recent finding that knockdown or inhibition of ENT1-mediated inosine reuptake was identified as a mechanism to promote brown adipocyte differentiation and therefore cardiometabolic health [7]. In addition to inosine, reuptake of its precursor adenosine is also ENT1-mediated, making the transporter an important regulator of extracellular adenosine concentrations [1]. As a result, inhibition of adenosine reuptake leads to altered downstream signaling of pathways implicated with adenosine receptors. Through this indirect effect on adenosine receptor signaling, multiple physiological events such as vasodilation and neurotransmission are associated with ENT1 function [2]. For instance, ENT1 inhibition, and thus modulation of adenosine homeostasis, is directly linked to mitigation of Tau pathologies, one of the major pathogenic hallmarks in Alzheimer’s disease [8]. Furthermore, as uptake of anticancer and antiviral drugs such as gemcitabine and ribavirin is mediated by hENT1, its expression levels are directly correlated to the efficacy of these nucleoside-derived therapies [9, 10]. In short, due to its involvement in the transport of nucleosides and closely related analogues, ENT1 inhibition is a relevant therapeutic strategy to combat various pathophysiological conditions.
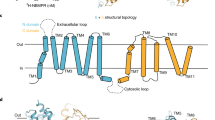
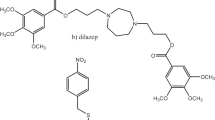
In the context of cardiac implications like ischemia and hypertension, direct inhibition of ENT1 to increase extracellular adenosine levels has been studied as a therapeutic strategy [5]. To this end, multiple efforts have been made to develop adenosine reuptake inhibitors, resulting in structure–activity relationships of various chemically diverse molecules like NBTI, draflazine and the marketed antithrombotic agents dipyridamole and dilazep (Fig. 1) [12]. In addition to classical characterization of the inhibitory potency, kinetic profiles have been established [11, 13] and multiple ENT1 molecular tools (fluorescent [14] and photoaffinity probes [15]) have been developed to study the function and structure of the transporter. With the use of X-ray crystallography, two hENT1 crystal structures were elucidated using a biochemically stable variant of the protein in complex with nucleoside-derived inhibitor NBTI and non-nucleoside inhibitor dilazep (Fig. 1) [16]. Dilazep, a symmetrical cycloalkyldiamine, which is clinically used as a vasodilator, is structurally different compared to the well investigated scaffolds such as NBTI and dipyridamole. In spite of the aforementioned crystal structure and earlier efforts investigating dilazep derivatives [17] and close analogues hexobendine and ST7092 (Fig. 1) [18], little is known about the structure–activity relationship or the binding mechanisms of these non-nucleoside ENT1 inhibitors. In this study, we report the synthesis and pharmacological evaluation of 39 new dilazep-like derivatives based on the structure of ST7092 (Fig. 1) to explore the chemical diversity tolerated in the hENT1 binding pocket. 17 of these derivatives were equipped with an electrophilic warhead to attempt irreversible interaction with amino acid residue C439 of the binding pocket. Additionally, predicted binding modes of several inhibitors were examined with the use of molecular docking to gain insight into the different binding profiles characterized by [3H]NBTI displacement assays. The binding affinity characterization and predicted binding mode investigation of these new non-nucleoside hENT1 inhibitors may contribute to a better understanding of the hENT1 binding pocket and will aid in the further development of non-nucleoside-derived molecular tools to study the transporters’ function and binding mechanisms.

Chemical structures of reported hENT1 (therapeutic) inhibitors dipyridamole and dilazep, and molecular tools NBTI, draflazine and ST7092 and their corresponding Ki values. Ki values are as previously described, with the exception of ST7092 (data unpublished) [11]
Materials and methods
Chemistry
General chemistry
All solvents and reagents were purchased from commercial sources and were of analytical grade. Demineralized water is referred to as H2O, as was used in all cases unless noted otherwise (e.g., brine). All reactions were routinely monitored with thin-layer chromatography (TLC), using aluminum silica gel coated 60 F254 plates from Merck (Darmstadt, Germany) and visualized by UV irradiation at 254 nm or staining with ninhydrin or KMnO4 solution. Purification by flash column chromatography was carried out with the use of silica gel irregular ZEOprep® particles (60–200 µm) from VWR (Amsterdam, The Netherlands) or using an Isolera™ One or Selekt automatic flash chromatography system from Biotage® (Uppsala, Sweden) with pre-packed cartridges (Phenomenex (Torrance, CA, USA) Gemini® Claricep™ (silica) or Biotage® Sfär C18 D Duo 100 Å 30 µm (C18)). Solutions were concentrated using a Heidolph (Schwabach, Germany) Hei-VAP Value rotary evaporator. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker (Billerica, MA, USA) AV-400 liquid spectrometer (1H NMR, 400 MHz and 13C NMR, 101 MHz) at ambient temperature and subsequently analyzed with MestReNova v14.1.0 software (Mestrelab Research S.L., Santiago de Compostela, Spain). Chemical shifts are reported in parts per million (ppm), designated by δ and corrected to the internal standard tetramethylsilane (δ = 0). Multiplicities are indicated by s, singlet; d, doublet; dd, doublet of doublets; ddd, doublet of doublet of doublets; td, triplet of doublets; t, triplet; dt, doublet of triplets; tt, triplet of triplets; q, quartet; p, pentet; m, multiplet; br s, broad singlet; br t, broad triplet. Coupling-constants (J) are reported in Hz. Mass and compound purity analyses were performed with liquid chromatography-mass spectrometry (LC–MS) using a LCMS-2020 system from Shimadzu (Kyoto, Japan) coupled to a Phenomenex Gemini® C18 110 Å column (50 mm × 3 mm × 3 μm). Samples were prepared by dissolving 0.3–0.8 mg of compound in 1 mL of a 1:1:1 mixture of CH3CN/H2O/tBuOH and were eluted using an isocratic system of H2O/CH3CN with 0.1% FA, using gradients from 100:0 to 60:40 and 90:10 to 10:90 in an elution time of 15 min. All tested compounds were determined to be of > 95% purity determined by HPLC.
Synthetic procedures
General procedure A:
To a stirred solution of the appropriate commercially available methylaminobenzoic acid (453 mg, 3.00 mmol, 1.0 equiv) in 1,4-dioxane (7.5 mL), was added dropwise Fmoc-Cl (0.72 mL, 3.60 mmol, 1.2 equiv). The mixture stirred for 10 min at 0 °C under N2 atmosphere followed by the addition of a suspension of K2CO3 (1.66 g, 12.0 mmol, 4.0 equiv) in 1,4-dioxane (7.5 mL) and the reaction mixture was allowed to stir at rt for 41 h. Subsequently, the mixture was diluted with H2O and neutralized to pH 7 with 0.5 M aqueous HCl. The aqueous phase was extracted with DCM (2 × 100 mL) and the combined organic phase was dried over MgSO4, filtrated and concentrated in vacuo. Flash column chromatography on silica gel using a gradient of 2 to 4% MeOH in DCM as mobile phase provided intermediates 1a and 1b.
General procedure B:
To a stirred solution of commercially available substituted benzoyl chloride (1.2 equiv) in DCM or 1,4-dioxane (0.2 M) at 0 °C under N2 atmosphere was added dropwise the appropriate bromoalcohol (1.0 equiv) and Et3N (2.0 equiv). The reaction mixture stirred for 0.5 h at 0 °C and overnight at rt. Subsequently, the mixture was quenched with H2O, diluted with DCM and the phases were separated. The aqueous phase was extracted with DCM (two times) and the combined organic phase was washed with brine, dried over MgSO4, filtrated and concentrated in vacuo. Flash column chromatography on silica gel using a gradient of EtOAc in PE as mobile phase provided intermediates 2a-2b, 2d-2f, 2h, 2k-2l and 2p.
General procedure C:
A stirred mixture of the appropriate substituted benzoic acid (1.2 equiv) and thionyl chloride (0.2 M) was refluxed at 75 °C for 4 h under N2 atmosphere. After cooling down to rt the thionyl chloride was evaporated in vacuo and the crude benzoyl chloride was used as described in general procedure B to provide intermediates 2c, 2g, 2i-2j, 2m and 8a-8e.
General procedure D:
To a stirred solution of the appropriate substituted benzoic acid (1.0 equiv) in DCM (0.2 M) was added DMAP (cat.) and EDC·HCl (2.0 equiv). The mixture stirred for 1.5 h at rt before 3-bromopropan-1-ol (1.2 equiv) was added and the reaction continued at rt overnight under N2 atmosphere. The mixture was diluted with DCM and washed with H2O (two times). The aqueous phase was extracted with DCM and the combined organic phase was dried over MgSO4, filtrated and concentrated in vacuo. Flash column chromatography on silica gel using a gradient of EtOAc in PE as mobile phase provided intermediates 2n-2o and 12a-12b.
General procedure E:
To a stirred solution of the appropriate bromopropyl benzoate (1.0 equiv) in anhydrous DMF (0.2 M) were added 1-Boc-piperazine (1.2 equiv) and K2CO3 (2.0 equiv). The reaction mixture was stirred for 72 h at room temperature (rt) under N2 atmosphere followed by addition of H2O and EtOAc. After separation, the organic phase was washed four times with H2O, once with brine, dried over MgSO4, filtrated and concentrated in vacuo. Flash column chromatography on silica gel using a gradient of MeOH in DCM as mobile phase provided intermediates 3a-3c. After NMR and LC–MS analyses, the formed intermediate was dissolved in DCM (0.2 M) and allowed to cool down to 0 °C after which TFA (20 equiv) was added dropwise. The mixture was stirred for 4 h, concentrated in vacuo and co-evaporated with toluene to remove excess TFA. The obtained oil was dissolved in EtOAc and subsequently co-evaporated with 4 N HCl in 1,4-dioxane to obtain intermediates 4a-4c as dihydrochloride salts.
General procedure F:
The appropriate bromopropyl benzoate (1.0 equiv), appropriate intermediate 4 (1.2 equiv) and K2CO3 (4.0 equiv) were dissolved in anhydrous DMF (0.2 M). The reaction mixture stirred for 68 h at rt under N2 atmosphere followed by 4 h at 50 °C. EtOAc and H2O were added and after separation the organic phase was washed four times with H2O, once with brine. The organic phase was dried over MgSO4, filtrated and concentrated in vacuo. Flash column chromatography on silica gel using a gradient of MeOH in DCM as mobile phase provided final compounds 6a-6k, 6u-6v, 9a, 9c-9f and intermediates 9h-9m, 13a-13b.
General procedure G:
The appropriate bromopropyl benzoate (3.0 equiv), piperazine (1.0 equiv) and K2CO3 (4.0 equiv) were dissolved in anhydrous DMF (0.2 M). The reaction mixture stirred for 68 h at rt under N2 atmosphere followed by 4 h at 50 °C. EtOAc and H2O were added and after separation the organic phase was washed four times with H2O, once with brine. The organic phase was dried over MgSO4, filtrated and concentrated in vacuo. Flash column chromatography on silica gel using a gradient of MeOH in DCM as mobile phase provided final compounds 6p-6t.
General procedure H:
To a stirred solution of intermediate 5 (1.0 equiv) in DCM (0.2 M) at 0 °C was added dropwise the commercially available substituted benzoyl chloride (1.1 equiv) and Et3N (3.0 equiv). The reaction mixture stirred for 1 h gradually warming up to rt under N2 atmosphere. Subsequently, the mixture was quenched with H2O, diluted with DCM and the phases were separated. The aqueous phase was extracted with DCM (two times) and the combined organic phase was washed with brine, dried over MgSO4, filtrated and concentrated in vacuo. Flash column chromatography on silica gel using a gradient of MeOH in DCM as mobile phase provided final compounds 6l and 6m.
General procedure I:
A stirred mixture of the appropriate substituted benzoic acid (1.2 equiv) and thionyl chloride (0.2 M) was refluxed at 75 °C for 4 h under N2 atmosphere. After cooling down to rt the thionyl chloride was evaporated in vacuo and the crude benzoyl chloride was used as described in general procedure H to provide final compounds 6n, 6o and 9b.
General procedure J:
To a stirred solution of the appropriate commercially available chlorosulfonylbenzoic acid (1.0 equiv) in 1,4-dioxane (0.4 M) was added dropwise an aqueous solution of KHF2 (3.0 equiv, 2 M). The mixture was stirred at rt for 1 h followed by dilution with EtOAc. The organic phase was washed with H2O, dried over MgSO4, filtrated and concentrated in vacuo to provide intermediates 7a and 7b.
General procedure K:
To a stirred mixture of the appropriate commercially available amino-substituted benzoic acid (1.0 equiv) in 1,4-dioxane and H2O (1:1 or 2:1) was added Et3N or NaOH (2.0 equiv) followed by slow addition of Boc2O (2.0 equiv). The reaction mixture was stirred for 17 h at rt and subsequently acidified by addition of 1 M aqueous HCl until no further precipitation was observed. The formed suspension was filtrated, the residue rinsed with H2O and subsequently dissolved in EtOH followed by concentration in vacuo to give intermediates 7d-7h.
General procedure L:
A stirred mixture of the appropriate substituted benzoic acid (1.2 equiv) in DCM or toluene (0.2 M) was allowed to cool down to 0 °C. Subsequently, oxalyl chloride (2.0 equiv) and two drops of DMF (cat.) were added and the reaction mixture was stirred for 4 h at 0 °C under N2 atmosphere. The solvent was evaporated in vacuo and the crude benzoyl chloride was used as described in general procedure B to provide intermediates 8f-8k.
General procedure M:
To a stirred solution of 4-hydroxy-3,5-dimethoxybenzoic acid (1.0 equiv) in THF (0.2 M) at 0 °C was added tetrabutylammonium hydroxide (2.0 equiv) and the appropriate bromoalkylamine (1.5 equiv). The reaction mixture was stirred for two days under N2 atmosphere followed by concentration in vacuo. The obtained residue was dissolved in water and 4 M aqueous HCl was added until pH 4 was reached. The aqueous solution was extracted twice with DCM. The combined organic phase was dried over MgSO4, filtrated and concentrated in vacuo. Flash column chromatography on silica gel using a gradient of MeOH in DCM as mobile phase provided intermediates 11a and 11b.
General procedure N:
The appropriate Boc-protected intermediate was dissolved in DCM (0.2 M) and allowed to cool down to 0 °C after which TFA (20 equiv) was added dropwise. The mixture was stirred for 4 h, concentrated in vacuo and co-evaporated with toluene to remove the excess TFA. The obtained oil was dissolved in DCM (0.2 M) and the stirred solution was cooled to 0 °C. Acryloyl chloride (1.5 equiv) was added followed by Et3N (3.0 equiv), and the reaction mixture stirred for 2 h at 0 °C under N2 atmosphere. The mixture was diluted with DCM and subsequently washed twice with brine. The organic phase was dried over MgSO4, filtrated and concentrated in vacuo. Flash column chromatography on silica gel using a gradient of MeOH in DCM as mobile phase provided final compounds 10a-10f and 14a-14b.
3-((((9H-fluoren-9-yl)methoxy)carbonyl)(methyl)amino)benzoic acid (1a). Intermediate 1a was obtained from 3-(methylamino)benzoic acid as a white solid (312 mg, 0.83 mmol, 28%) following general procedure A. 1H NMR (400 MHz, CDCl3) δ 10.59 (br s, 1H), 8.04 – 7.93 (m, 2H), 7.68 (d, J = 7.6 Hz, 2H), 7.45 – 6.90 (m, 8H), 4.46 (d, J = 6.7 Hz, 2H), 4.12 (s, 1H), 3.31 (s, 3H). LC–MS (ESI +) m/z calcd. for C23H19NO4 [(M + H)]+: 374.14; found: 374.10. HPLC tR: 10.989 min.
4-((((9H-fluoren-9-yl)methoxy)carbonyl)(methyl)amino)benzoic acid (1b). Intermediate 1b was obtained from 4-(methylamino)benzoic acid as a white solid (428 mg, 1.15 mmol, 38%) following general procedure A. 1H NMR (400 MHz, CDCl3) δ 8.15 – 8.01 (m, 2H), 7.80 – 7.69 (m, 2H), 7.48 – 7.33 (m, 4H), 7.30 – 7.11 (m, 4H), 4.53 (d, J = 6.3 Hz, 2H), 4.18 (t, J = 6.4 Hz, 1H), 3.32 (s, 3H). LC–MS (ESI +) m/z calcd. for C23H19NO4 [(M + H)]+: 374.14; found: 374.10. HPLC tR: 10.922 min.
3-bromopropyl benzoate (2a). Intermediate 2a was obtained from benzoyl chloride following general procedure B. Column chromatography with 3% EtOAc in PE as mobile phase. Transparent oil (520 mg, 2.14 mmol, 86%). 1H NMR (400 MHz, CDCl3) δ 8.04 – 7.99 (m, 2H), 7.55 – 7.48 (m, 1H), 7.43 – 7.36 (m, 2H), 4.41 (t, J = 6.1 Hz, 2H), 3.50 (t, J = 6.6 Hz, 2H), 2.25 (p, J = 6.3 Hz, 2H). LC–MS (ESI +) m/z calcd. for C10H11BrO2 [(M + H)]+: 243.00; found: 242.90. HPLC tR: 10.765 min.
3-bromopropyl 4-methylbenzoate (2b). Intermediate 2b was obtained from 4-methylbenzoyl chloride following general procedure B. Column chromatography with 0 to 4% EtOAc in PE as mobile phase. Transparent oil (556 mg, 2.16 mmol, 86%). 1H NMR (400 MHz, CDCl3) δ 7.96 – 7.90 (m, 2H), 7.26 – 7.21 (m, 2H), 4.45 (t, J = 6.0 Hz, 2H), 3.55 (t, J = 6.6 Hz, 2H), 2.41 (s, 3H), 2.31 (p, J = 6.6, 6.2 Hz, 2H). LC–MS (ESI +) m/z calcd. for C11H13BrO2 [(M + H)]+: 257.02; found: 256.95. HPLC tR: 11.284 min.
3-bromopropyl 3,4-dimethybenzoate (2c). Intermediate 2c was obtained from 3,4-dimethybenzoic acid following general procedure C. Column chromatography with 20 to 50% DCM in PE as mobile phase. Transparent oil (513 mg, 1.89 mmol, 76%). 1H NMR (400 MHz, CDCl3) δ 7.78 (d, J = 1.9 Hz, 1H), 7.75 (dd, J = 7.9, 1.9 Hz, 1H), 7.16 (d, J = 7.8 Hz, 1H), 4.41 (t, J = 6.0 Hz, 2H), 3.53 (t, J = 6.6 Hz, 2H), 2.33 – 2.23 (m, 8H). LC–MS (ESI +) m/z calcd. for C12H15BrO2 [(M + H)]+: 271.03; found: 270.95. HPLC tR: 11.974 min.
3-bromopropyl 3,5-dimethylbenzoate (2d). Intermediate 2d was obtained from 3,5-dimethylbenzoyl chloride following general procedure B. Column chromatography with 0 to 5% EtOAc in PE as mobile phase. Transparent oil (871 mg, 3.21 mmol, quant.). 1H NMR (400 MHz, CDCl3) δ 7.67 – 7.60 (m, 2H), 7.17 – 7.11 (m, 1H), 4.41 (t, J = 6.0 Hz, 2H), 3.52 (t, J = 6.6 Hz, 2H), 2.38 – 2.31 (m, 6H), 2.27 (p, J = 6.3 Hz, 2H). LC–MS (ESI +) m/z calcd. for C12H15BrO7 [(M + H)]+: 271.03; found: 270.95. HPLC tR: 12.128 min.
3-bromopropyl 4-chlorobenzoate (2e). Intermediate 2e was obtained from 4-chlorobenzoyl chloride following general procedure B. Column chromatography with 2 to 5% EtOAc in PE as mobile phase. Transparent oil (765 mg, 2.75 mmol, quant.). 1H NMR (400 MHz, CDCl3) δ 7.95 (d, J = 8.6 Hz, 2H), 7.39 (d, J = 8.6 Hz, 2H), 4.45 (t, J = 6.1 Hz, 2H), 3.54 (t, J = 6.5 Hz, 2H), 2.31 (p, J = 6.3 Hz, 2H). LC–MS (ESI +) m/z calcd. for C10H10BrClO2 [(M + H)]+: 276.96; found: 276.85. HPLC tR: 11.559 min.
3-bromopropyl 3,4-dichlorobenzoate (2f). Intermediate 2f was obtained from 3,4-dichlorobenzoyl chloride following general procedure B. Column chromatography with 2 to 4% EtOAc in PE as mobile phase. Yellow oil (703 mg, 2.25 mmol, 90%). 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 2.0 Hz, 1H), 7.86 (dd, J = 8.4, 2.0 Hz, 1H), 7.53 (d, J = 8.3 Hz, 1H), 4.48 (t, J = 6.1 Hz, 2H), 3.54 (t, J = 6.5 Hz, 2H), 2.33 (p, J = 6.4 Hz, 2H).
3-bromopropyl 3-(dimethylamino)benzoate (2g). Intermediate 2g was obtained from 3-(dimethylamino)benzoic acid following general procedure C. Column chromatography with 20 to 50% DCM in PE as mobile phase. Transparent oil (211 mg, 0.74 mmol, 21%). 1H NMR (400 MHz, CDCl3) δ 7.42 – 7.34 (m, 2H), 7.28 (t, J = 7.9 Hz, 1H), 6.91 (ddd, J = 8.3, 2.8, 1.1 Hz, 1H), 4.45 (t, J = 6.0 Hz, 2H), 3.54 (t, J = 6.6 Hz, 2H), 2.98 (s, 6H), 2.31 (p, J = 6.4 Hz, 2H). LC–MS (ESI +) m/z calcd. for C12H16BrNO2 [(M + H)]+: 286.04; found: 285.95. HPLC tR: 10.793 min.
3-bromopropyl 4-(dimethylamino)benzoate (2h). Intermediate 2h was obtained from 4-(dimethylamino)benzoyl chloride following general procedure B. Column chromatography with 5 to 25% EtOAc in PE as mobile phase. Yellow oil (488 mg, 1.71 mmol, 68%). 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 9.1 Hz, 2H), 6.62 (d, J = 9.1 Hz, 2H), 4.38 (t, J = 6.0 Hz, 2H), 3.53 (t, J = 6.7 Hz, 2H), 3.01 (s, 6H), 2.27 (p, J = 6.3 Hz, 2H). LC–MS (ESI +) m/z calcd. for C12H16BrNO2 [(M + H)]+: 286.04; found: 285.90. HPLC tR: 11.085 min.
3-bromopropyl 3-((((9H-fluoren-9-yl)methoxy)carbonyl)(methyl)amino)benzoate (2i). Intermediate 2i was obtained from 1a following general procedure C. Column chromatography with 10 to 25% EtOAc in PE as mobile phase. Yellow oil (297 mg, 0.71 mmol, 84%). 1H NMR (400 MHz, CDCl3) δ 7.95 – 7.86 (m, 2H), 7.69 (d, J = 7.6 Hz, 2H), 7.48 – 7.04 (m, 8H), 4.53 – 4.34 (m, 4H), 4.12 (s, 1H), 3.50 (t, J = 6.5 Hz, 2H), 3.30 (s, 3H), 2.29 (p, J = 6.3 Hz, 2H). LC–MS (ESI +) m/z calcd. for C26H24BrNO4 [(M + H)]+: 494.10; found: 494.10. HPLC tR: 12.951 min.
3-bromopropyl 4-((((9H-fluoren-9-yl)methoxy)carbonyl)(methyl)amino)benzoate (2j). Intermediate 2j was obtained from 1b following general procedure C. Column chromatography with 0 to 25% EtOAc in PE as mobile phase. Yellow oil (422 mg, 0.85 mmol, 29%). 1H NMR (400 MHz, CDCl3) δ 8.01 – 7.95 (m, 2H), 7.76 – 7.69 (m, 2H), 7.46 – 7.33 (m, 4H), 7.28 – 7.16 (m, 4H), 4.61 – 4.46 (m, 4H), 4.16 (t, J = 6.5 Hz, 1H), 3.57 (t, J = 6.5 Hz, 2H), 3.30 (s, 3H), 2.34 (p, J = 6.3 Hz, 2H). LC–MS (ESI +) m/z calcd. for C26H24BrNO4 [(M + H)]+: 494.10; found: 494.10. HPLC tR: 12.978 min.
3-bromopropyl 2-methoxybenzoate (2k). Intermediate 2k was obtained from 2-methoxybenzoyl chloride following general procedure B. Column chromatography with 5 to 15% EtOAc in PE as mobile phase. Transparent oil (652 mg, 2.39 mmol, 95%). 1H NMR (400 MHz, CDCl3) δ 7.77 (dd, J = 8.0, 1.8 Hz, 1H), 7.45 (ddd, J = 7.9, 7.1, 1.8 Hz, 1H), 7.01 – 6.91 (m, 2H), 4.41 (t, J = 6.0 Hz, 2H), 3.86 (s, 3H), 3.55 (t, J = 6.6 Hz, 2H), 2.26 (p, J = 6.3 Hz, 2H). LC–MS (ESI +) m/z calcd. for C11H13BrO3 [(M + H)]+: 273.01; found: 272.90. HPLC tR: 10.530 min.
3-bromopropyl 3-methoxybenzoate (2l). Intermediate 2l was obtained from 3-methoxybenzoyl chloride following general procedure B. Column chromatography with 5 to 15% EtOAc in PE as mobile phase. Transparent oil (1.27 g, 4.66 mmol, 78%). 1H NMR (400 MHz, CDCl3) δ 7.63 – 7.60 (m, 1H), 7.54 (dd, J = 2.8, 1.5 Hz, 1H), 7.33 (t, J = 8.0 Hz, 1H), 7.09 (ddd, J = 8.2, 2.7, 1.0 Hz, 1H), 4.44 (t, J = 6.1 Hz, 2H), 3.83 (s, 3H), 3.53 (t, J = 6.6 Hz, 2H), 2.30 (p, J = 6.3 Hz, 2H). LC–MS (ESI +) m/z calcd. for C11H13BrO3 [(M + H)]+: 273.01; found: 272.90. HPLC tR: 11.110 min.
3-bromopropyl 4-methoxybenzoate (2m). Intermediate 2m was obtained from 4-methoxybenzoic acid following general procedure C. Column chromatography with 2 to 8% EtOAc in PE as mobile phase. Yellow oil (1.15 g, 4.21 mmol, 84%). 1H NMR (400 MHz, CDCl3) δ 8.02 – 7.96 (m, 2H), 6.95 – 6.89 (m, 2H), 4.44 (t, J = 6.0 Hz, 2H), 3.87 (s, 3H), 3.55 (t, J = 6.6 Hz, 2H), 2.31 (p, J = 6.2 Hz, 2H). LC–MS (ESI +) m/z calcd. for C11H13BrO3 [(M + H)]+: 273.01; found: 272.95. HPLC tR: 10.938 min.
3-bromopropyl 3,4-dimethoxybenzoate (2n). Intermediate 2n was obtained from 3,4-dimethoxybenzoic acid following general procedure D. Column chromatography with 10 to 20% EtOAc in PE as mobile phase. Transparent oil (513 mg, 1.69 mmol, 28%). 1H NMR (400 MHz, CDCl3) δ 7.68 (dd, J = 8.4, 2.0 Hz, 1H), 7.54 (d, J = 2.0 Hz, 1H), 6.89 (d, J = 8.4 Hz, 1H), 4.45 (t, J = 6.0 Hz, 2H), 3.94 (s, 3H), 3.94 (s, 3H), 3.55 (t, J = 6.5 Hz, 2H), 2.32 (p, J = 6.3 Hz, 2H). LC–MS (ESI +) m/z calcd. for C12H15BrO4 [(M + H)]+: 303.02; found: 302.95. HPLC tR: 10.466 min.
3-bromopropyl 3,5-dimethoxybenzoate (2o) Intermediate 2o was obtained from 3,5-dimethoxybenzoic acid following general procedure D. Column chromatography with 40 to 70% DCM in PE as mobile phase. Transparent oil (1.09 g, 3.60 mmol, 60%). 1H NMR (400 MHz, CDCl3) δ 7.16 (d, J = 2.4 Hz, 2H), 6.64 (t, J = 2.4 Hz, 1H), 4.45 (t, J = 6.0 Hz, 2H), 3.82 (s, 6H), 3.54 (t, J = 6.5 Hz, 2H), 2.31 (p, J = 6.3 Hz, 2H). LC–MS (ESI +) m/z calcd. for C12H15BrO4 [(M + H)]+: 303.02; found: 302.95. HPLC tR: 11.275 min.
3-bromopropyl 3,4,5-trimethoxybenzoate (2p). Intermediate 2p was obtained from 3,4,5-trimethoxybenzoyl chloride following general procedure B. Column chromatography with 5 to 15% EtOAc in PE as mobile phase. White solid (6.66 g, 18.2 mmol, 91%). 1H NMR (400 MHz, CDCl3) δ 7.29 (s, 2H), 4.47 (t, J = 6.1 Hz, 2H), 3.92 (s, 9H), 3.54 (t, J = 6.5 Hz, 2H), 2.34 (p, J = 6.3 Hz, 2H). LC–MS (ESI +) m/z calcd. for C13H17BrO5 [(M + H)]+: 333.03; found: 333.00. HPLC tR: 10.538 min.
tert-butyl 4-(3-((2-methoxybenzoyl)oxy)propyl)piperazine-1-carboxylate (3a). Intermediates 3a and 4a were obtained from 2k following general procedure E. Column chromatography with 0 to 1.5% MeOH in DCM as mobile phase. Transparent oil (275 mg, 0.73 mmol, 53%). 1H NMR (400 MHz, CDCl3) δ 7.78 (dd, J = 7.9, 1.8 Hz, 1H), 7.47 (ddd, J = 8.4, 7.4, 1.8 Hz, 1H), 7.02 – 6.95 (m, 2H), 4.36 (t, J = 6.4 Hz, 2H), 3.90 (s, 3H), 3.44 (t, J = 4.7 Hz, 4H), 2.51 (t, J = 7.5, 7.1 Hz, 2H), 2.41 (t, J = 5.1 Hz, 4H), 1.99 – 1.90 (m, 2H), 1.46 (s, 9H). LC–MS (ESI +) m/z calcd. for C20H30N2O5 [(M + H)]+: 379.22; found: 379.20. HPLC tR: 6.521 min. 3-(piperazin-1-yl)propyl 2-methoxybenzoate dihydrochloride (4a). White solid (quant.) used without further analyses.
tert-butyl 4-(3-((3,5-dimethoxybenzoyl)oxy)propyl)piperazine-1-carboxylate (3b). Intermediates 3b and 4b were obtained from 2o following general procedure E. Column chromatography with 1 to 3% MeOH in DCM as mobile phase. Transparent oil (1.16 g, 2.85 mmol, 80%). 1H NMR (400 MHz, CDCl3) δ 7.18 (d, J = 2.4 Hz, 2H), 6.65 (t, J = 2.4 Hz, 1H), 4.37 (t, J = 6.5 Hz, 2H), 3.83 (s, 6H), 3.43 (t, J = 5.1 Hz, 4H), 2.50 (t, J = 7.1 Hz, 2H), 2.40 (t, J = 5.0 Hz, 4H), 2.02 – 1.90 (m, 2H), 1.46 (s, 9H). 3-(piperazin-1-yl)propyl 3,5-dimethoxybenzoate dihydrochloride (4b). White solid (954 mg, 2.50 mmol, 88%) used without further analyses.
tert-butyl 4-(3-((3,4,5-trimethoxybenzoyl)oxy)propyl)piperazine-1-carboxylate (3c). Intermediates 3c and 4c were obtained from 2p following general procedure E. Column chromatography with 2% MeOH in DCM as mobile phase. Transparent oil (4.95 g, 11.3 mmol, 88%). 1H NMR (400 MHz, CDCl3) δ 7.30 (s, 2H), 4.39 (t, J = 6.6, 2.1 Hz, 2H), 3.92 (s, 9H), 3.52 – 3.34 (m, 4H), 2.51 (t, J = 7.0 Hz, 2H), 2.47 – 2.33 (m, 4H), 1.98 (p, J = 6.7, 6.3 Hz, 2H), 1.47 (s, 9H). LC–MS (ESI +) m/z calcd. for C22H34N2O7 [(M + H)]+: 439.24; found: 439.15. HPLC tR: 6.746 and 7.023 min. 3-(piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate dihydrochloride (4c). White solid (4.29 g, 10.4 mmol, 91%) used without further analyses.
3-(4-(3-hydroxypropyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (5). To a stirred solution of intermediate 4c (1.23 g, 3.00 mmol, 1.0 equiv) in anhydrous DMF (15.0 mL) was added 3-bromopropan-1-ol (0.29 mL, 3.30 mmol, 1.1 equiv) and K2CO3 (1.24 g, 9.00 mmol, 3.0 equiv). The reaction mixture was allowed to stir overnight at rt under N2 atmosphere followed by filtration and concentration in vacuo. The acquired oil was dissolved in EtOAc (50 mL) and subsequently washed with H2O (50 mL). The aqueous phase was extracted with EtOAc (50 mL) and the combined organic phases were washed with brine (50 mL), dried over MgSO4, filtrated and concentrated in vacuo. The crude product was purified using flash column chromatography on silica gel with 10% MeOH in DCM as mobile phase to obtain intermediate 5 (773 mg, 1.95 mmol, 65%) as a transparent oil. 1H NMR (400 MHz, MeOD) δ 7.32 (s, 2H), 4.36 (t, J = 6.4 Hz, 2H), 3.88 (s, 6H), 3.82 (s, 3H), 3.61 (t, J = 6.2 Hz, 2H), 2.60 – 2.39 (m, 12H), 2.03 – 1.94 (m, 2H), 1.79 – 1.68 (m, 2H). LC–MS (ESI +) m/z calcd. for C20H32N2O6 [(M + H)]+: 396.23; found: 397.20. HPLC tR: 0.818 min.
3-(4-(3-(benzoyloxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (6a). Final compound 6a was obtained from intermediates 2a and 4c following general procedure F. Column chromatography with 2 to 4.5% MeOH in DCM as mobile phase. Yellow oil (884 mg, 1.77 mmol, 83%). 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 7.2 Hz, 2H), 7.55 (tt, J = 7.4, 1.6 Hz, 1H), 7.43 (t, J = 7.9 Hz, 2H), 7.30 (s, 2H), 4.38 (t, J = 6.5 Hz, 4H), 3.91 (s, 9H), 2.59 – 2.41 (m, 12H), 2.03 – 1.93 (m, 4H). 13C NMR (101 MHz, CDCl3) 166.4, 166.0, 152.8, 142.0, 132.7, 130.2, 129.4, 128.2, 125.2, 106.6, 63.4, 63.3, 60.7, 56.1, 55.0, 53.1, 53.1, 26.2, 26.1. LC–MS (ESI +) m/z calcd. for C27H36N2O7 [(M + H)]+: 501.26; found: 501.15. HPLC tR: 8.096 min.
3-(4-(3-((4-methylbenzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (6b). Final compound 6b was obtained from intermediates 2b and 4c following general procedure F. Column chromatography with 2 to 4% MeOH in DCM as mobile phase. Yellow oil (272 mg, 0.53 mmol, 66%). 1H NMR (400 MHz, CDCl3) δ 7.92 (d, J = 8.2 Hz, 2H), 7.30 (s, 2H), 7.22 (d, J = 8.0 Hz, 2H), 4.45 – 4.29 (m, 4H), 3.90 (s, 9H), 2.57 – 2.45 (m, 12H), 2.38 (s, 3H), 2.03 – 1.89 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 166.2, 165.8, 152.6, 143.2, 141.9, 129.3, 128.8, 127.3, 125.1, 106.5, 63.3, 62.9, 60.5, 55.9, 54.8, 52.9, 52.9, 26.0, 21.3. LC–MS (ESI +) m/z calcd. for C28H38N2O7 [(M + H)]+: 515.28; found: 515.35. HPLC tR: 7.544 min.
3-(4-(3-((3,4-dimethylbenzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (6c). Final compound 6c was obtained from intermediates 2c and 4c following general procedure F. Automatic column chromatography with 1 to 6% MeOH in DCM as mobile phase on Biotage Isolera One. Transparent oil (40.4 mg, 0.08 mmol, 14%). 1H NMR (400 MHz, CDCl3) δ 7.82 – 7.78 (m, 1H), 7.76 (dd, J = 7.8, 1.9 Hz, 1H), 7.29 (s, 2H), 7.19 (d, J = 7.9 Hz, 1H), 4.41 – 4.31 (m, 4H), 3.91 (s, 9H), 2.84 – 2.39 (m, 12H), 2.31 (s, 3H), 2.30 (s, 3H), 2.05 – 1.89 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 167.0, 166.3, 153.0, 142.4, 136.8, 130.7, 129.8, 128.0, 127.2, 125.5, 106.9, 63.7, 63.3, 61.1, 56.4, 55.3, 55.2, 53.3, 53.2, 26.4, 26.4, 20.1, 19.8. LC–MS (ESI +) m/z calcd. for C29H40N2O7 [(M + H)]+: 529.29; found: 529.30. HPLC tR: 7.807 min.
3-(4-(3-((3,5-dimethylbenzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (6d). Final compound 6d was obtained from intermediates 2d and 4c following general procedure F. Automatic column chromatography with 2 to 6% MeOH in DCM as mobile phase on Biotage Isolera One. Yellow oil (241 mg, 0.46 mmol, 57%). 1H NMR (400 MHz, CDCl3) δ 7.68 – 7.62 (m, 2H), 7.30 (s, 2H), 7.20 – 7.14 (m, 1H), 4.44 – 4.30 (m, 4H), 3.91 (s, 9H), 2.86 – 2.36 (m, 12H), 2.36 (s, 6H), 2.04 – 1.89 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 166.9, 166.2, 152.9, 142.2, 138.0, 134.5, 130.2, 127.2, 125.4, 106.8, 63.6, 63.3, 60.9, 56.2, 55.1, 55.1, 53.2, 53.2, 26.3, 21.2. LC–MS (ESI +) m/z calcd. for C29H40N2O7 [(M + H)]+: 528.29; found: 529.30. HPLC tR: 7.898 min.
3-(4-(3-((4-chlorobenzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (6e). Final compound 6e was obtained from intermediates 2e and 4c following general procedure F. Automatic column chromatography with 0 to 6% MeOH in DCM as mobile phase on Biotage Isolera One. Yellow oil (306 mg, 0.57 mmol, 71%). 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 8.6 Hz, 2H), 7.42 (d, J = 8.5 Hz, 2H), 7.29 (s, 2H), 4.37 (t, J = 6.5 Hz, 4H), 3.91 (s, 9H), 2.80 – 2.23 (m, 12H), 2.03 – 1.92 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 166.4, 165.9, 153.1, 139.5, 131.1, 128.8, 125.5, 106.9, 63.8, 63.7, 61.1, 56.4, 55.3, 55.2, 53.3, 26.4, 26.4. LC–MS (ESI +) m/z calcd. for C27H35ClN2O7 [(M + H)]+: 535.22; found: 535.15. HPLC tR: 7.716 min.
3-(4-(3-((3,4-dichlorobenzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (6f). Final compound 6f was obtained from intermediates 2f and 4c following general procedure F. Automatic column chromatography with 2 to 6% MeOH in DCM as mobile phase on Biotage Isolera One. Orange oil (301 mg, 0.53 mmol, 66%). 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 2.0 Hz, 1H), 7.85 (dd, J = 8.4, 2.0 Hz, 1H), 7.53 (d, J = 8.4 Hz, 1H), 7.29 (s, 2H), 4.42 – 4.34 (m, 4H), 3.91 (s, 9H), 2.80 – 2.31 (m, 12H), 2.03 – 1.92 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 166.3, 164.9, 153.0, 142.3, 137.7, 133.0, 131.6, 130.7, 130.3, 128.8, 125.5, 106.9, 64.2, 63.7, 61.1, 56.4, 55.2, 55.1, 53.3, 26.4, 26.3. LC–MS (ESI +) m/z calcd. for C27H34Cl2N2O7 [(M + H)]+: 569.18; found: 569.10. HPLC tR: 8.162 min.
3-(4-(3-((3-(dimethylamino)benzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (6g). Final compound 6g was obtained from intermediates 2g and 4c following general procedure F. Column chromatography with 6% MeOH in EtOAc as mobile phase. Yellow oil (227 mg, 0.42 mmol, 58%). 1H NMR (400 MHz, CDCl3) δ 7.42 – 7.34 (m, 2H), 7.29 (s, 2H), 7.30 – 7.28 (m, 1H), 6.90 (ddd, J = 8.2, 2.8, 1.0 Hz, 1H), 4.44 – 4.28 (m, 4H), 3.91 (s, 9H), 2.99 (s, 6H), 2.78 – 2.13 (m, 12H), 2.04 – 1.87 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 167.3, 166.3, 153.0, 150.5, 142.2, 131.0, 129.0, 125.4, 117.5, 116.8, 113.2, 106.8, 63.7, 63.3, 61.0, 56.3, 55.2, 55.2, 53.2, 53.2, 40.6, 26.3. LC–MS (ESI +) m/z calcd. for C29H41N3O7 [(M + H)]+: 544.30; found: 544.25. HPLC tR: 7.108 and 7.398 min.
3-(4-(3-((4-(dimethylamino)benzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (6h). Final compound 6h was obtained from intermediates 2h and 4c following general procedure F. Column chromatography with 1 to 3.5% MeOH in DCM as mobile phase. Transparent oil (103 mg, 0.19 mmol, 24%). 1H NMR (400 MHz, CDCl3) δ 7.90 (d, J = 9.1 Hz, 2H), 7.29 (s, 2H), 6.64 (d, J = 9.1 Hz, 2H), 4.37 (t, J = 6.5 Hz, 2H), 4.31 (t, J = 6.4 Hz, 2H), 3.91 (s, 6H), 3.91 (s, 3H), 3.04 (s, 6H), 2.81 – 2.25 (m, 12H), 2.04 – 1.88 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 167.0, 166.3, 153.3, 153.0, 142.2, 131.3, 125.4, 117.1, 110.7, 106.8, 63.7, 62.6, 61.0, 56.3, 55.3, 55.2, 53.3, 53.2, 40.1, 26.5, 26.3. LC–MS (ESI +) m/z calcd. for C29H41N3O7 [(M + H)]+: 544.30; found: 544.20. HPLC tR: 7.617 min.
3-(4-(3-((4-(methylamino)benzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (6i). Final compound 6i was obtained from intermediates 2i and 4c following general procedure F. Column chromatography with 2 to 6% MeOH in DCM as mobile phase. Yellow oil (64.0 mg, 0.12 mmol, 48%). 1H NMR (400 MHz, CDCl3) δ 7.86 (d, J = 8.9 Hz, 2H), 7.29 (s, 2H), 6.55 (d, J = 9.0 Hz, 2H), 4.37 (t, J = 6.5 Hz, 2H), 4.30 (t, J = 6.4 Hz, 2H), 4.28 – 4.21 (m, 1H), 3.91 (s, 9H), 2.88 (d, J = 4.9 Hz, 3H), 2.83 – 2.21 (m, 12H), 2.05 – 1.87 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 167.0, 166.3, 153.0, 153.0, 142.2, 131.6, 125.5, 118.4, 111.1, 106.8, 63.7, 62.7, 61.0, 56.3, 55.3, 55.2, 53.3, 53.3, 30.2, 26.5, 26.4. LC–MS (ESI +) m/z calcd. for C28H39N3O7 [(M + H)]+: 530.29; found: 530.25. HPLC tR: 9.641 min.
3-(4-(3-((3-(methylamino)benzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (6j). Final compound 6j was obtained from intermediates 2j and 4c following general procedure F. Column chromatography with 2 to 6% MeOH in DCM as mobile phase. Yellow oil (147 mg, 0.28 mmol, 46%). 1H NMR (400 MHz, CDCl3) δ 7.39 – 7.34 (m, 1H), 7.30 (s, 2H), 7.26 (dd, J = 2.5, 1.5 Hz, 1H), 7.23 (t, J = 7.9 Hz, 1H), 6.77 (ddd, J = 8.1, 2.6, 1.0 Hz, 1H), 4.46 – 4.28 (m, 4H), 3.91 (m, 10H), 2.86 (s, 3H), 2.78 – 2.24 (m, 12H), 2.03 – 1.90 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 167.1, 166.2, 152.9, 149.3, 142.1, 131.2, 129.1, 125.4, 118.2, 116.8, 112.8, 106.7, 63.6, 63.3, 60.9, 56.2, 55.1, 55.1, 53.2, 53.2, 30.7, 26.3. LC–MS (ESI +) m/z calcd. for C28H39N3O7 [(M + H)]+: 530.29; found: 530.30. HPLC tR: 6.675 and 6.981 min.
3-(4-(3-((2-methoxybenzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (6k). Final compound 6k was obtained from intermediates 2k and 4c following general procedure F. Automatic column chromatography with 0 to 5% MeOH in DCM as mobile phase on Biotage Isolera One. Orange oil (230 mg, 0.43 mmol, 54%). 1H NMR (400 MHz, CDCl3) δ 7.78 (ddd, J = 8.0, 2.0, 0.8 Hz, 1H), 7.51 – 7.43 (m, 1H), 7.30 (s, 2H), 7.02 – 6.94 (m, 2H), 4.47 – 4.27 (m, 4H), 3.91 (s, 9H), 3.90 (s, 3H), 3.34 – 2.17 (m, 12H), 2.05 – 1.89 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 166.3, 166.3, 159.2, 153.0, 142.2, 133.5, 131.6, 125.4, 120.3, 120.1, 112.0, 106.8, 63.6, 63.3, 61.0, 56.3, 56.0, 55.2, 55.2, 53.2, 53.2, 26.3, 26.3. LC–MS (ESI +) m/z calcd. for C28H38N2O8 [(M + H)]+: 531.27; found: 531.25. HPLC tR: 7.156 min.
3-(4-(3-((3-methoxybenzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (6l). Final compound 6l was obtained from intermediate 5 and 3-methoxybenzoyl chloride following general procedure H. Column chromatography with 3 to 6% MeOH in DCM as mobile phase. Transparent oil (36.6 mg, 0.07 mmol, 14%). 1H NMR (400 MHz, CDCl3) δ 7.63 (dt, J = 7.7, 1.2 Hz, 1H), 7.55 (dd, J = 2.8, 1.5 Hz, 1H), 7.35 (t, J = 8.0 Hz, 1H), 7.29 (s, 2H), 7.10 (ddd, J = 8.3, 2.7, 1.0 Hz, 1H), 4.37 (t, J = 6.3 Hz, 4H), 3.91 (s, 9H), 3.85 (s, 3H), 2.66 – 2.41 (m, 12H), 2.04 – 1.92 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 166.6, 166.4, 159.7, 153.1, 142.3, 131.8, 129.5, 125.5, 122.1, 119.4, 114.2, 106.9, 63.7, 63.6, 61.1, 56.4, 55.6, 55.2, 53.3, 26.4, 26.4. LC–MS (ESI +) m/z calcd. for C28H38N2O8 [(M + H)]+: 531.27; found: 531.25. HPLC tR: 7.338 min.
3-(4-(3-((4-methoxybenzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (6m). Final compound 6m was obtained from intermediate 5 and 4-methoxybenzoyl chloride following general procedure H. Column chromatography with 3 to 6% MeOH in DCM as mobile phase. Transparent oil (285 mg, 0.47 mmol, 94%). 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 9.1 Hz, 2H), 7.30 (s, 2H), 6.92 (d, J = 9.1 Hz, 2H), 4.37 (t, J = 6.5 Hz, 2H), 4.34 (t, J = 6.5 Hz, 2H), 3.91 (s, 6H), 3.90 (s, 3H), 3.85 (s, 3H), 2.69 – 2.48 (m, 12H), 2.06 – 1.90 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 166.2, 166.1, 163.3, 152.9, 142.1, 131.5, 125.3, 122.6, 113.5, 106.7, 63.4, 62.9, 60.8, 56.2, 55.4, 55.02, 54.97, 52.90, 52.90, 26.13, 26.09. LC–MS (ESI +) m/z calcd. for C28H38N2O8 [(M + H)]+: 531.27; found 531.25. HPLC tR: 7.291 min.
3-(4-(3-((3,4-dimethoxybenzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (6n). Final compound 6n was obtained from intermediate 5 and 3,4-dimethoxybenzoic acid following general procedure I. Column chromatography with 3 to 6% MeOH in DCM as mobile phase. Transparent oil (36.9 mg, 0.07 mmol, 33%). 1H NMR (400 MHz, CDCl3) δ 7.67 (dd, J = 8.4, 2.0 Hz, 1H), 7.54 (d, J = 2.0 Hz, 1H), 7.29 (s, 2H), 6.89 (d, J = 8.5 Hz, 1H), 4.36 – 4.33 (m, 4H), 3.94 (s, 3H), 3.93 (s, 3H), 3.91 (s, 6H), 3.91 (s, 3H), 2.75 – 2.37 (m, 12H), 2.05 – 1.93 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 166.5, 166.3, 153.1, 153.0, 148.7, 142.3, 125.4, 123.6, 122.9, 112.0, 110.3, 106.9, 63.7, 63.3, 61.0, 56.4, 56.1, 56.1, 55.23, 55.19, 53.2, 26.4, 26.3. LC–MS (ESI +) m/z calcd. for C29H40N2O9 [(M + H)]+: 561.28; found: 561.25. HPLC tR: 7.093 min.
3-(4-(3-((3,5-dimethoxybenzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (6o). Final compound 6o was obtained from intermediate 5 and 3,5-dimethoxybenzoic acid following general procedure I. Column chromatography with 3 to 6% MeOH in DCM as mobile phase. Transparent oil (82.0 mg, 0.15 mmol, 73%). 1H NMR (400 MHz, CDCl3) δ 7.29 (s, 2H), 7.18 (d, J = 2.4 Hz, 2H), 6.65 (t, J = 2.4 Hz, 1H), 4.37 (t, J = 6.3 Hz, 2H), 4.37 (t, J = 6.3 Hz, 2H), 3.91 (s, 6H), 3.91 (s, 3H), 3.83 (s, 6H), 2.70 – 2.39 (m, 12H), 2.05 – 1.92 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 166.4, 166.3, 160.7, 153.0, 142.2, 132.3, 125.4, 107.2, 106.8, 105.5, 63.6, 61.0, 56.3, 55.6, 55.2, 55.1, 53.2, 53.2, 26.3, 26.3. LC–MS (ESI +) m/z calcd. for C29H40N2O9 [(M + H)]+: 561.28; found: 561.25. HPLC tR: 7.540 min.
piperazine-1,4-diylbis(propane-3,1-diyl) bis(2-methoxybenzoate) (6p). Final compound 6p was obtained from intermediate 2k following general procedure G. Column chromatography with 3 to 5% MeOH in DCM as mobile phase. Transparent oil (492 mg, 1.05 mmol, 47%). 1H NMR (400 MHz, CDCl3) δ 7.78 (dd, J = 7.9, 1.8 Hz, 2H), 7.49 – 7.41 (m, 2H), 6.99 – 6.92 (m, 4H), 4.34 (t, J = 6.4 Hz, 4H), 3.87 (s, 6H), 2.70–2.40 (m, 12H), 1.94 (p, J = 6.8 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ 165.9, 158.8, 133.2, 131.2, 119.9, 119.8, 111.7, 62.9, 55.6, 54.8, 52.8, 25.9. LC–MS (ESI +) m/z calcd. for C26H34N2O6 [(M + H)]+: 471.25; found: 471.15. HPLC tR: 6.734 min.
piperazine-1,4-diylbis(propane-3,1-diyl) bis(3-methoxybenzoate) (6q). Final compound 6q was obtained from intermediate 2l following general procedure G. Column chromatography with 3 to 5% MeOH in DCM as mobile phase. Transparent oil (450 mg, 0.96 mmol, 45%). 1H NMR (400 MHz, CDCl3) δ 7.63 (dt, J = 7.6, 1.2 Hz, 2H), 7.55 (dd, J = 2.7, 1.5 Hz, 2H), 7.33 (t, J = 8.0 Hz, 2H), 7.08 (ddd, J = 8.2, 2.7, 1.0 Hz, 2H), 4.36 (t, J = 6.5 Hz, 4H), 3.83 (s, 6H), 2.60 – 2.41 (m, 12H), 1.96 (p, J = 7.1 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ 166.2, 159.4, 131.5, 129.2, 121.7, 119.1, 114.0, 63.4, 55.2, 54.9, 53.0, 26.1. LC–MS (ESI +) m/z calcd. for C26H34N2O6 [(M + H)]+: 471.25; found: 471.25. HPLC tR: 7.020 and 7.365 min.
piperazine-1,4-diylbis(propane-3,1-diyl) bis(4-methoxybenzoate) (6r). Final compound 6r was obtained from intermediate 2m following general procedure G. Automatic column chromatography with 3 to 5% MeOH in DCM as mobile phase on Biotage Isolera One. Transparent oil (365 mg, 0.78 mmol, 55%). 1H NMR (400 MHz, CDCl3) 8.02 – 7.95 (m, 4H), 6.96 – 6.88 (m, 4H), 4.34 (t, J = 6.5 Hz, 4H), 3.85 (s, 6H), 2.64 – 2.31 (m, 12H), 2.02 – 1.85 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 166.4, 163.4, 131.6, 122.8, 113.6, 63.2, 55.5, 55.2, 53.2, 26.4. LC–MS (ESI +) m/z calcd. for C26H34N2O6 [(M + H)]+: 471.25; found: 471.15. HPLC tR: 7.050 and 7.294 min.
piperazine-1,4-diylbis(propane-3,1-diyl) bis(3,4-dimethoxybenzoate) (6s). Final compound 6s was obtained from intermediate 2n following general procedure G. Column chromatography with 0.5 to 4% MeOH in DCM as mobile phase. Transparent oil (59.7 mg, 0.77 mmol, 15%). 1H NMR (400 MHz, CDCl3) δ 7.68 (dd, J = 8.4, 2.0 Hz, 2H), 7.54 (d, J = 2.0 Hz, 2H), 6.89 (d, J = 8.4 Hz, 2H), 4.36 (t, J = 6.5 Hz, 4H), 3.94 (s, 6H), 3.94 (s, 6H), 2.66 – 2.37 (m, 12H), 2.03 – 1.91 (m, 4H).13C NMR (101 MHz, CDCl3) δ 166. 5, 153.0, 148.7, 123.6, 122.9, 112.0, 110.3, 63.3, 56.1, 56.1, 55.2, 53.2, 26.4. LC–MS (ESI+) m/z calcd. for C28H38N2O8 [(M + H)]+: 531.27; found: 531.25. HPLC tR: 6.736 and 6.928 min.
piperazine-1,4-diylbis(propane-3,1-diyl) bis(3,5-dimethoxybenzoate) (6t). Final compound 6t was obtained from intermediate 2o following general procedure G. Column chromatography with 0.5 to 4% MeOH in DCM as mobile phase. Transparent oil (501 mg, 0.94 mmol, 58%). 1H NMR (400 MHz, CDCl3) δ 7.17 (d, J = 2.4 Hz, 4H), 6.62 (t, J = 2.4 Hz, 2H), 4.36 (t, J = 6.5 Hz, 4H), 3.81 (s, 12H), 2.66 – 2.36 (m, 12H), 1.95 1.96 (p, J = 7.6, 6.5 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ 166.0, 160.4, 132.0, 106.9, 105.2, 63.4, 55.3, 54.9, 53.0, 26.0. LC–MS (ESI +) m/z calcd. for C28H38N2O8 [(M + H)]+: 531.27; found: 531.25. HPLC tR: 7.833 min.
3-(4-(3-((2-methoxybenzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4-dimethoxybenzoate (6u). Final compound 6j was obtained from intermediates 2n and 4a following general procedure F. Column chromatography with 3 to 5% MeOH in DCM as mobile phase. Transparent oil (120 mg, 0.24 mmol, 43%). 1H NMR (400 MHz, CDCl3) δ 7.78 (dd, J = 7.9, 1.8 Hz, 1H), 7.68 (dd, J = 8.4, 2.0 Hz, 1H), 7.54 (d, J = 1.9 Hz, 1H), 7.47 (ddd, J = 8.5, 7.4, 1.8 Hz, 1H), 7.04 – 6.94 (m, 2H), 6.89 (d, J = 8.5 Hz, 1H), 4.41 – 4.31 (m, 4H), 3.94 (s, 3H), 3.93 (s, 3H), 3.90 (s, 3H), 2.61 – 2.40 (m, 12H), 2.01 – 1.90 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 166.4, 166.3, 159.1, 152.9, 148.6, 133.5, 131.6, 123.5, 122.9, 120.3, 120.1, 112.0, 112.0, 110.2, 63.3, 63.3, 56.0, 56.0, 55.9, 55.2, 53.2, 26.4, 26.2. LC–MS (ESI +) m/z calcd. for C27H36N2O7 [(M + H)]+: 501.26; found: 501.20. HPLC tR: 2.491 min.
3-(4-(3-((3,5-dimethoxybenzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4-dimethoxybenzoate (6v). Final compound 6j was obtained from intermediates 2n and 4b following general procedure F. Automatic column chromatography with 0 to 5% MeOH in DCM as mobile phase on Biotage Isolera One. Transparent oil (676 mg, 1.27 mmol, 87%). 1H NMR (400 MHz, CDCl3) δ 7.66 (dd, J = 8.4, 2.0 Hz, 1H), 7.53 (d, J = 2.0 Hz, 1H), 7.16 (d, J = 2.4 Hz, 2H), 6.87 (d, J = 8.5 Hz, 1H), 6.62 (t, J = 2.4 Hz, 1H), 4.42 – 4.30 (m, 4H), 3.92 (s, 3H), 3.90 (s, 3H), 3.80 (s, 6H), 2.62 – 2.38 (m, 12H), 2.00 – 1.90 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 165.8, 165.7, 160.2, 152.5, 148.2, 131.8, 123.0, 122.4, 111.5, 109.8, 106.7, 63.2, 62.8, 55.5, 55.5, 55.03, 54.7, 54.6, 52.8, 25.9, 25.8. LC–MS (ESI +) m/z calcd. for C28H38N2O8 [(M + H)]+: 531.27; found: 531.25. HPLC tR: 7.045 and 7.432 min.
3-(fluorosulfonyl)benzoic acid (7a). Intermediate 7a was obtained from 3-(chlorosulfonyl)benzoic acid following general procedure J. White solid (602 mg, 2.94 mmol, 98%). 1H NMR (400 MHz, DMSO-d6) δ 13.87 (br s, 1H), 8.50 – 8.43 (m, 2H), 8.42 – 8.38 (m, 1H), 7.95 (t, J = 7.8 Hz, 1H). LC–MS (ESI-) m/z calcd. for C7H5FO4S [(M-H)]−: 202.98; found: 202.95. HPLC tR: 8.100 min.
4-(fluorosulfonyl)benzoic acid (7b). Intermediate 7b was obtained from 4-(chlorosulfonyl)benzoic acid following general procedure J. White solid (961 mg, 4.71 mmol, 94%). 1H NMR (400 MHz, CDCl3) δ 8.38 – 8.32 (m, 2H), 8.18 – 8.12 (m, 2H). LC–MS (ESI-) m/z calcd. for C7H5FO4S [(M-H)]−: 202.98; found: 203.00. HPLC tR: 8.464 min.
4-isothiocyanatobenzoic acid (7c). A stirred solution of thiophosgene (0.15 mL, 2.00 mmol, 1.0 equiv) in acetone (2.00 mL) was allowed to cool down to 0 °C followed by addition of a solution of 4-aminobenzoic acid (274 mg, 2.00 mmol, 1.0 equiv) in acetone (2.00 mL). The mixture was allowed to stir at 0 °C for 3 h and at rt for 16 h. The reaction mixture was concentrated in vacuo, dissolved in EtOAc (20 mL) and washed with sat. NaHCO3 solution (20 mL). The aqueous phase was acidified to pH 3 by dropwise addition of 3 M aqueous HCl and extracted three times with EtOAc (20 mL). The combined organic phase was dried over MgSO4, filtrated and concentrated in vacuo to provide intermediate 7c as a yellow solid. (136 mg, 0.76, 38%). 1H NMR (400 MHz, DMSO-d6) δ 7.98 (d, J = 8.4 Hz, 2H), 7.53 (d, J = 8.7 Hz, 2H). LC–MS (ESI-) m/z calcd. for C8H5NO2S [(M-H)]−: 178.00; found: 177.90. HPLC tR: 9.494 min.
3-((tert-butoxycarbonyl)amino)benzoic acid (7d). Intermediate 7d was obtained from 3-aminobenzoic acid following general procedure K with Et3N. White solid (quant.). 1H NMR (400 MHz, CDCl3) δ 8.00 (t, J = 2.0 Hz, 1H), 7.79 – 7.70 (m, 2H), 7.41 (t, J = 7.9 Hz, 1H), 6.65 (br s, 1H), 1.54 (s, 9H). LC–MS (ESI-) m/z calcd. for C12H15NO4 [(M-H)]−: 236.09; found: 236.05. HPLC tR: 9.131 min.
4-((tert-butoxycarbonyl)amino)benzoic acid (7e). Intermediate 7e was obtained from 4-aminobenzoic acid following general procedure K with Et3N. White solid (quant.). 1H NMR (400 MHz, CDCl3) δ 8.16 – 7.93 (m, 2H), 7.64 – 7.34 (m, 2H), 6.83 (br s, 1H), 1.54 (s, 9H). LC–MS (ESI-) m/z calcd. for C12H15NO4 [(M-H)]−: 236.09; found: 236.05. HPLC tR: 9.117 min.
4-((tert-butoxycarbonyl)amino)-3-methoxybenzoic acid (7f). Intermediate 7f was obtained from 4-amino-3-methoxybenzoic acid following general procedure K with NaOH. Brown solid (1.86 g, 6.95 mmol, 70%). 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 8.4 Hz, 1H), 7.97 (br s, 1H), 7.63 (dd, J = 8.4, 1.9 Hz, 1H), 7.57 (d, J = 1.8 Hz, 1H), 3.93 (s, 3H), 1.53 (s, 9H). LC–MS (ESI-) m/z calcd. for C13H17NO5 [(M-H)]−: 266.10; found: 266.00. HPLC tR: 9.926 min.
4-(((tert-butoxycarbonyl)amino)methyl)benzoic acid (7g). Intermediate 7g was obtained from 4-(aminomethyl)benzoic acid following general procedure K with NaOH. White solid (1.77 g, 7.06 mmol, 88%). 1H NMR (400 MHz, MeOD) δ 7.98 (d, J = 8.0 Hz, 2H), 7.37 (d, J = 8.0 Hz, 2H), 4.29 (s, 2H), 1.46 (s, 9H). LC–MS (ESI-) m/z calcd. for C13H17NO4 [(M-H)]−: 250.11; found: 250.10. HPLC tR: 8.717 min.
4-(2-((tert-butoxycarbonyl)amino)ethyl)benzoic acid (7h). Intermediate 7h was obtained from 4-(2-aminoethyl)benzoic acid hydrochloride following general procedure K with NaOH. White solid (945 mg, 3.56 mmol, 89%). 1H NMR (400 MHz, MeOD) δ 7.97 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 8.2 Hz, 2H), 3.30 (t, J = 7.4 Hz, 2H), 2.85 (t, J = 7.3 Hz, 2H), 1.43 (s, 9H). LC–MS (ESI-) m/z calcd. for C14H19NO4 [(M-H)]−: 264.12; found: 264.10. HPLC tR: 9.064 min.
2-bromoethyl 3-(fluorosulfonyl)benzoate (8a). Intermediate 8a was obtained from 7a and 2-bromoethan-1-ol following general procedure C. Column chromatography with 30% EtOAc in PE as mobile phase. Transparent oil (259 mg, 0.83 mmol, 82%). 1H NMR (400 MHz, CDCl3) δ 8.69 (t, J = 1.8 Hz, 1H), 8.48 (dt, J = 7.9, 1.5 Hz, 1H), 8.23 (ddd, J = 7.9, 2.0, 1.2 Hz, 1H), 7.79 (t, J = 7.7 Hz, 1H), 4.71 (t, J = 6.0 Hz, 2H), 3.69 (t, J = 6.0 Hz, 2H).
4-bromobutyl 3-(fluorosulfonyl)benzoate (8b). Intermediate 8b was obtained from 7a and 4-bromobutan-1-ol following general procedure C. Column chromatography with 20% EtOAc in PE as mobile phase. Transparent oil (294 mg, 0.87 mmol, 83%). 1H NMR (400 MHz, CDCl3) δ 8.65 (t, J = 1.8 Hz, 1H), 8.46 (dt, J = 7.9, 1.4 Hz, 1H), 8.21 (ddd, J = 7.9, 2.0, 1.2 Hz, 1H), 7.79 (t, J = 8.0, 0.7 Hz, 1H), 4.45 (t, J = 6.1 Hz, 2H), 3.51 (t, J = 6.2 Hz, 2H), 2.14 – 1.94 (m, 4H).
2-bromoethyl 4-(fluorosulfonyl)benzoate (8c). Intermediate 8c was obtained from 7b and 2-bromoethan-1-ol following general procedure C. Column chromatography with 10% to 20% EtOAc in PE as mobile phase. Transparent oil (503 mg, 1.62 mmol, 50%). 1H NMR (400 MHz, CDCl3) δ 8.37 – 8.30 (m, 2H), 8.16 – 8.08 (m, 2H), 4.72 (t, J = 6.1 Hz, 2H), 3.71 (t, J = 6.0 Hz, 2H).
3-bromopropyl 4-(fluorosulfonyl)benzoate (8d). Intermediate 8d was obtained from 7h and 3-bromopropan-1-ol following general procedure C. Column chromatography with 20% EtOAc in PE as mobile phase. Transparent oil (756 mg, 2.33 mmol, 99%). 1H NMR (400 MHz, CDCl3) δ 8.33 – 8.25 (m, 2H), 8.11 (d, J = 8.5 Hz, 2H), 4.56 (t, J = 6.1 Hz, 2H), 3.57 (t, J = 6.4 Hz, 2H), 2.37 (p, J = 6.3 Hz, 2H).
4-bromobutyl 4-(fluorosulfonyl)benzoate (8e). Intermediate 8e was obtained from 7b and 4-bromobutan-1-ol following general procedure C. Column chromatography with 10% EtOAc in PE as mobile phase. Transparent oil (227 mg, 0.67 mmol, 41%). 1H NMR (400 MHz, CDCl3) δ 8.32 – 8.26 (m, 2H), 8.13 – 8.07 (m, 2H), 4.44 (t, J = 6.1 Hz, 2H), 3.50 (t, J = 6.3 Hz, 2H), 2.11 – 1.94 (m, 4H).
3-bromopropyl 3-((tert-butoxycarbonyl)amino)benzoate (8f). Intermediate 8f was obtained from 7d and 3-bromopropan-1-ol following general procedure L. Column chromatography with 5 to 15% EtOAc in PE as mobile phase. Yellow oil (157 mg, 0.44 mmol, 15%). 1H NMR (400 MHz, CDCl3) δ 7.94 – 7.89 (m, 1H), 7.78 – 7.70 (m, 1H), 7.70 (dt, J = 7.7, 1.3 Hz, 1H), 7.37 (t, J = 7.9 Hz, 1H), 6.79 (s, 1H), 4.46 (t, J = 6.0 Hz, 2H), 3.54 (t, J = 6.6 Hz, 2H), 2.37 – 2.19 (m, 2H), 1.53 (s, 9H).
3-bromopropyl 4-((tert-butoxycarbonyl)amino)benzoate (8g). Intermediate 8g was obtained from 7e and 3-bromopropan-1-ol following general procedure L. Column chromatography with 5 to 15% EtOAc in PE as mobile phase. Yellow oil (231 mg, 0.65 mmol, 22%). 1H NMR (400 MHz, CDCl3) δ 8.00 – 7.92 (m, 2H), 7.49 – 7.42 (m, 2H), 6.89 (s, 1H), 4.44 (t, J = 6.1 Hz, 2H), 3.54 (t, J = 6.6 Hz, 2H), 2.31 (p, J = 6.3 Hz, 2H), 1.52 (s, 9H).
4-bromobutyl 4-((tert-butoxycarbonyl)amino)benzoate (8h). Intermediate 8h was obtained from 7e and 4-bromobutan-1-ol following general procedure L. Column chromatography with 5 to 10% EtOAc in PE as mobile phase. White solid (332 mg, 0.89 mmol, 30%). 1H NMR (400 MHz, CDCl3) δ 8.01 – 7.92 (m, 2H), 7.54 – 7.45 (m, 2H), 7.28 (br s, 1H), 4.33 (t, J = 6.2 Hz, 2H), 3.47 (t, J = 6.5 Hz, 2H), 2.05 – 1.97 (m, 2H), 1.97 – 1.88 (m, 2H), 1.51 (s, 9H).
3-bromopropyl 4-((tert-butoxycarbonyl)amino)-3-methoxybenzoate (8i). Intermediate 8i was obtained from 7f and 3-bromopropan-1-ol following general procedure L. Column chromatography with 5 to 15% EtOAc in PE as mobile phase. Yellow oil (321 mg, 0.83 mmol, 28%). 1H NMR (400 MHz, CDCl3) δ 8.17 (d, J = 8.5 Hz, 1H), 7.65 (dd, J = 8.5, 1.8 Hz, 1H), 7.50 (d, J = 1.8 Hz, 1H), 7.30 (br s, 1H), 4.44 (t, J = 6.1 Hz, 2H), 3.93 (s, 3H), 3.54 (t, J = 6.6 Hz, 2H), 2.32 (p, J = 6.3 Hz, 2H), 1.53 (s, 9H).
3-bromopropyl 4-(((tert-butoxycarbonyl)amino)methyl)benzoate (8j). Intermediate 8j was obtained from 7g and 3-bromopropan-1-ol following general procedure L in the presence of pyridine (6.0 equiv). Column chromatography with 5 to 10% EtOAc in PE as mobile phase. White solid (743 mg, 2.00 mmol, 57%). 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 8.3 Hz, 2H), 7.35 (d, J = 8.2 Hz, 2H), 5.16 (br s, 1H), 4.45 (t, J = 6.0 Hz, 2H), 4.34 (d, J = 6.6 Hz, 2H), 3.54 (t, J = 6.5 Hz, 2H), 2.31 (p, J = 6.3 Hz, 2H), 1.46 (s, 9H).
3-bromopropyl 4-(2-((tert-butoxycarbonyl)amino)ethyl)benzoate (8k). Intermediate 8k was obtained from 7h and 3-bromopropan-1-ol following general procedure L in the presence of pyridine (6.0 equiv). Column chromatography with 5 to 10% EtOAc in PE as mobile phase. White solid (192 mg, 0.50 mmol, 17%). 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 8.4 Hz, 2H), 7.27 (d, J = 8.2 Hz, 2H), 4.72 (br t, J = 6.2 Hz, 1H), 4.45 (t, J = 6.0 Hz, 2H), 3.55 (t, J = 6.6 Hz, 2H), 3.39 (q, J = 6.8 Hz, 2H), 2.87 (t, J = 7.0 Hz, 2H), 2.32 (p, J = 6.3 Hz, 2H), 1.43 (s, 9H).
3-(4-(2-((3-(fluorosulfonyl)benzoyl)oxy)ethyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (9a). Final compound 9a was obtained from intermediates 8a and 4c following general procedure F. Column chromatography with 6 to 10% MeOH in EtOAc as mobile phase. Transparent oil (13.2 mg, 0.02 mmol, 4%). 1H NMR (400 MHz, CDCl3) δ 8.66 (t, J = 1.8 Hz, 1H), 8.43 (dt, J = 7.9, 1.4 Hz, 1H), 8.20 (ddd, J = 7.9, 2.0, 1.2 Hz, 1H), 7.76 (t, J = 8.4 Hz, 1H), 7.29 (s, 2H), 4.52 (t, J = 5.9 Hz, 2H), 4.37 (t, J = 6.6 Hz, 2H), 3.91 (s, 9H), 2.81 (t, J = 6.0 Hz, 2H), 2.72–2.40 (m, 10H), 2.00 (p, J = 6.8 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 166.4, 164.2, 153.0, 136.5, 132.3, 130.2, 129.7, 125.4, 106.9, 63.6, 63.4, 61.1, 56.6, 56.4, 55.2, 53.4, 53.2, 26.3. LC–MS (ESI +) m/z calcd. for C26H33FN2O9S [(M + H)]+: 569.20; found: 569.15. HPLC tR: 7.638 min.
3-(4-(3-((3-(fluorosulfonyl)benzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (9b). Final compound 9b was obtained from intermediate 5 and 7a following general procedure I. Column chromatography with 3% MeOH in DCM as mobile phase. Transparent oil (30.3 mg, 0.05 mmol, 5%).1H NMR (400 MHz, CDCl3) δ 8.65 (t, J = 1.6 Hz, 1H), 8.43 (dt, J = 8.0, 1.6 Hz, 1H), 8.20 (ddd, J = 8.0, 2.0, 1.2 Hz, 1H), 7.75 (t, J = 7.8 Hz, 1H), 7.29 (s, 2H), 4.45 (t, J = 6.6 Hz, 2H), 4.37 (t, J = 6.6 Hz, 2H), 3.91 (s, 6H), 3.91 (s, 3H), 2.85 – 2.24 (m, 12H), 2.09 – 1.87 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 166.3, 164.2, 153.0, 136.4, 132.4, 132.3, 130.2, 129.6, 125.5, 106.9, 64.7, 63.7, 61.1, 56.4, 55.2, 55.0, 53.3, 26.4, 26.2. LC–MS (ESI +) m/z calcd. for C27H35FN2O9S [(M + H)]+: 583.21; found: 583.25. HPLC tR: 7.585 min.
3-(4-(4-((3-(fluorosulfonyl)benzoyl)oxy)butyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (9c). Final compound 9c was obtained from intermediates 8b and 4c following general procedure F. Column chromatography with 4 to 8% MeOH in DCM as mobile phase. Transparent oil (40.1 mg, 0.06 mmol, 13%). 1H NMR (400 MHz, CDCl3) δ 8.65 (t, J = 1.8 Hz, 1H), 8.44 (dt, J = 7.9, 1.5 Hz, 1H), 8.20 (ddd, J = 7.9, 2.0, 1.2 Hz, 1H), 7.75 (t, J = 7.9 Hz, 1H), 7.29 (s, 2H), 4.41 (t, J = 6.6 Hz, 2H), 4.37 (t, J = 6.6 Hz, 2H), 3.91 (s, 9H), 2.63 – 2.33 (m, 12H), 2.01 – 1.93 (m, 2H), 1.84 (p, J = 8.0, 7.0, 6.5 Hz, 2H), 1.70 – 1.60 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 166.3, 164.2, 153.0, 136.4, 133.9, 132.4, 132.2, 130.1, 129.6, 125.5, 106.9, 66.1, 63.7, 61.0, 58.1, 56.4, 55.2, 53.3, 26.8, 26.4, 23.5. LC–MS (ESI +) m/z calcd. for C28H37FN2O9S [(M + H)]+: 597.23; found: 597.35. HPLC tR: 7.675 min.
3-(4-(2-((4-(fluorosulfonyl)benzoyl)oxy)ethyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (9d). Final compound 9d was obtained from intermediates 8c and 4c following general procedure F. Column chromatography with 4 to 8% MeOH in DCM as mobile phase. Transparent oil (21.6 mg, 0.03 mmol, 4%). 1H NMR (400 MHz, CDCl3) δ 8.32 – 8.24 (m, 2H), 8.13 – 8.07 (m, 2H), 7.29 (s, 2H), 4.52 (t, J = 5.9 Hz, 2H), 4.37 (t, J = 6.6 Hz, 2H), 3.91 (s, 9H), 2.81 (t, J = 5.9 Hz, 2H), 2.76 – 2.29 (m, 10H), 1.97 (p, J = 6.8 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 166.4, 164.5, 153.0, 136.7, 130.8, 128.7, 125.5, 106.9, 63.7, 63.6, 61.1, 56.6, 56.4, 55.2, 53.5, 53.3, 26.4. LC–MS (ESI +) m/z calcd. for C26H33FN2O9S [(M + H)]+: 569.19; found: 569.60. HPLC tR: 7.677 min.
3-(4-(3-((4-(fluorosulfonyl)benzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (9e). Final compound 9e was obtained from intermediates 8d and 4c following general procedure F. Column chromatography with 2 to 3% MeOH in DCM as mobile phase. Transparent oil (12.8 mg, 0.02 mmol, 1%). 1H NMR (400 MHz, CDCl3) δ 8.27 (d, J = 7.9 Hz, 2H), 8.10 (d, J = 8.7 Hz, 2H), 7.29 (s, 2H), 4.45 (t, J = 6.5 Hz, 2H), 4.37 (t, J = 6.6 Hz, 2H), 3.91 (s, 9H), 2.87 – 2.22 (m, 12H), 2.08 – 1.91 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 166.4, 164.6, 153.1, 136.8, 130.8, 128.7, 125.4, 106.9, 64.6, 63.6, 61.1, 56.4, 55.2, 55.0, 53.2, 26.3, 26.2. LC–MS (ESI +) m/z calcd. for C27H35FN2O9S [(M + H)]+: 583.21; found: 583.20. HPLC tR: 7.636 min.
3-(4-(4-((4-(fluorosulfonyl)benzoyl)oxy)butyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (9f). Final compound 9f was obtained from intermediates 8e and 4c following general procedure F. Column chromatography with 4 to 8% MeOH in DCM as mobile phase. Transparent oil (23.3 mg, 0.04 mmol, 10%). 1H NMR (400 MHz, CDCl3) δ [ppm] 8.31 – 8.25 (m, 2H), 8.13 – 8.06 (m, 2H), 7.29 (s, 2H), 4.41 (t, J = 6.5 Hz, 2H), 4.37 (t, J = 6.6 Hz, 2H), 3.91 (s, 9H), 2.84 – 2.27 (m, 12H), 1.98 (p, J = 6.8 Hz, 2H), 1.83 (p, J = 7.1 Hz, 2H), 1.72 – 1.58 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 166.4, 164.6, 153.0, 136.9, 130.8, 128.6, 125.5, 106.9, 66.1, 63.7, 61.1, 58.1, 56.4, 55.2, 53.3, 26.8, 26.4, 23.5. LC–MS (ESI +) m/z calcd. for C28H37FN2O9S [(M + H)]+: 597.23; found: 597.25. HPLC tR: 7.762 min.
3-(4-(3-((4-isothiocyanatobenzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (9g). To a stirred solution of benzoic acid 7c (136 mg, 0.76 mmol, 1.0 equiv) in DCM (7.5 mL) was added EDC·HCl (291 mg, 1.62 mmol, 2.0 equiv) and DMAP (cat.). Subsequently intermediate 5 (301 mg, 0.76 mmol, 1.0 equiv) was added and the reaction mixture was stirred for 16 h at rt under N2 atmosphere. The mixture was diluted with DCM (20 mL) after which the organic phase was washed twice with H2O (20 mL). The combined aqueous phases were extracted with DCM (20 mL) and the combined organic phases were dried over MgSO4, filtrated and concentrated in vacuo. Flash column chromatography on silica gel using a gradient of 0 to 5% MeOH in DCM as mobile phase provided final compound 9g. Yellow oil (34.2 mg, 0.06 mmol, 8%). 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 8.7 Hz, 2H), 7.29 (s, 2H), 7.31 – 7.23 (m, 2H), 4.37 (t, J = 6.6 Hz, 4H), 3.91 (s, 6H), 3.91 (s, 3H), 2.68 – 2.41 (m, 12H), 2.04 – 1.91 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 166.3, 165.5, 153.0, 142.3, 137.9, 135.7, 131.1, 128.9, 125.8, 125.4, 106.9, 63.9, 63.7, 61.0, 56.4, 55.2, 55.1, 53.2, 26.3, 26.3. LC–MS (ESI +) m/z calcd. for C28H35N3O7S [(M + H)]+: 558.22; found: 558.15. HPLC tR: 8.163 min.
3-(4-(3-((3-((tert-butoxycarbonyl)amino)benzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (9h). Intermediate 9h was obtained from intermediates 8f and 4c following general procedure F. Column chromatography with 0 to 4% MeOH in DCM as mobile phase. Yellow oil (134 mg, 0.22 mmol, 59%). 1H NMR (400 MHz, CDCl3) δ 7.91 (t, J = 1.9 Hz, 1H), 7.76 – 7.65 (m, 2H), 7.36 (t, J = 8.0 Hz, 1H), 7.29 (s, 2H), 6.77 (s, 1H), 4.37 (td, J = 6.5, 4.1 Hz, 4H), 3.91 (s, 9H), 2.72 – 2.37 (m, 12H), 2.02 – 1.90 (m, 4H), 1.52 (s, 9H). LC–MS (ESI +) m/z calcd. for C32H45N3O9 [(M + H)]+: 616.32; found: 616.30. HPLC tR: 7.955 min.
3-(4-(3-((4-((tert-butoxycarbonyl)amino)benzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (9i). Intermediate 9i was obtained from intermediates 8g and 4c following general procedure F. Column chromatography with 0 to 4% MeOH in DCM as mobile phase. Yellow oil (222 mg, 0.36 mmol, 84%). 1H NMR (400 MHz, CDCl3) δ 8.00 – 7.92 (m, 2H), 7.46 (d, J = 8.7 Hz, 2H), 7.29 (s, 2H), 7.06 (s, 1H), 4.36 (dt, J = 11.3, 6.5 Hz, 4H), 3.91 (s, 9H), 2.77 – 2.31 (m, 12H), 2.06 – 1.91 (m, 4H), 1.52 (s, 9H). LC–MS (ESI +) m/z calcd. for C32H45N3O9 [(M + H)]+: 616.32; found: 616.35. HPLC tR: 7.955 min.
3-(4-(4-((4-((tert-butoxycarbonyl)amino)benzoyl)oxy)butyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (9j). Intermediate 9j was obtained from intermediates 8h and 4c following general procedure F. Column chromatography with 0 to 4% MeOH in DCM as mobile phase. Yellow oil (226 mg, 0.36 mmol, 60%). 1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 8.7 Hz, 2H), 7.47 (d, J = 8.7 Hz, 2H), 7.29 (s, 2H), 7.20 (br s, 1H), 4.38 (t, J = 6.5 Hz, 2H), 4.31 (t, J = 6.4 Hz, 2H), 3.91 (s, 3H), 3.90 (s, 6H), 2.86 – 2.22 (m, 12H), 2.03 – 1.94 (m, 2H), 1.84 – 1.73 (m, 2H), 1.71 – 1.61 (m, 2H), 1.51 (s, 9H). LC–MS (ESI +) m/z calcd. for C33H47N3O9 [(M + H)]+: 630.34; found: 630.30. HPLC tR: 8.078 min.
3-(4-(3-((4-((tert-butoxycarbonyl)amino)-3-methoxybenzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (9k). Intermediate 9k was obtained from intermediates 8i and 4c following general procedure F. Column chromatography with 0 to 4% MeOH in DCM as mobile phase. Yellow oil (255 mg, 0.40 mmol, 72%). 1H NMR (400 MHz, CDCl3) δ 8.16 (d, J = 8.5 Hz, 1H), 7.66 (dd, J = 8.5, 1.8 Hz, 1H), 7.51 (d, J = 1.7 Hz, 1H), 7.29 (s, 3H), 4.36 (q, J = 6.8 Hz, 4H), 3.93 (s, 3H), 3.91 (s, 6H), 3.91 (s, 3H), 2.70 – 2.43 (m, 12H), 2.05 – 1.92 (m, 4H), 1.53 (s, 9H). LC–MS (ESI +) m/z calcd. for C33H47N3O10 [(M + H)]+: 646.33; found: 646.35. HPLC tR: 8.326 min.
3-(4-(3-((4-(((tert-butoxycarbonyl)amino)methyl)benzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (9l). Intermediate 9l was obtained from intermediates 8j and 4c following general procedure F. Column chromatography with 1 to 3% MeOH in DCM as mobile phase. Yellow oil (254 mg, 0.40 mmol, 40%). 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 8.1 Hz, 2H), 7.35 (d, J = 8.3 Hz, 2H), 7.29 (s, 2H), 5.41 (t, J = 6.1 Hz, 1H), 4.43 – 4.29 (m, 6H), 3.91 (s, 6H), 3.90 (s, 3H), 2.72 – 2.38 (m, 12H), 2.03 – 1.92 (m, 4H), 1.46 (s, 9H). LC–MS (ESI +) m/z calcd. for C33H47N3O9 [(M + H)]+: 630.34; found: 630.30. HPLC tR: 7.890 min.
3-(4-(3-((4-(2-((tert-butoxycarbonyl)amino)ethyl)benzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (9m). Intermediate 9m was obtained from intermediates 8k and 4c following general procedure F. Column chromatography with 0 to 4% MeOH in DCM as mobile phase. Yellow oil (132 mg, 0.21 mmol, 62%). 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 8.2 Hz, 2H), 7.29 (s, 2H), 7.27 (d, J = 8.0 Hz, 2H), 4.69 (t, J = 6.0 Hz, 1H), 4.42 – 4.34 (m, 4H), 3.91 (s, 6H), 3.91 (s, 3H), 3.39 (q, J = 6.8 Hz, 2H), 2.86 (t, J = 7.1 Hz, 2H), 2.69 – 2.42 (m, 12H), 2.04 – 1.91 (m, 4H), 1.43 (s, 9H). LC–MS (ESI +) m/z calcd. for C34H49N3O9 [(M + H)]+: 644.35; found: 644.35. HPLC tR: 8.014 min.
3-(4-(3-((3-acrylamidobenzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (10a). Final compound 10a was obtained from intermediate 9h following general procedure N. Column chromatography with 3 to 5% MeOH in DCM as mobile phase. Transparent oil (10.3 mg, 0.02 mmol, 8%). 1H NMR (400 MHz, CDCl3) δ 8.29 – 7.98 (m, 3H), 7.76 (dt, J = 7.8, 1.4 Hz, 1H), 7.41 (t, J = 8.2 Hz, 1H), 7.29 (s, 2H), 6.47 (dd, J = 16.9, 1.7 Hz, 1H), 6.37 (dd, J = 16.8, 9.9 Hz, 1H), 5.78 (dd, J = 9.8, 1.7 Hz, 1H), 4.41 – 4.30 (m, 4H), 3.91 (s, 9H), 2.95 – 2.48 (m, 12H), 2.10 – 1.96 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 166.4, 166.2, 163.9, 153.0, 142.2, 138.4, 131.1, 131.0, 129.3, 128.3, 125.4, 124.6, 120.9 106.9, 63.7, 63.5, 61.0, 56.3, 55.1, 55.0, 53.0, 52.9, 26.2, 26.1. LC–MS (ESI +) m/z calcd. for C30H39N3O8 [(M + H)]+: 570.28; found: 570.25. HPLC tR: 7.006 min.
3-(4-(3-((4-acrylamidobenzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (10b). Final compound 10b was obtained from intermediate 9i following general procedure N. Column chromatography with 1 to 6% MeOH in DCM as mobile phase. Transparent oil (80.7 mg, 0.14 mmol, 55%). 1H NMR (400 MHz, CDCl3) δ 8.17 (s, 1H), 7.99 (d, J = 8.7 Hz, 2H), 7.72 (d, J = 8.9 Hz, 2H), 7.29 (s, 2H), 6.47 (dd, J = 16.8, 1.4 Hz, 1H), 6.33 (dd, J = 16.9, 10.1 Hz, 1H), 5.79 (dd, J = 10.1, 1.4 Hz, 1H), 4.40 – 4.32 (m, 4H), 3.91 (s, 3H), 3.90 (s, 6H), 2.78 – 2.44 (m, 12H), 2.04 – 1.94 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 166.4, 166.2, 163.9, 153.0, 142.4, 131.0, 130.9, 128.7, 125.8, 125.3, 119.2, 106.8, 63.5, 63.2, 61.0, 56.3, 55.0, 52.7, 26.0. LC–MS (ESI +) m/z calcd. for C30H39N3O8 [(M + H)]+: 570.28; found: 570.30. HPLC tR: 7.039 min.
3-(4-(4-((4-acrylamidobenzoyl)oxy)butyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (10c). Final compound 10c was obtained from intermediate 9j following general procedure N. Column chromatography with 3 to 5% MeOH in DCM as mobile phase. Transparent oil (164 mg, 0.28 mmol, 78%). 1H NMR (400 MHz, CDCl3) δ 8.74 (s, 1H), 7.97 (d, J = 8.8 Hz, 2H), 7.76 (d, J = 8.8 Hz, 2H), 7.29 (s, 2H), 6.52 – 6.36 (m, 2H), 5.76 (dd, J = 8.9, 2.6 Hz, 1H), 4.37 (t, J = 6.5 Hz, 2H), 4.30 (t, J = 6.1 Hz, 2H), 3.91 (s, 3H), 3.90 (s, 6H), 2.87 – 2.43 (m, 12H), 2.00 (p, J = 6.7 Hz, 2H), 1.84 – 1.66 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 166.3, 166.2, 164.1, 152.9, 142.6, 142.2, 131.1, 130.7, 128.5, 125.6, 125.3, 119.2, 106.8, 64.5, 63.4, 61.0, 57.8, 56.3, 54.8, 52.7, 52.3, 26.6, 26.0, 22.8. LC–MS (ESI +) m/z calcd. for C31H41N3O8 [(M + H)]+: 584.30; found: 584.25. HPLC tR: 7.149 min.
3-(4-(3-((4-acrylamido-3-methoxybenzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (10d). Final compound 10d was obtained from intermediate 9k following general procedure N. Column chromatography with 3% MeOH in DCM as mobile phase. Transparent oil (83.5 mg, 0.14 mmol, 48%). 1H NMR (400 MHz, CDCl3) δ 8.56 (d, J = 8.5 Hz, 1H), 8.07 (s, 1H), 7.69 (dd, J = 8.5, 1.8 Hz, 1H), 7.55 (d, J = 1.8 Hz, 1H), 7.29 (s, 2H), 6.46 (dd, J = 16.8, 1.3 Hz, 1H), 6.32 (dd, J = 16.9, 10.1 Hz, 1H), 5.81 (dd, J = 10.1, 1.3 Hz, 1H), 4.41 – 4.33 (m, 4H), 3.96 (s, 3H), 3.91 (s, 6H), 3.91 (s, 3H), 2.93 – 2.38 (m, 12H), 2.03 – 1.93 (m, 4H). 13C NMR (126 MHz, CDCl3) δ 166.3, 166.3, 163.5, 153.0, 147.5, 142.2, 131.9, 131.3, 128.4, 125.4, 125.4, 123.4, 118.9, 110.7, 106.8, 63.6, 63.5, 61.0, 56.3, 56.1, 55.1, 53.1, 26.3. LC–MS (ESI +) m/z calcd. for C31H41N3O9 [(M + H)]+: 600.29; found: 600.30. HPLC tR: 6.878 and 7.263 min.
3-(4-(3-((4-(acrylamidomethyl)benzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (10e). Final compound 10e was obtained from intermediate 9l following general procedure N. Column chromatography with 3 to 5% MeOH in DCM as mobile phase. Transparent oil (40.3 mg, 0.07 mmol, 17%). 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 8.3 Hz, 2H), 7.35 (d, J = 8.4 Hz, 2H), 7.28 (s, 2H), 6.34 (dd, J = 17.0, 1.5 Hz, 1H), 6.30 (t, J = 6.2 Hz, 1H), 6.17 (dd, J = 17.0, 10.2 Hz, 1H), 5.69 (dd, J = 10.2, 1.5 Hz, 1H), 4.57 (d, J = 6.0 Hz, 2H), 4.39–4.33 (m, 4H), 3.91 (s, 6H), 3.90 (s, 3H), 2.79 – 2.51 (m, 12H), 2.05–1.95 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 166.4, 166.3, 165.7, 153.0, 143.6, 142.3, 130.5, 130.0, 129.5, 127.7, 127.3, 125.3, 106.9, 63.4, 63.2, 61.0, 56.4, 55.0, 54.9, 52.7, 43.3, 26.0. LC–MS (ESI +) m/z calcd. for C31H41N3O8 [(M + H)]+: 584.30; found: 584.25. HPLC tR: 6.806 min.
3-(4-(3-((4-(2-acrylamidoethyl)benzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (10f). Final compound 10f was obtained from intermediate 9m following general procedure N. Column chromatography with 3 to 6% MeOH in DCM as mobile phase. Transparent oil (34.8 mg, 0.06 mmol, 28%). 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 8.3 Hz, 2H), 7.30 – 7.26 (m, 4H), 6.27 (dd, J = 17.0, 1.4 Hz, 1H), 6.04 (dd, J = 17.0, 10.3 Hz, 1H), 5.67 (t, J = 5.9 Hz, 1H), 5.64 (dd, J = 10.3, 1.4 Hz, 1H), 4.39 – 4.34 (m, 4H), 3.91 (s, 6H), 3.91 (s, 3H), 3.62 (q, J = 6.8 Hz, 2H), 2.93 (t, J = 7.0 Hz, 2H), 2.64 – 2.48 (m, 12H), 2.03 – 1.93 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 166.5, 166.3, 165.7, 153.0, 144.4, 142.3, 130.7, 130.0, 128.9, 128.8, 126.8, 125.4, 106.9, 63.6, 63.4, 61.1, 56.4, 55.2, 53.1, 40.5, 35.8, 26.3. LC–MS (ESI +) m/z calcd. for C32H43N3O8 [(M + H)]+: 598.31; found: 598.30. HPLC tR: 6.968 min.
4-(2-((tert-butoxycarbonyl)amino)ethoxy)-3,5-dimethoxybenzoic acid (11a). Intermediate 11a was obtained from tert-butyl (2-bromoethyl)carbamate following general procedure M. Column chromatography with 1 to 4% MeOH in DCM as mobile phase. Transparent oil (785 mg, 2.30 mmol, 77%). 1H NMR (400 MHz, CDCl3) δ 10.40 (s, 1H), 7.37 (s, 2H), 5.97 (t, J = 5.5 Hz, 1H), 4.15 (t, J = 4.9 Hz, 2H), 3.93 (s, 6H), 3.44 (q, J = 5.1 Hz, 2H), 1.47 (s, 9H). LC–MS (ESI-) m/z calcd. for C16H23NO7 [(M-H)]−: 340.14; found: 340.10. HPLC tR: 9.356 min.
4-(3-((tert-butoxycarbonyl)amino)propoxy)-3,5-dimethoxybenzoic acid (11b). Intermediate 11b was obtained from tert-butyl (3-bromopropyl)carbamate following general procedure M. Column chromatography with 1 to 4% MeOH in DCM as mobile phase. Transparent oil (1.25 g, 3.52 mmol, 88%). 1H NMR (400 MHz, CDCl3) δ 9.91 (s, 1H), 7.37 (s, 2H), 5.95 (br t, J = 6.0 Hz, 1H), 4.14 (t, J = 5.8 Hz, 2H), 3.92 (s, 6H), 3.49 – 3.36 (m, 2H), 2.04 – 1.89 (m, 2H), 1.48 (s, 9H). LC–MS (ESI-) m/z calcd. for C17H25NO7 [(M-H)]−: 354.16; found: 354.10. HPLC tR: 9.600 min.
3-bromopropyl 4-(2-((tert-butoxycarbonyl)amino)ethoxy)-3,5-dimethoxybenzoate (12a). Intermediate 12a was obtained from intermediate 9a following general procedure D. Column chromatography with 10 to 15% EtOAc in PE as mobile phase. Transparent oil (286 mg, 0.62 mmol, 45%). 1H NMR (400 MHz, CDCl3) δ 7.30 (s, 2H), 5.84 (t, J = 5.6 Hz, 1H), 4.51 – 4.44 (m, 2H), 4.12 (t, J = 4.9 Hz, 2H), 3.92 (s, 6H), 3.54 (t, J = 6.5 Hz, 2H), 3.40 (q, J = 5.2 Hz, 2H), 2.34 (p, J = 6.3 Hz, 2H), 1.45 (s, 9H). LC–MS (ESI +) m/z calcd. for C19H28BrNO7 [(M + H)]+: 462.11; found: 462.10. HPLC tR: 11.734 min.
3-bromopropyl 4-(3-((tert-butoxycarbonyl)amino)propoxy)-3,5-dimethoxybenzoate (12b). Intermediate 12b was obtained from intermediate 9b following general procedure D. Column chromatography with 0.5 to 1.5% MeOH in DCM as mobile phase. Transparent oil (800 mg, 1.68 mmol, 39%). 1H NMR (400 MHz, CDCl3) δ 7.30 (s, 2H), 5.73 (br s, 1H), 4.47 (t, J = 6.1 Hz, 2H), 4.10 (t, J = 5.7 Hz, 2H), 3.92 (s, 6H), 3.55 (td, J = 6.5, 4.6 Hz, 4H), 2.34 (p, J = 6.3 Hz, 2H), 2.10 (p, J = 6.2 Hz, 2H), 1.46 (s, 9H). LC–MS (ESI +) m/z calcd. for C20H30BrNO7 [(M + H)]+: 476.13; found: 476.10. HPLC tR: 11.876 min.
3-(4-(3-((4-(2-((tert-butoxycarbonyl)amino)ethoxy)-3,5-dimethoxybenzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (13a). Intermediate 13a was obtained from intermediates 12a and 4c following general procedure F. Automatic column chromatography with 3 to 5% MeOH in DCM as mobile phase on Biotage Isolera One. Yellow oil (136 mg, 0.19 mmol, 37%). 1H NMR (400 MHz, CDCl3) δ 7.29 (s, 4H), 5.86 (t, J = 5.5 Hz, 1H), 4.38 (t, J = 6.6, 1.4 Hz, 4H), 4.12 (t, J = 4.9 Hz, 2H), 3.92 (s, 6H), 3.91 (s, 6H), 3.91 (s, 3H), 3.40 (q, J = 5.1 Hz, 2H), 2.75 – 2.36 (m, 12), 2.04 – 1.92 (m, 4H), 1.46 (s, 9H). LC–MS (ESI +) m/z calcd. for C36H53N3O12 [(M + H)]+: 720.37; found: 720.30. HPLC tR: 8.033 min.
3-(4-(3-((4-(3-((tert-butoxycarbonyl)amino)propoxy)-3,5-dimethoxybenzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (13b). Intermediate 13b was obtained from intermediates 12b and 4c following general procedure F. Automatic column chromatography with 3 to 5% MeOH in DCM as mobile phase on Biotage Isolera One. Yellow oil (305 mg, 0.42 mmol, 53%). 1H NMR (400 MHz, CDCl3) δ 7.30 (s, 2H), 7.30 (s, 2H), 5.79 (t, J = 5.8 Hz, 1H), 4.38 (t, J = 6.5 Hz, 4H), 4.11 (t, J = 5.7 Hz, 2H), 3.92 (s, 6H), 3.91 (s, 6H), 3.91 (s, 3H), 3.42 (q, J = 6.1 Hz, 2H), 2.81 – 2.31 (m, 12H), 2.07 – 1.87 (m, 6H), 1.47 (s, 9H). LC–MS (ESI +) m/z calcd. for C37H55N3O12 [(M + H)]+: 734.39; found: 734.40. HPLC tR: 8.259 min.
3-(4-(3-((4-(2-acrylamidoethoxy)-3,5-dimethoxybenzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (14a). Final compound 14a was obtained from intermediate 13a following general procedure N. Column chromatography with 4 to 8% MeOH in DCM as mobile phase. Transparent oil (65.6 mg, 0.09 mmol, 15%). 1H NMR (400 MHz, CDCl3) δ 7.31 (s, 2H), 7.29 (s, 2H), 6.87 (br t, J = 5.4 Hz, 1H), 6.30 (dd, J = 17.0, 1.5 Hz, 1H), 6.15 (dd, J = 17.0, 10.2 Hz, 1H), 5.67 (dd, J = 10.2, 1.6 Hz, 1H), 4.41 – 4.35 (m, 4H), 4.17 (t, J = 5.0 Hz, 2H), 3.92 (s, 6H), 3.91 (s, 9H), 3.63 (q, J = 5.2 Hz, 2H), 2.70 – 2.41 (m, 12H), 1.99 (p, J = 6.8 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ 166.3, 166.1, 165.5, 153.0, 152.9, 142.2, 140.5, 131.2, 126.2, 126.0, 125.4, 106.8, 72.7, 63.8, 63.6, 61.0, 56.4, 56.3, 55.1, 53.2, 39.4, 26.3. LC–MS (ESI +) m/z calcd. for C34H47N3O11 [(M + H)]+: 674.33; found: 674.35. HPLC tR: 7.183 min.
3-(4-(3-((4-(3-acrylamidopropoxy)-3,5-dimethoxybenzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (14b). Final compound 14b was obtained from intermediate 13b following general procedure N. Column chromatography with 3 to 6% MeOH in DCM as mobile phase. Transparent oil (105 mg, 0.15 mmol, 25%). 1H NMR (400 MHz, DMSO-d6) δ 8.14 (t, J = 5.6 Hz, 1H), 7.26 (s, 2H), 7.26 (s, 2H), 6.21 (dd, J = 17.1, 10.1 Hz, 1H), 6.06 (dd, J = 17.1, 2.3 Hz, 1H), 5.56 (dd, J = 10.1, 2.3 Hz, 1H), 4.34 (t, J = 6.1 Hz, 4H), 3.97 (t, J = 6.3 Hz, 2H), 3.85 (s, 6H), 3.84 (s, 6H), 3.73 (s, 3H), 3.33 – 3.23 (m, 14H), 2.27 – 2.12 (m, 4H), 1.79 (p, J = 6.6 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.3, 165.3, 164.6, 152.9, 152.8, 141.9, 140.9, 131.8, 124.9, 124.6, 124.6, 106.7, 106.7, 70.7, 62.0, 60.2, 56.2, 56.1, 35.8, 29.8. LC–MS (ESI +) m/z calcd. for C35H49N3O11 [(M + H)]+: 688.34; found: 688.30. HPLC tR: 7.254 min.
3-(4-(3-((4-(3-isothiocyanatopropoxy)-3,5-dimethoxybenzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (14c). Intermediate 13b was dissolved in DCM (0.2 M) and allowed to cool down to 0 °C after which TFA (20 equiv) was added dropwise. The mixture was stirred for 4 h, concentrated in vacuo and co-evaporated with toluene to remove the excess TFA. The obtained deprotected amine (171 mg, 0.18 mmol, 1.0 equiv) was dissolved in THF (1.8 mL) and the stirred solution was allowed to cool down to 0 °C. Subsequently triethylamine (0.07 mL, 0.53 mmol, 3.0 equiv) and thiophosgene (0.02 mL, 0.19 mmol, 1.1 equiv) were added dropwise and the reaction mixture stirred for 0.5 h at 0 °C under N2 atmosphere. The mixture was diluted with DCM (40 mL) and subsequently washed with H2O (40 mL). The aqueous phase was extracted twice with DCM (40 mL) after which the organic phases were combined, dried over MgSO4, filtrated and concentrated in vacuo. Flash column chromatography on silica gel using a gradient of 3 to 5% MeOH in DCM as mobile phase provided final compound 14c. Yellow oil (12.0 mg, 0.02 mmol, 10%). 1H NMR (400 MHz, CDCl3) δ 7.29 (s, 2H), 7.28 (s, 2H), 4.41 – 4.33 (m, 4H), 4.12 (t, J = 5.6 Hz, 2H), 3.91 (s, 15H), 3.91 – 3.85 (m, 2H), 2.54 – 2.40 (m, 12H), 2.14 – 2.04 (m, 2H), 2.03 – 1.92 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 166.4, 166.3, 153.2, 153.1, 140.7, 125.9, 125.5, 106.9, 106.7, 69.2, 63.8, 63.7, 61.1, 56.4, 56.4, 55.2, 53.3, 41.9, 30.8, 26.4. LC–MS (ESI +) m/z calcd. for C33H45N3O10S [(M + H)]+: 676.29; found: 676.25. HPLC tR: 8.371 min.
3-(4-(3-((3,5-dimethoxy-4-(3-(vinylsulfonamido)propoxy)benzoyl)oxy)propyl)piperazin-1-yl)propyl 3,4,5-trimethoxybenzoate (14d). Intermediate 13b was dissolved in DCM (0.2 M) and allowed to cool down to 0 °C after which TFA (20 equiv) was added dropwise. The mixture was stirred for 4 h, concentrated in vacuo and co-evaporated with toluene to remove the excess TFA. The obtained deprotected amine (240 mg, 0.38 mmol, 1.0 equiv) was dissolved in DCM (8.0 mL) and the stirred solution was allowed to cool to 0 °C. Triethylamine (0.26 mL, 1.89 mmol, 5.0 equiv) and 2-chloroethane-1-sulfonyl chloride (0.05 mL, 0.45 mmol, 1.2 equiv) were added and the reaction mixture stirred for 0.5 h at 0 °C. The reaction was quenched with 20 mL of sat. NH4Cl solution and subsequently extracted three times with DCM (30 mL). The combined organic phases were dried over MgSO4, filtrated and concentrated in vacuo. Automatic column chromatography (C18) with 0 to 90% CH3CN in H2O + 0.1% TFA as mobile phase on Biotage® Selekt provided final compound 14d. Transparent oil (58.0 mg, 0.08 mmol, 21%). 1H NMR (400 MHz, CDCl3) δ 7.26 (s, 2H), 7.25 (s, 2H), 6.54 (dd, J = 16.6, 9.9 Hz, 1H), 6.24 (d, J = 16.6 Hz, 1H), 6.19 (br s, 1H), 5.92 (d, J = 9.9 Hz, 1H), 4.40 (t, J = 5.9 Hz, 4H), 4.18 (t, J = 5.5 Hz, 2H), 3.92 (s, 6H), 3.91 (s, 9H), 3.63 (s, 8H), 3.34 – 3.27 (m, 2H), 3.25 – 3.17 (m, 4H), 2.31 – 2.19 (m, 4H), 1.99 (p, J = 5.8 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 166.2, 166.0, 163.0, 162.6, 153.1, 152.8, 142.7, 141.0, 136.3, 126.2, 124.7, 124.4, 117.6, 114.7, 72.1, 61.5, 61.4, 61.1, 56.4, 56.3, 54.5, 54.5, 48.9, 48.8, 41.6, 29.1, 23.9. LC–MS (ESI +) m/z calcd. for C34H49N3O12S [(M + H)]+: 724.31; found: 724.25. HPLC tR: 7.548 min.
[3H]NBTI displacement assays
Dilazep and ST7092 were obtained from ASTA-Werke AG (Bielefeld, Germany) and Chemie Linz AG (Linz, Austria), respectively. Radioligand displacement assays were performed as previously described [11]. In brief, the experiments were performed on erythrocyte membranes (Sanquin Bloedvoorziening, Amsterdam, The Netherlands) using a single concentration of 1 µM or multiple concentrations (ranging from 10–10 to 10–6) of unlabeled inhibitors in the presence of 4 nM [3H]NBTI at 10 °C for 1 h in assay buffer (50 mM Tris–HCl, pH 7.4, 0.1% w/v CHAPS). Non-specific binding (NSB) was assessed with 10 µM unlabeled NBTI. To characterize time-dependent affinity, erythrocyte membranes were preincubated with potentially covalent inhibitors for 4 h at 10 °C. Subsequently, [3H]NBTI was added to the preincubated and control samples (containing erythrocyte membranes, unlabeled inhibitor and radioligand) and both types of samples were incubated for another hour. Incubation was terminated by rapid vacuum filtration over Whatman™ UniFilter™ 96-well GF/C microplates using a FilterMate™ Universal Harvester (PerkinElmer, Groningen, The Netherlands). Subsequently, the plates were washed ten times with ice-cold wash buffer (50 mM Tris–HCl, pH 7.4). Thereafter, the filters were dried for 45 min at 55 °C and 25 µL of Microscint (PerkinElmer) was added per well. The filter-bound radioactivity was determined by scintillation spectrometry using a MicroBeta2 2450 Microplate Counter (PerkinElmer).
Wash-out [3H]NBTI displacement assays
Wash-out assays (as described previously [11]) were performed by incubation of competing inhibitors at a final concentration of 10 × Ki (as determined in displacement assays) with erythrocyte membranes at 10 °C for 1 h while gently shaking. Subsequently the samples were centrifuged at 13,200 rpm (16,100 g) at 4 °C for 5 min and the supernatant containing the unbound inhibitor was aspirated. Pellets were resuspended in 1 mL of assay buffer (50 mM Tris–HCl, pH 7.4, 0.1% w/v CHAPS), and samples were incubated at 10 °C for 10 min. After four cycles of centrifugation and washing, the supernatant was discarded and the membranes were resuspended in a total volume of 400 μL containing 4 nM [3H]NBTI and subsequently incubated at 10 °C for 60 min. Incubations were terminated by rapid vacuum filtration through GF/C filters using a Brandel harvester (Brandel, Gaithersburg, MD, USA). Filters were washed three times with ice-cold wash buffer (50 mM Tris–HCl, pH 7.4) and collected in tubes. Emulsifier-Safe™ scintillation fluid (PerkinElmer) was added and samples were counted by scintillation spectrometry using a Tri-Carb 2900TR liquid scintillation analyzer (PerkinElmer, Boston, MA, USA). All experimental data was analyzed using GraphPad Prism 10.0.2 software (GraphPad Software Inc., San Diego, CA, USA). [3H]NBTI assays were baseline-corrected with NSB and normalized to this value (0%) and washed or unwashed TB (100%), respectively. Data shown represent the mean ± SD of three individual experiments each performed in duplicate. Differences between unwashed and washed was determined in an unpaired Student’s t-test with Welch’s correction. Significant differences are displayed as * p < 0.05.
Data analysis
For data analysis, dilazep and ST7092 were used as positive controls. All experimental data was analyzed using GraphPad Prism 10.0.2 software (GraphPad Software Inc., San Diego, CA, USA). [3H]NBTI assays were baseline-corrected with NSB and normalized to this value (0%) and TB (100%). The radioligand displacement curves were fitted to a one-site binding model by nonlinear regression from which the pIC50 values were obtained. Additionally, pKi values were calculated from pIC50 values and the saturation KD value (1.1 nM [11]) via the Cheng-Prusoff equation [19]: Ki = IC50/(1 + [radioligand]/KD). Data shown represent the mean ± SD of at least two or mean ± SEM of at least three individual experiments each performed in duplicate.
Molecular docking
The binding poses of dilazep derivatives on hENT1 were predicted using molecular docking with ICM-Pro version 3.9-2c (Molsoft LLC, San Diego). Protein and ligand preparation was previously done by converting ENT1 X-ray structure co-crystalized with NBTI (PDB: 6OB6) and dilazep (PDB: 6OB7) to ICM objects. The proteins were prepared by adding and optimizing the position of hydrogen atoms, as well as the orientation and protonation states of histidine and cysteine residues and the orientation of glutamine and asparagine residues. Moreover, the stabilizing mutations F168L and A175P were reverted. Prior to docking, 6OB6 and 6OB7 were aligned based on their 3D structure to superimpose the location of NBTI and dilazep. Protonation states of the ligands to be docked (dilazep, ST7092, 6m, 6n, 10b, 10e, and 14b) were computed, resulting in monoprotonated and unprotonated (homo)piperazine rings. All protonation states of the homopiperazine and piperazine rings were kept for docking, but only the most biologically active unprotonated state [18] was considered for further analysis. Docking was performed on ICM-Pro with default settings after generating receptor maps with both dilazep and NBTI as the center of the binding pocket in the 6OB7 structure. This resulted in a binding pocket of 48 residues. Three poses were generated for each of the ligands with a thoroughness of 3.00. Furthermore, compound 10b was covalently docked in ICM-Pro with custom covalent reactions targeting C439. The results from docking were subsequently filtered and analyzed considering the predicted docking scores and interactions, as well as the available experimental data. The best poses in terms of docking score that maintained the general binding mode of dilazep were selected for further analysis and were visualized using PyMOL version 2.5.2 [20]. For visualization purposes, residues 440–451 in TM2 were hidden in the representations.
Results
Design and synthesis
Several compounds were designed and synthesized with different substitution patterns on one or both phenyl rings of parent structure ST7092 (Scheme 1). Additionally, several potentially covalent inhibitors were designed and synthesized (Scheme 2). From early analysis of the hENT1 binding pocket, C439 emerged as potential target amino acid residue for covalent binding (supplementary Figure S1b). In order to establish an irreversible interaction, a variety of electrophilic warheads were installed at one site of the trimethoxy benzoate. All compounds synthesized in this study, generally followed a convergent synthesis approach similar as described by Playa et al. [17].

Synthesis of dilazep derivatives 6a-6v. Reagents and conditions: a) Fmoc-Cl, K2CO3, dioxane, 0 °C, 21–41 h, 28–38%; b) 3-bromopropan-1-ol, Et3N, DCM, 0 °C to rt, 6–24 h, 68%-quant.; c) i. SOCl2, reflux, 3 h; ii. 3-bromopropan-1-ol, Et3N, DCM, 0 °C to rt, 17–29 h, 21–84%; d) EDC·HCl, DMAP, DCM, rt, 20–22 h, 28–60%; e) 1-Boc-piperazine, K2CO3, KI, anhydrous DMF, rt, 3 days, 53–89%; f) TFA, DCM, 0 °C, 4 h, quant.; g) 3-bromopropan-1-ol, K2CO3, KI, anhydrous DMF, rt to 50 °C, 16 h, 65%; h) 2a-2k or 2n, K2CO3, KI, anhydrous DMF, rt to 50 °C, 3 days, 14–87%; i) piperazine, K2CO3, KI, anhydrous DMF, rt to 50 °C, 3 days, 15–78%; j) corresponding R2-benzoyl chloride, Et3N, DCM, 0 °C to rt, 2 h, 14–94%. k) i. corresponding R2-benzoic acid, SOCl2, reflux, 3 h; ii. Et3N, DCM, 0 °C to rt, 4 h, 33–73%

Synthesis of warhead-containing dilazep derivatives. Reagents and conditions: a) 2 M KHF2 (aqueous), dioxane, rt, 1 h, 94–98%; b) Boc2O, Et3N or NaOH, dioxane/water, rt, 17–25 h, 70%-quant.; c) CSCl2, acetone, 0 °C to rt, 16 h, 10–38%; d) i. SOCl2, reflux, 3 h; ii. corresponding bromoalcohol, Et3N, DCM, 0 °C to rt, 17–29 h, 41–100%; e) i. oxalyl chloride, cat. DMF, DCM, 0 °C, 4 h; ii. corresponding bromoalcohol, Et3N, anhydrous dioxane or DCM, rt, 16–23 h, 15–57%; f) 4c (from Scheme 1), K2CO3, KI, anhydrous DMF, rt, 16 h-4 days, 2–84%; g) i. SOCl2, reflux, 3 h; ii. 5 (from Scheme 1), Et3N, DCM, 0 °C to rt, 16 h, 5%; h) 5 (from Scheme 1), EDC·HCl, DMAP, DCM, rt, 16 h, 8%; i) TBAOH, 2-(Boc-amino)ethyl bromide or 3-(Boc-amino)propyl bromide, THF, 0 °C, 2 days, 59–88%; j) EDC·HCl, DMAP, DCM, rt, 4–17 h, 39–45%; k) i. TFA, DCM, 0 °C to rt, 4 h, quant. ii. acryloyl chloride, Et3N, DCM, 0 °C to rt, 0.5–19 h, 8–78%; l) i. TFA, DCM, 0 °C to rt, 4 h, quant. ii. 2-chloroethanesulfonyl chloride, Et3N, DCM, 0 °C, 0.5 h, 21%
The synthesis of non-covalent compounds either started with substituted benzoyl chlorides or, where substituted benzoyl chlorides were not commercially available, the in situ conversion of substituted benzoic acids into their corresponding acyl chlorides using SOCl2, followed by a nucleophilic substitution by 3-bromopropan-1-ol to obtain bromopropyl benzoates 2a-2m and 2p. In the case of 2i and 2j, commercially available methylamino benzoic acids were first protected using Fmoc-Cl to yield intermediate 1a and 1b prior to the aforementioned acylation. Lastly, bromopropyl benzoates 2n and 2o were synthesized from their corresponding benzoic acids via a Steglich esterification. Three of these building blocks (2k, 2o and 2p) were subsequently substituted with 1-Boc-piperazine, followed by removal of the Boc group using TFA to give secondary amines 4a-4c. With the use of intermediates 2a-2k, alkylation of secondary amine 4c resulted in the synthesis of final compounds 6a-6k. Similarly, final compounds 6u and 6v were synthesized from alkylation of intermediates 4a and 4b using bromopropyl benzoate 2n. Towards final compounds 6l-6o, piperazine intermediate 4c was first alkylated with 3-bromopropan-1-ol to give intermediate 5. This was subsequently acylated with either commercially available or in situ synthesized benzoyl chlorides to give the final compounds. Finally, bis-alkylation of piperazine with the corresponding bromopropyl benzoates resulted in the synthesis of symmetrical ST7092 derivatives 6p-6t.
Similar to the non-covalent inhibitors, the potentially covalent inhibitors started with the synthesis of different bromopropyl benzoate building blocks (Scheme 2). Firstly, fluorosulfonyl-warhead-substituted benzoic acids (7a and 7b) were prepared from the commercially available chlorosulfonylbenzoic acids through treatment with an aqueous solution of 2 M KHF2. Subsequently, intermediates 7a and 7b, were used in similar conditions as mentioned before, using in situ acyl chloride-formation by thionyl chloride, followed by substitution with the corresponding bromoalcohol to synthesize bromoalkyl benzoates 8a-8e. Alternatively, 7a was also directly acylated onto intermediate 5 (Scheme 1) to furnish final compound 9b. Using bromoalkyl benzoates 8a-8e in a nucleophilic substitution on intermediate 4c (Scheme 1) gave final compounds 9a and 9c-9f. Additionally, several commercially available amine-substituted benzoic acids were Boc-protected to give intermediates 7d-7h in high yields, which were likewise converted to their corresponding bromoalkyl benzoates 8f-8k through oxalyl chloride and a catalytic amount of DMF followed by bromoalcohol substitution. Alkylation of secondary amine 4c (Scheme 1) with the use of intermediates 8f-8k, gave potentially covalent inhibitor precursors 9h-9n which were deprotected using TFA. Subsequently, an acrylamide warhead was installed on the formed free amine to obtain final compounds 10a-10f. In the presence of thiophosgene, 4-aminobenzoic acid was converted to the isothiocyanate-substituted intermediate 7c, which was used to obtain final compound 9g from intermediate 5 (Scheme 1) by a Steglich esterification in presence of EDC and DMAP.
To include potentially covalent inhibitors with warheads installed on one of the existing methoxy substituents of ST7092, syringic acid was alkylated with the appropriate Boc-protected bromoalkylamine in a chemoselective manner using a solution of tetrabutylammonium hydroxide to give intermediates 11a and 11b [21]. Subsequent Steglich esterification of benzoic acids 11a and 11b with bromopropan-1-ol resulted in the bromopropyl benzoates 12a and 12b. Nucleophilic substitution of intermediates 12a and 12b with the use of secondary amine 4c (Scheme 1) provided precursors 13a and 13b. To obtain acrylamide substituted final compounds 14a and 14b, the aforementioned conditions with TFA and acryloyl chloride were used. Finally, intermediate 13b was similarly deprotected, followed by treatment with thiophosgene or 2-chloroethanesulfonyl chloride to provide final compounds 14c-14d, bearing an isothiocyanate and vinylsulfonamide warhead, respectively.
Pharmacological characterization
To determine the affinities of the non-covalent hENT1 inhibitors, a [3H]NBTI displacement assay was performed on erythrocyte cell membranes endogenously expressing the transporter. All compounds were initially screened at a concentration of 1 µM to provide the percentage displacement. If more than 75% displacement was observed, compounds were subsequently characterized in full concentration-effect curves to determine their affinity.
Dilazep and close analogue ST7092 both displayed high affinity (pKi values of 9.39 ± 0.06 vs. 8.75 ± 0.04, respectively) as was observed previously in literature (Table 1) [18]. Exchanging one of the trimethoxy benzoate moieties of ST7092 with hydrophobic or electron-withdrawing substituents (compounds 6a-6f) completely abolished inhibitory activity of hENT1. Therefore, the influence of hydrogen bond donating and accepting groups was examined with the use of compounds 6g-6j. These para- and meta-substituted dimethylamine and methylamine inhibitors, showed low [3H]NBTI displacement (6% to 21%), indicating the necessity of having methoxy substituents on both sides of the molecule. Hence, the minimal requirements for methoxy substitutions were investigated since removal of one of the trimethoxy benzoates resulted in major loss of inhibitory activity of these compounds. Symmetrical compounds 6p-6r as well as asymmetrical compounds 6k, 6l, 6t and 6u showed little to no displacement of [3H]NBTI. Other asymmetrical methoxy substitution patterns (6m, 6s and 6v) displayed around 30% [3H]NBTI displacement, whereas removal of one of the methoxy substituents in compounds 6n and 6o led to submicromolar affinities with pKi values of 6.87 ± 0.03 and 6.84 ± 0.06, respectively.
Next, the potentially covalent inhibitors that we synthesized were subjected to the aforementioned radioligand displacement assay. To study the existence of a covalent interaction with hENT1, an additional preincubation step was included. The compounds of interest were either preincubated with the erythrocyte cell membranes for 4 h or not to determine the time-dependent shift in affinity expected upon covalent binding with C439.
Substitution of a fluorosulfonyl warhead on the para or meta position in compounds 9a-9f, in combination with linker lengths between 2 and 4 carbon atoms, resulted in low [3H]NBTI displacement (a maximum of 61%) that seemed to slightly increase with linker length. No noteworthy differences between 4 h preincubation and without preincubation were found (Table 2). Introduction of an isothiocyanate (9g) resulted in decent, albeit similar affinity values with and without preincubation (7.31 ± 0.04 and 7.53 ± 0.10, respectively). Next an acrylamide warhead was introduced onto the ST7092 scaffold in various orientations (10a-10f). Binding of compound 10b resulted in an pKi value of 7.48 ± 0.15 without preincubation and a similar affinity with 4 h preincubation (7.36 ± 0.22), while close analogues 10a and 10d were only able to displace around 20% of [3H]NBTI binding. The absence of a pKi shift for compound 10b led to the design of compounds 10c, 10e and 10f to accommodate a close proximity to C439. Unfortunately, increasing linker length at the alkyl chain between the piperazine- and acrylamide-substituted benzoate (10c) and the phenyl ring and acrylamide warhead (10e and 10f) decreased hENT1 binding (between 7 and 47% displacement). In order to investigate the importance of all methoxy substituents to accommodate irreversible interaction with hENT1, an acrylamide warhead was introduced via ether linkage (compound 14a) resulting in similar binding affinities and no observable pKi shift compared to compounds 9g and 10b.
Next, compounds 14b-14d, with increased linker length and varying warheads, were tested (Fig. 2). While all three compounds showed good binding, 14b and 14c (Fig. 2a and 2b) containing an acrylamide and isothiocyanate warhead displayed single digit nanomolar affinities (pKi values of 8.23 ± 0.09 and 8.97 ± 0.05, respectively). However, since no leftward shift in the dose response curves with 4 h preincubation was observed, no indication of irreversible binding was found for either of the three high affinity compounds. All inhibitors with an affinity below 50 nM were additionally tested in a wash-out radioligand binding assay to further investigate irreversibility of binding. All six inhibitors demonstrated a recovery of 80 to 100% of [3H]NBTI binding after four washing steps, indicating no covalent interaction (Fig. 3).

Affinity characterization of inhibitors 14b, 14c and 14d. Displacement of specific [3H]NBTI binding by increasing concentrations of dilazep derivatives 14b (a), 14c (b) and 14d (c) in erythrocyte cell membranes endogenously expressing hENT1 at 10 °C with or without 4 h preincubation. Data are normalized to 100% of the total binding and represent the mean ± SEM of three individual experiments performed in duplicate

Recovery of specific [3H]NBTI binding after washing. Erythrocyte cell membranes endogenously expressing hENT1 were preincubated with a 10 × Ki concentration of ENT1 inhibitors 9g, 10b and 14a-d for 1 h at 10 °C followed by no (unwashed) or 4 times (washed) washing. Data are shown as mean ± SD from three independent experiments performed in duplicate. Differences between unwashed and washed were determined in an unpaired Student’s t-test with Welch’s correction. Significant differences are displayed as * p < 0.05
Molecular docking
With the use of the published hENT1 crystal structure (PDB: 6OB7), possible binding modes of compounds ST7092, 6m and 6n were investigated by molecular docking.
When dilazep was redocked in the hENT1 structure, the key interactions originally described in the crystal structure were maintained, thus validating the suitability of the structure for docking of analogs (Fig. 4a). These interactions include hydrogen bonds with N338 and Q158, respectively, via the methoxy groups and a hydrogen bond with W29 via the carbonyl group that goes deeper into the pocket. As originally described, π-π stacking was observed with F334 and F307. Of note, we docked dilazep with an unprotonated homopiperazine ring, in agreement with the originally described structure. The two mono-protonated isomers also conserved the original interactions, albeit with lower docking scores. The piperazine derivative of dilazep, ST7092, maintained a similar binding pose, conserving the interactions with N338, Q158 and π-stacking at the top of the pocket (Fig. 4b). However, the change of a homopiperazine to a piperazine ring affected the torsion angles of the ring substituents leading to slightly weaker polar contacts with W29 and Q158. This was also observed in a lower binding affinity and docking score (Table 1 and Table S1). Similarly, the di-substituted piperazine analogue 6n was able to conserve all interactions albeit with worse docking scores either with the substituted ring interacting with N338 (most energetically favorable pose, Fig. 4d), or flipped (second most favorable pose, not shown). The effect on torsion angles driven by the ring substitution meant that interactions at the bottom of the pocket were only kept when additional methoxy groups were available for interacting with Q158. When this was not the case, the interaction was lost, as it was observed for the mono-substituted analog 6m (Fig. 4c), which was not able to interact with Q158 in any of the analyzed poses.

Binding poses of dilazep piperazine analogs with decreasing number of methoxy substitutions including their docking scores. hENT1 PDB: 6OB7 (grey) with (re)docked dilazep and co-crystallized structure inhibitor in a thin line for reference (a), ST7092 (b), 6m (c), and 6n (d). Polar contacts (hydrogen bonds) are represented as dashed yellow lines. Target nucleophilic residue C439 (TM2) is colored in orange for reference
Six out of the seventeen compounds equipped with a warhead displayed submicromolar affinity (compounds 9g, 10b, 14a-14d). In order to investigate their ability to displace [3H]NBTI, but their inability to bind covalently to C439 of hENT1, their potential binding mode was examined.
Compound 10b was used as starting point as in theory it was able to keep key interactions of dilazep with W29 and N338 while forming a covalent bond (Fig. 5b). However, given the experimental data we hypothesize that instead a hydrogen bond was formed between the warhead and Q158 (Fig. 5c). Compounds such as 10e, with an additional carbon atom before the warhead, were not able to maintain all the key interactions while occupying the bottom part of the pocket (Figs. 5d-f). In contrast, the compounds with the warhead placed directly on the trimethoxy benzoate group, such as 14b, showed the highest binding affinity (Table 2). From a structural point of view, this can be explained by the fact that the warhead can be accommodated at different locations in the pocket (Fig. 5g-i), including the NBTI pocket (Fig. 5i) while maintaining the canonical interactions or any other way occupying the bottom part of the pocket. Unfortunately, while the location of the warheads seemed optimal for covalent binding in several compounds (Fig. 5c, 5f, 5h), covalent interaction was not observed. Further investigation of the binding pocket to find potential influences on C439’s ability to binding covalently showed that this residue is situated in a highly hydrophobic area (supplementary Figure S1c) which consists of its adjacent neighboring residues L438 and L440 and multiple other apolar amino acids such as glycines, alanines and phenylalanines.

Binding poses of dilazep piperazine analogues substituted with electrophilic warheads. hENT1 (grey) crystal structure 6OB7 in complex with dilazep (a) and different docking poses of 10b (b, covalent and c, non-covalent), 10e (d-f) and 14b (g-i). Polar contacts (hydrogen bonds) are represented as dashed yellow lines. Target nucleophilic residue C439 (TM2) is colored in orange for reference
Discussion and conclusion
As mentioned in the introduction, hENT1, an important therapeutic target, plays a key role in transporting adenosine, purines and nucleoside-derived drugs [22, 23]. While high affinity adenosine reuptake inhibitors like NBTI, draflazine and dipyridamole have been thoroughly investigated, studies regarding derivatives of the vasodilator dilazep are limited [24]. Moreover, molecular tools for studying the protein’s function and binding mechanisms focus mainly on NBTI- and dipyridamole-based tools [25, 26]. Hence, this study focused on the design, synthesis, pharmacological and computational evaluation of 39 novel dilazep-derived inhibitors of which17 derivatives were equipped with an electrophilic warhead aimed to establish covalent interaction with hENT1 amino acid residue C439. With the use of a [3H]NBTI binding assay, the binding affinities or percentage displacement of all synthesized inhibitors were determined. Pentamethoxy-substituted compounds 6n and 6o displayed an 80-fold decrease in affinity (Table 1) compared to ST7092, whereas further removal of methoxy groups (compounds 6k-6m and 6q-6r, Table 1) completely diminished [3H]NBTI displacement. This indicates that at least two methoxy substituents are needed at the benzoate site where alterations in the substitution pattern are made. Moreover, taken together with other investigated substitution patterns (Table 1) this demonstrates the small range of chemical diversity that is tolerated for the hENT1 as was shown previously for dilazep derivatives [17]. Although from previous research it was postulated that a single methoxy substitution on both benzoates would still be feasible [17], our research shows that symmetrical compounds with mono- or di-methoxy substitution patterns lost inhibitory activity. Furthermore, the previous study showed that substituting the trimethoxy pattern on both phenyl rings in the parent structure with a single electron withdrawing group such as fluor on the para-positions, maintained inhibitory properties at 10 µM whilst on the contrary the 4-Cl substituted 6e was not able to inhibit [3H]NBTI binding at 1 µM. Additionally, the limited tolerance for chemical diversity accounts for hENT1 inhibitors in general which was found using kinetic and thermodynamic radioligand binding studies [27]. This previous research has shown that various inhibitors (dilazep, dipyridamole and NBTI) maintained a single binding mode at different temperatures which was favored by polar interactions suggesting a low tolerability for chemically diverse inhibitors which would favor different interactions. This supports our finding that most changes in the substitution pattern of ST7092 dramatically decreased affinity.
In order to gain more insight into the loss of affinity as a result of different substitution patterns on one of the ST7092 phenyl rings, compounds ST7092, 6m and 6n were docked into the hENT1 structure co-crystallized with dilazep (PDB: 6OB7, Fig. 4b-d) [16]. Compared to the binding mode of dilazep and ST7092, compound 6n, where one of the ortho-methoxy groups of ST7092 was removed, retained similar interactions, however with weaker polar interactions with W29 and Q158 (Fig. 4d). In addition, para-methoxy substituted 6m (Fig. 4c), lost interaction with Q158 in all analyzed binding poses. This compound also showed the weakest affinity, suggesting that a relatively strong interaction with Q158 is needed to displace NTBI, which uses this glutamine as its anchor point [16], followed by interactions with the other aforementioned residues (W29, F307, F334 and N338), as has been previously described for dilazep [16, 28, 29]. Accordingly, earlier mutagenesis studies have shown that mutations of these highly conserved residues significantly decreased the inhibitory properties of dilazep as well as other inhibitors NBTI, dipyridamole and draflazine [30, 31].
In multiple other protein families as well as SLC subfamilies, the use of covalent inhibitors has shown to be of great use in the field of chemical biology to determine protein expression levels and elucidate protein structures [32, 33]. Additionally, multiple covalent therapies are on the market showing advantages such as improved efficacy and decreased drug resistance [34]. Therefore, several potentially covalent inhibitors were synthesized to target cysteine 439 (TM2) in the central cavity of the binding pocket (supplementary Figure S1). To establish a first indication of covalent binding, a time-dependent radioligand binding assay was performed as previously described for other radioligand binding assays [35]. When comparing apparent affinity with 4 h preincubation to affinity values without preincubation, the apparent affinity of covalent inhibitors should increase as a result of increased transporter occupancy over time. Unfortunately, none of the potential covalent inhibitors displayed any increase in [3H]NBTI displacement over time at 1 µM (Table 2). However, inhibitors 14b, 14c and 14d were able to displace [3H]NBTI at high affinity (Fig. 2). The high affinity of compound 14b can be explained by the accommodation of the warhead in various manners (Fig. 5g-i) such as the occupation of an additional space in the binding pocket by the warhead (Fig. 5i), previously described as opportunistic site 2 [16]. This unshared binding site between inhibitors dilazep and NBTI accommodates the thioinosine moiety of NBTI through hydrophobic interactions. Unfortunately, when incorporating a 4 h preincubation step prior to the radioligand displacement assay, no significant increase of the affinity was observed for inhibitors 14b, 14c and 14d (Fig. 2), indicating solely non-covalent interactions between the inhibitors and hENT1. A further series of experiments, in which we introduced washing steps to differentiate between covalently and reversibly bound inhibitors, did not yield evidence for covalent interactions either (Fig. 3).
In order to successfully design a covalent inhibitor, multiple factors should be considered (as has been extensively reviewed). Particularly important are a high affinity scaffold, a nucleophilic target amino acid residue and warhead reactivity [36]. With the rational design of the dilazep-derived covalent inhibitors multiple substitutions were made to establish a close proximity between the warhead and C439 while maintaining the interactions needed for high affinity. Elongation of the inhibitor in various ways (compounds 9c, 9f, 10c, 10e, 10f, 14a and 14b) was introduced to bridge the 4.3 Å distance between the warhead substitution side and the nucleophilic side of the cysteine (supplementary Figure S1b). Although the addition of an extra linker should reach across the distance to the target anchor point, there was no indication of covalent binding (Table 2), which suggested a mismatch between the reactivity of the warhead and of C439. Covalent interactions with cysteine have been used in a plethora of protein studies because of its high intrinsic nucleophilicity (pKa ≈ 8.5) [37]. With the use of Michael acceptors such as acrylamide and dithiocarbamate formation by isothiocyanates, reversible-covalent interactions can be formed with cysteine residues [37, 38]. Therefore, the non-covalent binding compounds 10a-10f, 14a and 14b directed us to the design of covalent compounds bearing vinylsulfonamide and fluorosulfonyl warheads with an increased reactivity towards cysteines compared to the aforementioned [39]. However, the inability of the compounds with increased warhead reactivity to bind covalently to C439 could indicate that this amino acid residue is not intrinsically nucleophilic enough to establish covalent binding. As has been described for many other protein families, pKa values of cysteines can widely vary depending on their spatial environment in the protein [40], which in our case is mainly hydrophobic (supplementary Figure S1c) and therefore might contribute to a reduction in reactivity [41]. Unfortunately, in silico calculations of cysteine pKa values often show a lack of accuracy and generate false positives, making valuable predictions to be used in covalent inhibitor design challenging [42, 43]. Therefore, in further development of covalent dilazep-derived inhibitors the reactivity of other target nucleophilic amino acid residues as well as a matched warhead in terms of reactivity should be considered. Additionally, this research shows that, despite the presence of a high-affinity scaffold and a sufficiently reactive warhead, establishing covalent interactions with non-catalytic nucleophilic amino acid residues is not trivial.
In conclusion, different substitution patterns of the trimethoxy benzoates of dilazeps’ close analogue ST7092 led to decreased interactions in the binding pocket and therefore diminished hENT1 affinity. Conversely, compounds 14b, 14c and 14d displayed high affinities for the transporter mainly by occupying the opportunistic site 2 without binding covalently to amino acid residue C439. Taken together, this study reported new dilazep derivatives active as hENT1 inhibitors and the first high affinity dilazep derivatives equipped with an electrophilic warhead that did not induce covalent binding in the hENT1 binding pocket. These results will aid in the rational and structure-based design of novel inhibitors as well as pharmacological tools to further study hENT1 function, binding mechanisms and (patho)physiological implications.
Data Availability
Data is contained within the article or supplementary material. Data not shown is available from the corresponding authors, upon reasonable request.
Abbreviations
- ENT1:
-
Equilibrative nucleoside transporter 1
- NBTI:
-
S-(4-Nitrobenzyl)-6-thioinosine
- NSB:
-
Non-specific binding
- SLC:
-
Solute carrier
- TB:
-
Total binding
References
Pastor-Anglada M, Pérez-Torras S (2018) Emerging Roles of Nucleoside Transporters. Front Pharmacol 9:1–8. https://doi.org/10.3389/fphar.2018.00606
Molina-Arcas M, Casado FJ, Pastor-Anglada M (2009) Nucleoside Transporter Proteins. Curr Vasc Pharmacol 7:426–434. https://doi.org/10.2174/157016109789043892
Young JD (2016) The SLC28 (CNT) and SLC29 (ENT) nucleoside transporter families: a 30-year collaborative odyssey. Biochem Soc Trans 44:869–876. https://doi.org/10.1042/BST20160038
Boswell-Casteel RC, Hays FA (2017) Equilibrative nucleoside transporters—A review. Nucleosides, Nucleotides Nucleic Acids 36:7–30. https://doi.org/10.1080/15257770.2016.1210805
King AE, Ackley MA, Cass CE et al (2006) Nucleoside transporters: from scavengers to novel therapeutic targets. Trends Pharmacol Sci 27:416–425. https://doi.org/10.1016/j.tips.2006.06.004
Baldwin SA, Beal PR, Yao SYM et al (2004) The equilibrative nucleoside transporter family, SLC29. Pflügers Arch - Eur J Physiol 447:735–743. https://doi.org/10.1007/s00424-003-1103-2
Niemann B, Haufs-Brusberg S, Puetz L et al (2022) Apoptotic brown adipocytes enhance energy expenditure via extracellular inosine. Nature 609:361–368. https://doi.org/10.1038/s41586-022-05041-0
Chang C-P, Chang Y-G, Chuang P-Y et al (2021) Equilibrative nucleoside transporter 1 inhibition rescues energy dysfunction and pathology in a model of tauopathy. Acta Neuropathol Commun 9:112. https://doi.org/10.1186/s40478-021-01213-7
Farrell JJ, Elsaleh H, Garcia M et al (2009) Human Equilibrative Nucleoside Transporter 1 Levels Predict Response to Gemcitabine in Patients With Pancreatic Cancer. Gastroenterology 136:187–195. https://doi.org/10.1053/j.gastro.2008.09.067
Iikura M, Furihata T, Mizuguchi M et al (2012) ENT1, a Ribavirin Transporter, Plays a Pivotal Role in Antiviral Efficacy of Ribavirin in a Hepatitis C Virus Replication Cell System. Antimicrob Agents Chemother 56:1407–1413. https://doi.org/10.1128/AAC.05762-11
Vlachodimou A, Konstantinopoulou K, IJzerman AP, Heitman LH (2020) Affinity, binding kinetics and functional characterization of draflazine analogues for human equilibrative nucleoside transporter 1 (SLC29A1). Biochem Pharmacol 172:113747. https://doi.org/10.1016/j.bcp.2019.113747
Buolamwini JK (1997) Nucleoside Transport Inhibitors: Structure-Activity Relationships and Potential Therapeutic Applications. Curr Med Chem 4:35–66. https://doi.org/10.2174/0929867304666220309201038
Vlachodimou A, Bouma J, De Cleyn M et al (2023) Kinetic profiling of novel spirobenzo-oxazinepiperidinone derivatives as equilibrative nucleoside transporter 1 inhibitors. Purinergic Signal. https://doi.org/10.1007/s11302-023-09948-9
Sui B, Chen J, Ge D et al (2023) Assembly Characterization of Human Equilibrium Nucleoside Transporter 1 (hENT1) by Inhibitor Probe-Based dSTORM Imaging. Anal Chem 95:9207–9218. https://doi.org/10.1021/acs.analchem.3c00596
Addo JK, Buolamwini JK (2004) Design, Synthesis, and Evaluation of 5‘-S-Aminoethyl-N6-azidobenzyl-5‘-thioadenosine Biotin Conjugate: A Bifunctional Photoaffinity Probe for the es Nucleoside Transporter. Bioconjug Chem 15:536–540. https://doi.org/10.1021/bc034165j
Wright NJ, Lee S-Y (2019) Structures of human ENT1 in complex with adenosine reuptake inhibitors. Nat Struct Mol Biol 26:599–606. https://doi.org/10.1038/s41594-019-0245-7
Playa H, Lewis TA, Ting A et al (2014) Dilazep analogues for the study of equilibrative nucleoside transporters 1 and 2 (ENT1 and ENT2). Bioorg Med Chem Lett 24:5801–5804. https://doi.org/10.1016/j.bmcl.2014.10.026
IJzerman AP, Voorsehuur AH, (1990) The relationship between ionization and affinity of nucleoside transport inhibitors. Naunyn Schmiedebergs Arch Pharmacol 342:336–341. https://doi.org/10.1007/BF00169446
Yung-Chi C, Prusoff WH (1973) Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22:3099–3108. https://doi.org/10.1016/0006-2952(73)90196-2
The PyMOL Molecular Graphics System, Version 2.5.2, Schrödinger, LLC, New York, NY, 2021
Liu P, Huang L, Faul MM (2007) A simple method for chemoselective phenol alkylation. Tetrahedron Lett 48:7380–7382. https://doi.org/10.1016/j.tetlet.2007.08.030
Baldwin SA, Mackey JR, Cass CE, Young JD (1999) Nucleoside transporters: molecular biology and implications for therapeutic development. Mol Med Today 5:216–224. https://doi.org/10.1016/S1357-4310(99)01459-8
Molina-Arcas M, Pastor-Anglada M (2010) Role of Nucleoside Transporters in Nucleoside-Derived Drug Sensitivity. Nucleosides, Nucleotides Nucleic Acids 29:335–346. https://doi.org/10.1080/15257771003729823
Rehan S, Shahid S, Salminen TA et al (2019) Current Progress on Equilibrative Nucleoside Transporter Function and Inhibitor Design. SLAS Discov 24:953–968. https://doi.org/10.1177/2472555219870123
IJzerman AP, Menkveld GJ, Thedinga KH (1989) A refined method for the photoaffinity labelling of the nitrobenzylthioinosine-sensitive nucleoside transport protein: Application to cell membranes of calf lung tissue. Biochim Biophys Acta - Biomembr 979:153–156. https://doi.org/10.1016/0005-2736(89)90430-6
Lin W, Buolamwini JK (2011) Design, Synthesis, and Evaluation of 2-Diethanolamino-4,8-diheptamethyleneimino-2-(N-aminoethyl-N-ethanolamino)-6-(N, N-diethanolamino)pyrimido[5,4-d]pyrimidine-fluorescein Conjugate (8MDP-fluor), As a Novel Equilibrative Nucleoside Transporter Probe. Bioconjug Chem 22:1221–1227. https://doi.org/10.1021/bc2000758
Rehan S, Ashok Y, Nanekar R, Jaakola V-P (2015) Thermodynamics and kinetics of inhibitor binding to human equilibrative nucleoside transporter subtype-1. Biochem Pharmacol 98:681–689. https://doi.org/10.1016/j.bcp.2015.09.019
Wright NJ, Lee S-Y (2021) Toward a Molecular Basis of Cellular Nucleoside Transport in Humans. Chem Rev 121:5336–5358. https://doi.org/10.1021/acs.chemrev.0c00644
Wu Z, Han Z, Zhou W et al (2022) Insight into the nucleoside transport and inhibition of human ENT1. Curr Res Struct Biol 4:192–205. https://doi.org/10.1016/j.crstbi.2022.05.005
Visser F, Sun L, Damaraju V et al (2007) Residues 334 and 338 in Transmembrane Segment 8 of Human Equilibrative Nucleoside Transporter 1 Are Important Determinants of Inhibitor Sensitivity, Protein Folding, and Catalytic Turnover. J Biol Chem 282:14148–14157. https://doi.org/10.1074/jbc.M701735200
Paproski RJ, Visser F, Zhang J et al (2008) Mutation of Trp29 of human equilibrative nucleoside transporter 1 alters affinity for coronary vasodilator drugs and nucleoside selectivity. Biochem J 414:291–300. https://doi.org/10.1042/BJ20080074
Glukhova A, Thal DM, Nguyen AT et al (2017) Structure of the Adenosine A1 Receptor Reveals the Basis for Subtype Selectivity. Cell 168:867-877.e13. https://doi.org/10.1016/j.cell.2017.01.042
Napolitano L, Scalise M, Koyioni M et al (2017) Potent inhibitors of human LAT1 (SLC7A5) transporter based on dithiazole and dithiazine compounds for development of anticancer drugs. Biochem Pharmacol 143:39–52. https://doi.org/10.1016/j.bcp.2017.07.006
Sutanto F, Konstantinidou M, Dömling A (2020) Covalent inhibitors: a rational approach to drug discovery. RSC Med Chem 11:876–884. https://doi.org/10.1039/D0MD00154F
Doornbos MLJ, Wang X, Vermond SC et al (2019) Covalent Allosteric Probe for the Metabotropic Glutamate Receptor 2: Design, Synthesis, and Pharmacological Characterization. J Med Chem 62:223–233. https://doi.org/10.1021/acs.jmedchem.8b00051
Hartung IV, Rudolph J, Mader MM et al (2023) Expanding Chemical Probe Space: Quality Criteria for Covalent and Degrader Probes. J Med Chem 66:9297–9312. https://doi.org/10.1021/acs.jmedchem.3c00550
Gehringer M, Laufer SA (2019) Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J Med Chem 62:5673–5724. https://doi.org/10.1021/acs.jmedchem.8b01153
Ábrányi-Balogh P, Petri L, Imre T et al (2018) A road map for prioritizing warheads for cysteine targeting covalent inhibitors. Eur J Med Chem 160:94–107. https://doi.org/10.1016/j.ejmech.2018.10.010
Martin JS, MacKenzie CJ, Fletcher D, Gilbert IH (2019) Characterising covalent warhead reactivity. Bioorg Med Chem 27:2066–2074. https://doi.org/10.1016/j.bmc.2019.04.002
Harris RC, Liu R, Shen J (2020) Predicting Reactive Cysteines with Implicit-Solvent-Based Continuous Constant pH Molecular Dynamics in Amber. J Chem Theory Comput 16:3689–3698. https://doi.org/10.1021/acs.jctc.0c00258
Zhang W, Pei J, Lai L (2017) Statistical Analysis and Prediction of Covalent Ligand Targeted Cysteine Residues. J Chem Inf Model 57:1453–1460. https://doi.org/10.1021/acs.jcim.7b00163
Flanagan ME, Abramite JA, Anderson DP et al (2014) Chemical and Computational Methods for the Characterization of Covalent Reactive Groups for the Prospective Design of Irreversible Inhibitors. J Med Chem 57:10072–10079. https://doi.org/10.1021/jm501412a
Liu R, Yue Z, Tsai C-C, Shen J (2019) Assessing Lysine and Cysteine Reactivities for Designing Targeted Covalent Kinase Inhibitors. J Am Chem Soc 141:6553–6560. https://doi.org/10.1021/jacs.8b13248
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Conceptualization: M.A.D., A.P.IJ. and D.E.; Methodology: M.A.D., M.G.G., W.J., G.J.P.W., L.H.H., A.P.IJ., D.E.; Investigation and formal analysis: M.A.D., M.G.G., M.D.D., N.J.M., J.P.D.V., R.L.; Writing—original draft: M.A.D., M.G.G., A.P.IJ., D.E.; Writing—review and editing; M.A.D., M.G.G., W.J., G.J.P.W., L.H.H., A.P.IJ., D.E.; Supervision: A.P.IJ., D.E.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Competing interests
Adriaan P. IJzerman is on the editorial board of Purinergic Signalling.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dilweg, M.A., Gorostiola González, M., de Ruiter, M.D. et al. Exploring novel dilazep derivatives as hENT1 inhibitors and potentially covalent molecular tools. Purinergic Signalling (2024). https://doi.org/10.1007/s11302-024-10026-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11302-024-10026-x