
Abstract
Documenting species and population diversity is becoming increasingly important as the destruction and degradation of natural ecosystems are leading to a worldwide biodiversity loss. Despite the rapid development of genetic tools, many species remain undocumented and little is known about the diversity of individuals and populations, especially for tropical African plants. In this study, we aim to identify putative hidden species and/or differentiated populations in the tropical African tree Staudtia kamerunensis Warb. (Myristicaceae), a widespread species characterized by a high morphological diversity and a complex taxonomical history. Historical herbarium vouchers were sampled and leaf or cambium samples were collected in the field, dried in silica gel, and subsequently genotyped at 14 microsatellite loci (SSRs), as well as sequenced for two nuclear genes (At103, Agt1) and one plastid region (psbA-trnH). These genetic data were then analyzed using Bayesian clustering, population genetics, and the construction of haplowebs to assess genetic clustering patterns, the distribution of genetic diversity, and genetic differentiation among populations. Multiple genetically differentiated clusters were observed in parapatry throughout Central Africa. Genetic diversity was high and similar among these clusters, apart from the most differentiated populations in southeast Democratic Republic of the Congo (DR Congo), which showed lower levels of genetic diversity. The genetic breaks detected between S. kamerunensis populations are likely not indicative of hidden species but rather result from ancient rainforest fragmentation during cold and dry periods in the Pliocene and/or Pleistocene. The strong genetic divergence between populations in southeast DR Congo could be the result of an ongoing speciation linked to ecological niche differentiation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The increase of human-mediated destruction and degradation of natural ecosystems is leading to worldwide biodiversity loss, which in turn strongly impacts ecosystem functioning (Bradshaw et al. 2009; Scheffers et al. 2016). As species are going extinct at an alarming rate, cataloguing biodiversity and delimiting species are becoming ever more important, since it provides essential information for ecosystem management (Cazzolla Gatti et al. 2022), and for identifying valuable organisms for medicine, food, and other human uses (Bickford et al. 2007; Cheek et al. 2020). Furthermore, alpha taxonomy (i.e., the delimitation and naming of taxa) is crucial for conservation purposes because species that remain undiscovered are often the ones that are at a high risk of extinction (Cheek et al. 2020). Luckily, the rapid development of genetic tools is increasingly facilitating the scientific discovery of species, especially in cases where taxa cannot readily be distinguished morphologically (Struck et al. 2018).
Additionally, genetic diversity within species and populations (i.e., intraspecific diversity) is extremely important, since it strongly impacts ecosystem resilience and the ability of a species to adapt to environmental changes (Laikre et al. 2020; Hoban et al. 2021; Stange et al. 2021). Identification of intraspecific genetic diversity is essential for adequate conservation efforts, as it helps to assess population structure, connectivity, inbreeding, and demographic history, as well as to identify populations and units for conservation (e.g., evolutionary significant units or ESUs) (Hohenlohe et al. 2021; Willi et al. 2022). Indeed, even if a species does not go extinct, losing some of its differentiated populations can result in a net loss of genetic diversity and potentially relevant (adaptive) variants.
Over the last decade, efforts have been made to document the genetic diversity of tropical African tree species using population genetics methods (e.g., Hardy et al. 2013; Helmstetter et al. 2020a, b). These methods are valuable for estimating divergence between closely related taxa, as they enable the detection of the earliest stages of speciation (Daïnou et al. 2014). Furthermore, they can support species delimitation when taxonomic classification based on morphology alone proves difficult, which is often the case for polymorphic tree species in highly diverse tropical forests. For example, multiple hidden species have been revealed in taxonomically recognized tropical African trees using population genetics methods (Ikabanga et al. 2017; Lissambou et al. 2019; Ewédjè et al. 2020), even in well-known timber trees such as Milicia (Daïnou et al. 2016), Entandrophragma (Monthe et al. 2018), and Khaya (Bouka et al. 2022). These studies identified differentiated genetic clusters occurring in sympatry without showing signs of introgression, which suggest that they are reproductively isolated and hence correspond to distinct species according to the biological species concept (De Quieroz 2007). These findings indicate that multiple tropical African tree species remain undescribed, and that species richness is likely underestimated for tropical Africa. While differentiated genetic clusters can indicate the presence of hidden species, they can also represent conspecific populations (i.e., populations belonging to the same species) that diverged due to recent or ancient barriers to gene flow. Such conspecific populations often occur in parapatry and show low levels of genetic differentiation with an abundance of shared alleles and admixed individuals. Many tropical African tree species are characterized by genetic discontinuities throughout their distribution range which are likely caused by climatic fluctuations during the Pliocene and Pleistocene, when periods of low precipitation caused rainforests to retract, leading to forest fragmentation and genetic isolation of populations (Hardy et al. 2013; Helmstetter et al. 2020a, b; Piñeiro et al. 2021). The evolutionary responses to these climatic changes seem to be variable, asynchronous, and species dependent, and are driven by different life-history traits and environmental factors. Hence, the level of genetic population structure varies among species, and populations are characterized by differences in ecological niche, demographic history, genetic diversity, and gene flow (Piñeiro et al. 2017; Migliore et al. 2019; Demenou et al. 2020; Helmstetter et al. 2020a, b; Vanden Abeele et al. 2021). Identifying these differences in intraspecific genetic diversity and structure is essential to enhance our understanding of the evolutionary processes that shape the highly diverse tropical African forests, as well as for the implementation of appropriate conservation guidelines to protect this valuable ecosystem.
A tropical tree species that could greatly benefit from a molecular genetics study is Staudtia kamerunensis Warb. (Myristicaceae), an evergreen tree that occurs in various forest types in Central Africa, as the nomenclatural history of this species has not been recently revised (Fouilloy 1974). Staudtia kamerunensis was first described by Otto Warburg in 1897, based on two specimens collected in Cameroon (Warburg 1897). Later, four other (sub)species were described in Staudtia (Warburg 1904; Vermoesen 1923; Gilbert and Troupin 1951), which were later lumped together, with S. kamerunensis as the only recognized species today (Fouilloy 1974). Within this taxon, two varieties were described: var. kamerunensis (synonym: Staudtia kamerunensis var. macrocarpa) and var. gabonensis (synonyms: Staudtia gabonensis, Staudtia stipitata and Staudtia congensis), with the latter differing from the type specimen by having smaller fruits (Fouilloy 1974) and thinner valves (personal observation). The distribution of S. kamerunensis var. kamerunensis is probably limited to Lower Guinea (western side of the Central African rainforest block), while var. gabonensis occurs in Lower Guinea and Congolia (eastern side of the Central African rainforest block) (Doucet 2003). However, as fruits are often lacking from collected specimens, correct identifications have proven difficult, hampering comprehensive studies at population level. Moreover, fruit size is a difficult criterion to assess on herbarium vouchers, since fruits often shrink during the drying process or can be immature at the time of collection. Maturity of the fruits is impossible to verify from herbarium specimens since the characteristic yellow color of mature S. kamerunensis fruits is lost during the drying process. Also, the most recent taxonomic classification (Fouilloy 1974) is still debated and its implementation remains inconsistent. In Korup National Park (Cameroon), for example, the two S. kamerunensis varieties are still treated as distinct species because of their different morphology (Thomas et al. 2003). Moreover, the two varieties are described as sympatric in western Gabon (Doucet 2003), suggesting that they are reproductively isolated and may correspond to distinct species under the biological species concept.
A phylogeographic study of the plastomes of S. kamerunensis revealed five main genetic lineages occurring in parapatry, four of them in Lower Guinea and one in Congolia (Matvijev et al. 2022). The Congolian lineage had a more recent origin and showed traces of relatively recent range expansion (in the last 200,000 years), generating three parapatric sub-lineages, while the Lower Guinean lineages did not show traces of recent range expansion. Plastomes, which are usually maternally inherited, can provide information on a part of the colonization history of plants, yet are not adequate for species delimitation since chloroplast capture among closely related species is common in plants (Stegemann et al. 2012). Hence, data from the nuclear genome is needed to investigate species delimitation, notably since it provides information on allelic co-occurrence in heterozygous individuals (Doyle 1995; Flot et al. 2010).
In the current study, we searched for differentiated genetic clusters in the tropical African tree S. kamerunensis by examining the genetic diversity in samples collected across the Central African distribution range. By combining multiple molecular markers (microsatellites, chloroplast, and nuclear sequences), we addressed the following questions: (1) Can we identify genetically differentiated clusters within S. kamerunensis? If so, (2) are the genetic discontinuities linked to (ancient) fragmentation of the Central African rainforest or do they correspond to species or variety limits? (3) How does the genetic diversity compare between biogeographic regions? (4) Are the differentiated clusters characterized by a different demographic history?
Materials and methods
Taxon sampling and DNA extraction
Leaves or cambium from 403 trees were collected during field missions in Cameroon, Gabon, Republic of the Congo, and Democratic Republic of the Congo (DR Congo) and dried using silica gel. As fruits were mostly absent or unreachable, no attempt was made to identify intraspecific varieties and all samples were labelled as S. kamerunensis. Additionally, leaf material was collected from 161 herbarium vouchers stored at Meise Botanic Garden (BR), which hosts the largest collection of this species, seven from the herbarium of Université libre de Bruxelles (BRLU), 13 from the Royal Botanic Garden Edinburgh (E), and 12 from the Royal Botanic Gardens Kew (K). All herbarium samples were either identified to species level or as S. kamerunensis var. gabonensis. The absence of var. kamerunensis in such a large collection confirmed the problems with the revised taxonomic classification. Moreover, an online search through multiple catalogues of herbaria known for their African collections revealed only two specimens identified as S. kamerunensis var. kamerunensis. Alternatively, the lack of var. kamerunensis could be attributed to the fact that it is rare and only occurs at low densities (Doucet 2003; Thomas et al. 2003).
In total, 596 individuals of S. kamerunensis, covering most of the species’ distribution area, were sampled for molecular analyses. DNA was extracted using the NucleoSpin Plant II kit (Macherey–Nagel) for silica-dried samples, and either a cetyltrimethylammonium bromide (CTAB) protocol (Doyle and Doyle 1990; with an additional sorbitol washing step for herbarium specimens following Janssens et al. 2006) or following the protocol described in Cappellini et al. (2010).
Genotyping and sequencing
Fourteen microsatellite markers (also called short sequence repeats or SSRs) were amplified in S. kamerunensis and genotyped on an ABI 3730 DNA Analyzer (Applied Biosystems) following the protocol described by Vanden Abeele et al. (2018). Electropherograms were interpreted with Geneious 9.1.6 (Kearse et al. 2012). Only individuals for which at least seven of the fourteen loci successfully amplified were considered for further analyses. To avoid an overrepresentation of certain geographic areas (which can bias clustering analyses; Puechmaille 2016), subsampling was done to keep no more than 3 samples per square area with a side of 0.01° (approximately 1 km2), resulting in an SSR dataset of 400 individuals.
The chloroplast intergenic spacer psbA-trnH (psbAF as Sang et al. (1997); trnH2 as in Tate and Simpson (2003); and two nuclear sequences, At103 (Li et al. 2008) and Agt1 (modified for Myristicaceae species as in Steeves (2011), were amplified on 90 samples chosen to represent the genetic clusters detected using SSRs (see the “Genetic clustering using SSR data” section) and sent for sequencing at Macrogen Inc. (Seoul, South Korea).
Genetic diversity and differentiation in the SSR dataset
A factorial correspondence analysis with three dimensions (FCA 3D) was used in Genetix 4.05.2 (Belkhir et al. 2004) to visualize variation of the SSR dataset and look for potential outlier genotypes. The electropherograms of samples that deviated strongly were assessed and corrected if necessary. The final Genetix output file was then converted to an input file for the STRUCTURE software 2.3.4 (Pritchard et al. 2000) using PGDSpider 2.1.0.1 (Lischer and Excoffier 2012).
The Bayesian clustering algorithm implemented in STRUCTURE was used to identify genetic clusters. The complete SSR dataset (n = 400) was run using an admixture model, a burn-in period of 100,000 followed by 100,000 MCMC replicates, maximum number of clusters (Kmax) set between 1 and 10, and 10 iterations for each Kmax. As shown by Wang (2017), unbalanced sampling causes poor individual assignments to source populations and gives incorrect estimates of K when the default ancestry prior is used in STRUCTURE. Therefore, we used the uncorrelated allele frequency model and an alternative ancestry prior; a separate α value for each population and a starting α value of 0.2 (adjusted as recommended by Wang (2017) as the reciprocal of the number of genetic clusters, which was five according to preliminary runs). To account for the possible presence of null alleles, recessive alleles were declared for each locus, so that null allele frequencies were directly estimated by STRUCTURE for each cluster. To infer the most likely number of clusters K, the log-likelihood values LnP(D) were plotted against K (Pritchard et al. 2000) using Structure Harvester (Earl and vonHoldt 2012). The cluster assignment probability q for each sample was inferred from the run with the highest log-likelihood value. The STRUCTURE clustering results were summarized and visualized using CLUMPAK (Kopelman et al. 2015). The geographic distribution of the clusters was visualized using QGIS 3.4.5.
To summarize the genetic diversity among sampled individuals, a principal component analysis (PCA) was performed and visualized as a scatterplot in RStudio (RStudio Team 2019) using the packages adegenet (Jombart 2008) and ade4 (Chessel et al. 2007). The genetic clusters inferred in STRUCTURE were used as group assignments.
For each pair of inferred clusters, the proportion of admixed individuals was calculated as the number of individuals for which q1 < 0.8, q2 < 0.8, and q1 + q2 ≥ 0.8, divided by the number of individuals for which q1 + q2 ≥ 0.8 (where q1 and q2 are the assignment probabilities to the pair of clusters compared for an individual). Pairwise genetic differentiation between inferred clusters was assessed by computing FST (Weir and Cockerham 1984) and RST (Slatkin 1995). While FST is based on allele identity, RST is based on allele size and is better at detecting the contribution of stepwise mutations to genetic differentiation (Hardy et al. 2003). For this reason, we tested for a phylogeographic signal by comparing the observed RST to the expected RST value obtained from 10,000 permutations of allele sizes among alleles (Hardy et al. 2003). All calculations were done using SPAGeDi 1.5d (Hardy and Vekemans 2002).
For all genetic clusters, the number of alleles (NA), effective number of alleles (NAe), allelic richness (AR), expected heterozygosity (He), observed heterozygosity (Ho), and the inbreeding coefficient FIS were calculated using SPAGeDi 1.5d (Hardy and Vekemans 2002) by only including samples with an assignment probability q ≥ 0.8. Differences in diversity metrics between genetic clusters were assessed using paired t-tests of single-locus estimates using loci as replicates.
We performed a bottleneck test to determine if a recent demographic bottleneck occurred in any of our genetic clusters. When a population goes through a bottleneck, the increased genetic drift decreases the genetic diversity but affects more strongly the number of alleles at neutral loci than the expected heterozygosity (He, i.e., proportion of heterozygous genotypes expected under Hardy–Weinberg equilibrium), so that He will be higher than expected based on the number of alleles when assuming mutation-drift equilibrium (Heq; Piry et al. 1999). Thus, by comparing He and Heq, we can determine if a recent demographic bottleneck occurred (Cornuet et al. 1999). Here, we used the function “BOTTLENECK” in INEST v2.2 (Chybicki 2017), and tested each genetic cluster separately. We assumed the two-phase mutation model (TPM) and kept default values for the proportion of multi-step mutations (0.22) and the average multi-step mutation size (3.1), running the analysis for 10,000 coalescent simulations. A positive Z-score, based on the combined Z-scores with a significant p-value, indicates a recent bottleneck, while a significantly negative Z-score indicates a recent expansion.
Genetic diversity and differentiation of Agt1, At103, and psbA-trnH haplotypes
Forward and reverse DNA sequences were imported and assembled in Geneious 9.1.6 (Biomatters, New Zealand). The assemblies were manually trimmed and double peaks in nuclear sequences, indicating heterozygous individuals, were coded with the corresponding IUPAC ambiguity codes. Four psbA-trnH sequences previously amplified in S. kamerunensis (Parmentier et al. 2013) were downloaded from GenBank and added to the newly generated psbA-trnH sequences. The MAFFT (Katoh et al. 2002) plugin was used to separately align the Agt1, At103, and psbA-trnH sequences applying the E-INS-i algorithm, a 200PAM/k = 2 scoring matrix and a gap open penalty of 1.53. The PHASE software (Stephens et al. 2001) together with SeqPHASE (Flot 2010) was used to infer the haplotypes of the nuclear markers. The resulting sequences were submitted to GenBank (Online Resource 1).
Median-joining haplotype networks (Bandelt et al. 1999) were constructed for each gene marker and were modified into haplowebs (Flot et al. 2010) by adding connections between haplotypes co-occurring in heterozygous individuals using HaplowebMaker (Spöri and Flot 2020; available online at https://eeg-ebe.github.io/HaplowebMaker/). These connections were used to reveal gene pools or single-locus fields for recombination (sl-FFRs, conform Doyle 1995 and Flot et al. 2010), an approach that can help species delimitation by highlighting reproductive barriers. Haplotypes were represented with pie charts, colored according to the SSR cluster assignment of the corresponding individuals.
Results
Genetic clustering using SSR data
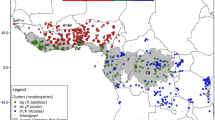
When using STRUCTURE to estimate the number of genetic clusters K in the SSR dataset, the mean log-likelihood of the data substantially increased up to K = 4, and then slightly increased to reach the highest mean value at K = 6 (Online Resource 2). The run with the highest likelihood overall was observed for one of the iterations at K = 5. Since none of the samples was separated into a sixth cluster in most of the iterations at K = 6 to K = 10 compared to K = 5 (major modes, only some samples with additional admixture) (Online Resource 3), and the clustering patterns delineated geographically coherent clusters at K = 5 (Fig. 1A), we opted for five as the most plausible number of genetic clusters in S. kamerunensis (Fig. 1B).

(A) Distribution of 400 collected Staudtia kamerunensis samples and their corresponding genetic cluster assignment, as inferred with STRUCTURE for the most likely scenario of K = 5. The map was made using QGIS 3.4 (QGIS.org 2021). Country codes are as follows: NGA Nigeria, CMR Cameroon, GNQ Equatorial Guinea, GAB Gabon, COG Republic of the Congo, CAR Central African Republic, and COD Democratic Republic of the Congo. (B) Bar plot showing the individual cluster assignments for the STRUCTURE run with the highest likelihood at K = 5.
The five inferred genetic clusters showed a mostly parapatric or allopatric distribution (Fig. 1A) that can be described in relation to Central African phytogeographic regions (White 1979, 1983). Two genetic clusters were restricted to Lower Guinea: “LG North” in south-eastern Nigeria, Cameroon, Equatorial Guinea, and northern Gabon, and “LG South” in south-western DR Congo, Congo, and Gabon. Individuals from each cluster co-occurred in eastern Gabon (between Lastoursville and Okondja). Two genetic clusters were restricted to Congolia: “Congo North” in north-western DR Congo and “Congo South” in the forest-savannah mosaics occurring eastwards (Kivu) and southwards (Katanga), with both clusters overlapping in the rainforest-savannah transition zone. The fifth genetic cluster “LG-Congo” spanned both biogeographic regions, and occurred in south-eastern Cameroon, Central African Republic (CAR), Congo, and DR Congo.
On the PCA scatterplots (Online Resource 4), unassigned samples (q < 0.8) occupied an intermediate position between the five genetic clusters inferred with STRUCTURE. All clusters appeared well separated when the first three principal components were considered.
Genetic differentiation and diversity in the SSR dataset
Using an assignment probability threshold q ≥ 0.8, 50 out of 400 samples were considered as unassigned to one of the five inferred genetic clusters (i.e., admixed samples). The proportion of admixed samples was highest between clusters “LG South” and “LG-Congo” (6.3%), followed by “Congo North” and “Congo South” (5.8%), and “LG North” and “LG South” (5.4%) (Table 1). Most of these samples occurred in the contact zones of the respective genetic clusters, with a substantial number of occurrences in central Gabon and central DR Congo. No admixed samples were found between “LG North” and “Congo South.”
Genetic differentiation as measured by FST (Table 1) was lowest between “LG-Congo” and “Congo North” (FST = 0.09), “LG North” (FST = 0.10), and “LG South” (FST = 0.10). Genetic differentiation was highest between “Congo South” and “LG North” (FST = 0.16), “LG South” (FST = 0.16), and “LG-Congo” (FST = 0.15). When accounting for allele size, genetic differentiation values (RST) were usually higher and ranged from 0.13 between “LG North” and “LG South” to 0.44 between “LG North” and “Congo South” (Table 1). Allele size permutation tests showed that RST was significantly higher than FST between most clusters (Table 1) but there was no significant difference between “LG North” and “LG South,” and between “LG South” and “LG-Congo.”
To estimate genetic diversity indices for the inferred clusters (Table 2), only samples with an assignment probability q ≥ 0.8 were considered. The number of effective alleles was highest in “Congo North” (NAe = 11.74) and “LG North” (NAe = 10.85), and was lowest in “Congo South” (NAe = 6.96). The rarefied allelic richness among 6 gene copies was similar among all genetic clusters, and ranged from 3.82 in “Congo South” to 4.44 in “Congo North.” Observed heterozygosity was highest in “Congo North” (Ho = 0.70) and “LG South” (Ho = 0.69), and lowest in “Congo South” (Ho = 0.55). The inbreeding coefficient Fi was highest in “Congo South” (Fi = 0.27), and lowest in “LG South” (Fi = 0.16) and “Congo North” (Fi = 0.15). However, none of these diversity indices differed significantly between two clusters according to paired t-tests.
All five clusters had a significantly negative Z-score (Table 2). The highest Z-score was observed for “Congo South” (− 1.6465), and the lowest for “LG South” (− 9.2829). These results indicate that none of the clusters recently underwent a demographic bottleneck, but rather expanded in population size.
Analyses of nuclear and chloroplast sequences
We obtained sequences from 79, 71, and 84 individuals for Agt1, At103, and psbA-trnH, respectively. The nuclear gene Agt1 yielded the longest alignment with a length of 731 bp and consisted of 24 haplotypes (polymorphic sites P = 27, number of heterozygous samples nHe = 43), with haplotype frequencies ranging from 1 to 18. The alignment of the nuclear gene At103 had a length of 438 bp and consisted of 22 haplotypes (P = 25, nHe = 44), with one overrepresented haplotype (frequency of 36). The chloroplast psbA-trnH intergenic region yielded the shortest and least diverse alignment with a length of 416 bp and 12 unique haplotypes (P = 18).
The median-joining haplotype network for the nuclear sequence Agt1 showed that haplotypes are mostly shared between samples from “LG-Congo” and “Congo North,” and to a lesser extent between those from “LG–Congo” and “LG South” (Fig. 2A). All the haplotypes observed in “LG North” samples were unique to the cluster, apart from one haplotype observed in two samples from east Gabon (between Lastoursville and Okondja), which was also observed in eight samples assigned to “LG South,” one assigned to “LG-Congo,” and seven unassigned samples. The one sample included from “Congo South” carried a unique Agt1 haplotype. The Agt1 haploweb, which visualizes the haplotypes co-occurring in heterozygous samples, revealed three distinct “fields for recombination” (FFRs) among the specimens sampled (Fig. 2B). The largest FFR comprised samples from all SSR clusters apart from “Congo South.” The latter cluster was represented by one homozygous sample and hence formed a separate FFR. The third gene pool consisted of two haplotypes from two samples collected in the Ejagham Forest Reserve in west Cameroon, assigned to “LG North.” Most of the haplotypes of the samples assigned to “LG North” (bottom left in Fig. 2) were only connected to the major gene pool through one admixed heterozygous sample from northeast Gabon, which carried the same allele as some samples from “LG South” and “LG-Congo.”

Haploweb for the nuclear marker Agt1 in Staudtia kamerunensis. Each circle corresponds to a haplotype and has a size proportional to the number of samples. Curved lines connect haplotypes co-occurring in heterozygous individuals, with the thickness proportional to the frequency of the co-occurrence. Each crossing line corresponds to one mutated position in the alignment. (A) Colors correspond to the five genetic clusters inferred with STRUCTURE. Samples that appeared admixed in the SSR analysis (highest assignment probability q < 0.8) are colored grey, and sequenced samples that were not included in the SSR analysis are colored black. (B) Colors correspond to the inferred single-locus fields for recombination (FFRs)
The median-joining haplotype network for At103 was characterized by a dominant haplotype in the middle of the network (Fig. 3A). This haplotype was shared between 36 samples from all SSR clusters (also including the only sample from “Congo South”). Other haplotypes were mostly unique to the different SSR clusters or shared between “LG-Congo” and “Congo North.” The At103 haploweb showed a major gene pool with connections between all but one haplotype (in a homozygous sample from “Congo North”) (Fig. 3B).

Haploweb for the nuclear marker At103 in Staudtia kamerunensis. Each circle corresponds to a haplotype and has a size proportional to the number of samples. Curved lines connect haplotypes co-occurring in heterozygous individuals, with the thickness proportional to the frequency of the co-occurrence. Each crossing line corresponds to one mutated position in the alignment. (A) Colors correspond to the five genetic clusters inferred with STRUCTURE. Samples that appeared admixed in the SSR analysis (highest assignment probability q < 0.8) are colored grey. (B) Colors correspond to the inferred single-locus fields for recombination (FFRs)
The median-joining haplotype network constructed for the chloroplast psbA-trnH sequences showed that the haplotypes were mostly unique to the different SSR clusters (Fig. 4). One haplotype was shared between multiple samples from “LG South,” “LG-Congo,” and two unassigned samples, while another haplotype was shared between multiple samples from “LG South,” two samples from “LG North” (collected in central east Gabon), and two unassigned samples (collected in central/north east Gabon). The only sample from “Congo South” had the same haplotype as samples from “Congo North.” The three psbA-trnH sequences downloaded from GenBank are grouped with a haplotype found in samples from “LG North” collected in south Cameroon, the same region of origin as the GenBank samples.

Haplotype network for the chloroplast marker psbA-trnH in Staudtia kamerunensis. Each circle corresponds to a haplotype and has a size proportional to the number of individuals. Colors correspond to the five genetic clusters inferred with STRUCTURE. Individuals that appeared admixed in the SSR analysis (highest assignment probability q smaller than 0.8) are colored grey, and sequenced samples that were not included in the SSR analysis are colored black
Discussion
We examined the genetic diversity and differentiation patterns in S. kamerunensis, a tropical tree that occurs in evergreen and semi-deciduous forests in Central Africa. Our results show multiple genetic discontinuities throughout the species’ distribution range, and relatively high diversity at the level of microsatellite markers, as well as at the level of nuclear and chloroplast sequences.
Genetic discontinuities: distinct species or relics of ancient forest fragmentation
The Bayesian clustering analysis reveals five differentiated genetic clusters with a para- or allopatric distribution within S. kamerunensis. Such genetic clusters can represent different species (interspecific diversity as in Daïnou et al. 2016; Ikabanga et al. 2017; Monthe et al. 2018; Lissambou et al. 2019; Ewédjè et al. 2020; Bouka et al. 2022), or they can correspond to conspecific populations that differentiated during past isolation and (recently) came into secondary contact (intraspecific diversity as in Hardy et al. 2013; Migliore et al. 2019; Demenou et al. 2020; Helmstetter et al. 2020a, b; Piñeiro et al. 2021; Vanden Abeele et al. 2021). Therefore, we considered additional genetic markers and differentiation analyses to conclude on the delineation of species boundaries.
Based on the chloroplast marker and the two nuclear markers, genetic divergence appears to be strongest between the populations in southwest Cameroon (majority of “LG North”) and the other ones. All individuals from this area carry distinct nuclear and chloroplast haplotypes (although for At103, the haplotype with the highest overall frequency is shared with other genetic clusters). Furthermore, the populations from southwest Cameroon almost seem to have reached mutual allelic exclusivity since they almost form a distinct field for recombination in the Agt1 haploweb, if not for one unassigned/admixed heterozygous individual from northeast Gabon. This strong genetic divergence could mean that each group corresponds to a distinct species, with occasional hybridization in contact zones in Gabon, resulting in a few shared haplotypes in individuals from this region. However, genetic differentiation at the level of the microsatellite markers appears weaker, as the proportion of admixed individuals between “LG North” and “LG South” (5.44%) is among the highest for all genetic cluster combinations, and estimates of genetic differentiation (FST and RST) are among the lowest we observed. Despite the differences in alleles frequencies detected by STRUCTURE, the scarcity of private alleles and the abundance of alleles shared between the different SSR clusters in the haplowebs suggest that they are not reproductively isolated and are therefore most likely conspecific.
The occurrence of differentiated genetic clusters is reminiscent of the two varieties described in S. kamerunensis, where var. kamerunensis is probably limited to Lower Guinea (western side of the Central African rainforest block) while var. gabonensis occurs in both Lower Guinea and Congolia (eastern side of the Central African rainforest block) (Doucet 2003). However, the genetic clusters identified in our study are parapatric, while the two varieties have been described as sympatric in western Lower Guinea (e.g., in Korup National Park (Thomas et al. 2003) and in western Gabon (Doucet 2003)). Therefore, we rather speculate that the genetic clusters all belong to S. kamerunensis var. gabonensis. Under this hypothesis, the kamerunensis variety is either not present in our sampling or so underrepresented that the Bayesian clustering algorithm failed to separate those few individuals in an additional SSR cluster. The latter could explain the relatively high number of unassigned individuals in central Gabon, of which some might correspond to var. kamerunensis and putative hybrids, since this region is reported to harbor both varieties (Doucet 2003). Additionally, some of the smaller FFRs observed in the Agt1 and At103 haplowebs might actually consist of (undetected) var. kamerunensis individuals. The fact that S. kamerunensis var. kamerunensis is missing or underrepresented in our dataset, despite an extensive sampling, could be explained by the rarity of this variety. In fact, (historical) botanical collections contain only two documented specimens of this variety (although this could also result from the difficulties associated with the morphological identification of dried specimens) and inventory studies report much lower densities of var. kamerunensis compared to var. gabonensis (Doucet 2003; Thomas et al. 2003). As shown before, the sampling scheme can determine whether hidden genetic diversity is detected or missed in delimitation studies on species with unequal densities. In the African timber genus Milicia for instance, a rare hidden species was detected using genetic methods only after obtaining a more homogeneous geographic sampling (Daïnou et al. 2014, 2016).
Impact of historical climatic fluctuations and biogeographic barriers
Strong genetic differentiation and sharp genetic breaks within African tree species are not uncommon and are usually interpreted as resulting from population fragmentation and retraction into forest refugia during Plio- and Pleistocene glaciations (Hardy et al. 2013; Helmstetter et al. 2020a, b; Piñeiro et al. 2021). Previous phylogeographic studies on African trees showed that rainforest fragmentation in Lower Guinea not necessarily occurred according to the most often considered refuge hypotheses (Maley 1996; Anhuf et al. 2006), and that the evolutionary response of Central African tree species to climatic fluctuations is variable and individualistic (Helmstetter et al. 2020a, b; Piñeiro et al. 2021). Our results confirm these findings for Lower Guinea, and show that such discrepancies between postulated refuge areas and current distribution of intraspecific diversity also occur in Congolia. Furthermore, we identify multiple differences between the location and orientation of the genetic breaks in S. kamerunensis and those observed in other Central African tree species.
Among S. kamerunensis populations, both east–west and north–south genetic breaks are observed. The genetic differentiation between east and west often coincides with the boundary between the biogeographic subregions Lower Guinea and Congolia, especially at the species level (Droissart et al. 2018), since intraspecific genetic data for Congolia is largely insufficient (but see Ahossou et al. 2020; Vanden Abeele et al. 2021). However, this seems not to be the case in S. kamerunensis, as one large genetic cluster (LG-Congo) spans both bioregions. Nonetheless, both bioregions harbor distinct genetic clusters, suggesting that arid periods during the Pliocene and/or Pleistocene caused retraction and fragmentation of rainforests in both Lower Guinea and Congolia.
In Lower Guinea, strong divergent populations in southwestern Cameroon are not uncommon, which highlights the importance of the Cameroon Volcanic Line (CVL) as a putative forest refuge during Pleistocene glacial periods and/or driver of (population) differentiation (Couvreur et al. 2021). In multiple species, this north–south break in Lower Guinea might be maintained by environmental differences across the climatic hinge between 0 and 3°N (Hardy et al. 2013), which could limit dispersal and prevent genetic homogenization of populations after secondary contact following expansion out of putative forest refugia (Helmstetter et al. 2020a, b). In S. kamerunensis however, the genetic break is situated further south, in Central Gabon, which suggests that individuals assigned to “LG North” have a wide ecological amplitude, as successful establishment of the species seems to be little impacted by the temperature and rainfall differences across the climatic inversion. Moreover, lineage dispersal across the inversion is likely frequent, as gene dispersal is expected to be extensive in S. kamerunensis populations, both through seed (nuclear and chloroplast genes) and pollen dispersal (nuclear genes). Indeed, the fleshy fruits of S. kamerunensis are dispersed by frugivores such as hornbills, so they can potentially travel over large distances (up to almost 7000 m; Holbrook and Smith 2000). While information on pollinators is currently lacking, pollen dispersal might be even more extensive, which is supported by the differences in genetic structure observed from nuclear and chloroplast markers. In eastern Gabon for example, between Lastoursville and Okondja, we observed individuals assigned to either the nuclear SSR cluster “LG North” or “LG South” (as well as many admixed individuals). However, “LG North” and “LG South” individuals in this area cannot be distinguished based on their psbA-trnH chloroplast haplotype (dispersed through seeds), which is shared with other “LG South” individuals that occur more southward. The fact that this east–west break in Gabon observed from chloroplast sequence data (also confirmed with S. kamerunensis plastome data; Matvijev et al. 2022) is partly erased at the level of the nuclear DNA indicates that gene flow through pollen is more extensive than through seeds. This discordance in nuclear-chloroplast genomic divergence is also observed in the Mayumbe region (i.e., different plastid lineages in southern Congo vs. southwestern DR Congo, but the same nuclear cluster assignment) and around the Cameroon Volcanic Line (i.e., different plastid lineages north and south of the CVL, but the same nuclear cluster assignment; Matvijev et al. 2022).
Interestingly, while admixture between the different genetic clusters is abundant in Gabon, no individuals with mixed “LG North” and “LG-Congo” ancestry are observed in Cameroon, although both clusters are geographically close. This is likely because secondary contact between both clusters in Cameroon is relatively recent, which is supported by phylogeographic analyses that reconstructed the dispersal history of S. kamerunensis plastomes (Matvijev et al. 2022). This phylogeographic reconstruction shows that the populations in southeast Cameroon are the result of Congolian populations that expanded into northern Congo and southern CAR, and subsequently into Cameroon. Additional sampling in this region is needed to verify if gene flow occurs between these clusters in areas where they co-occur, or if they are too genetically different to allow mixing. However, we observed relatively low genetic differentiation between both clusters, as well as some individuals with mixed “LG North” and “LG-Congo” ancestry in Gabon; thus, the east–west genetic break in Cameroon is unlikely to result from genetic incompatibility.
In addition to the divergent populations in Lower Guinea, our analyses reveal multiple genetic discontinuities among S. kamerunensis populations in Congolia, which suggests that Pliocene and/or Pleistocene glaciations also caused forest retraction and fragmentation in this region. The strongest genetic differentiation (based on FST and RST estimates) is observed between the genetic cluster in south-eastern DR Congo (Congo South) and all the other ones. This cluster mostly occurs in gallery forest in Miombo woodlands and dry evergreen Muhulu forests (south) or highland and transition forests (east), while the other ones are mostly associated with humid lowland forests (Shapiro et al. 2021). Hence, the genetic divergence could be the result of ongoing speciation, linked to ecological niche differentiation. However, the proportion of individuals with mixed “Congo North” and “Congo South” ancestry is relatively high in the transition zone between the different vegetation types, which indicates that gene flow between both clusters is frequent in their contact zone. Interestingly, the genetic break in south-eastern DR Congo we identified using nuclear SSR data is not observed in phylogeographic analyses using plastome data (Matvijev et al. 2022), since plastid lineages in southern and eastern DR Congo are genetically similar to those in northeast DR Congo. This disparity in plastid and nuclear lineage distribution could be caused by selection on the nuclear genome if the “Congo South” cluster developed new adaptations to its peculiar habitat. Additionally, the genetic break observed between plastid lineages in central DR Congo (Matvijev et al. 2022) is erased at the nuclear DNA, but the break between clusters in western DR Congo might be maintained because of a limited gene flow between populations in swamp forests (“LG-Congo”) and semi-deciduous forests (“Congo North”).
Distribution of genetic diversity and the demographic history of populations
Lower Guinea is characterized by a higher plant species diversity and endemism rate compared to Congolia (Sosef et al. 2017; Droissart et al. 2018), a pattern that is usually also observed in the distribution of intraspecific diversity of widespread tropical African species (although genetic data for Congolia remain scarce; Daïnou et al. 2016; Ahossou et al. 2020; Vanden Abeele et al. 2021). In S. kamerunensis however, this unequal distribution of intraspecific diversity is less pronounced. That is, each bioregion is characterized by two differentiated genetic clusters, with a fifth one spanning both Lower Guinea and Congolia, and the estimated nuclear genetic diversity is similar among most of these clusters. However, the south-eastern populations in DR Congo (Congo South) do show a lower nuclear genetic diversity, possibly because they are little connected to northern populations and because population densities are expected to be low in the drier woodlands, where the species is limited to gallery forests. Contrastingly, the S. kamerunensis populations in south-western Cameroon (LG North) are characterized by a higher genetic diversity, as they harbor the highest amount of (unique) Agt1 and psbA-trnH haplotypes. High intraspecific diversity around the CVL has also been documented for various other trees (Hardy et al. 2013; Dauby et al. 2014; Heuertz et al. 2014; Piñeiro et al. 2017; Migliore et al. 2019) or herbaceous species (Ley et al. 2014), which gives support to the hypothesis that the rainforest survived in this area during glacial maxima.
The five genetic clusters identified in this work coincide partially with the plastid lineages and sublineages described by Matvijev et al. (2022; Table 3): (1) the “LG North” cluster coincides with plastid lineages 1 and 2, (2) “LG South” coincides with plastid lineages 3 and 4, and (3) “Congo South,” “Congo North,” and “LG-Congo” tend to respectively coincide with plastid lineages 5a, 5b, and 5c (with multiple exceptions, especially for “Congo South”). It suggests that in the areas previously identified as relatively stable (no hint of recent range expansion according to plastome data, where plastid lineages 1, 2, 3, and 4 occur; Matvijev et al. 2022), pollen dispersal contributed to homogenize gene pools between some parapatric maternal lineages, while in areas showing a strong signature of range expansion (mostly Congolia, where plastid lineage 5 occurs) differentiated gene pools (nuclear clusters “LG-Congo,” “Congo North,” and “Congo South”) could have arisen from the same maternal origin through founder effects as distinct populations expanded in different directions. However, SSR data do not show signatures of different demographic histories between populations from Lower Guinea or Congolia. The correspondence between plastid lineages and nuclear genetic clusters supports the hypothesis that the genetic breaks result from population isolation (following habitat fragmentation or colonization of novel habitats) while their imperfect match indicates that there is probably no barrier to gene flow between these clusters or lineages once their ranges overlap (secondary contact zone).
Conclusions
Our analyses reveal multiple genetic discontinuities among S. kamerunensis populations throughout Central Africa, likely resulting from rainforest fragmentation during cold and dry glaciations in the Pliocene and/or Pleistocene. The most differentiated populations are found in southeast DR Congo (genetic cluster “Congo South”), which could be the result of ongoing speciation linked to ecological niche differentiation. That is, these differentiated populations mostly occur in gallery forest and dry evergreen Muhulu forests (south DR Congo) or highland and transition forests (east DR Congo), while all other S. kamerunensis populations are mostly associated with humid lowland forests. Since subtaxon identification proves to be difficult, it is possible that var. kamerunensis is underrepresented in our sampling (due to its low densities in the field), meaning that it remained undetected in the clustering analyses. To verify whether S. kamerunensis var. gabonensis and S. kamerunensis var. kamerunensis can be distinguished genetically, field observations and additional collections are needed from west Gabon and southwest Cameroon, where the occurrence of both varieties has been recorded.
References
Ahossou OD, Daïnou K, Janssens SB, Triest L and Hardy OJ (2020) Species delimitation and phylogeography of African tree populations of the genus Parkia (Fabaceae). Tree Genet Genomes 16:68. https://doi.org/10.1007/s11295-020-01463-x
Anhuf D, Ledru MP, Behling H, Da Cruz FW, Cordeiro RC, Van der Hammen T, Karmann I et al (2006) Paleo-environmental change in Amazonian and African rainforest during the LGM. Palaeogeogr Palaeoclimatol Palaeoecol 239:510–527
Bandelt HJ, Forster P, Rohl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Belkhir K, Borsa P, Chikhi L, Raufaste N and Bonhomme F (2004) GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations. Montpellier, France: Université de Montpellier II; 2004. https://doi.org/10.1159/000078211
Bickford D, Lohman DJ, Sodhi NS, Ng PKL, Meier R, Winker K, Ingram KK, Das I (2007) Cryptic species as a window on diversity and conservation. Trends Ecol Evol 22:148–155
Bouka GUD, Doumenge C, Ekué MRM, Daïnou K, Florence J, Degen B, Loumeto JJ et al (2022) Khaya revisited: genetic markers and morphological analysis reveal six species in the widespread taxon K. anthotheca. Taxon 71:814–832. https://doi.org/10.1002/tax.12720
Bradshaw CJA, Sodhi NS, Brook BW (2009) Tropical turmoil: a biodiversity tragedy in progress. Front Ecol Environ 7:79–87
Cappellini E, Gilbert MTP, Geuna F, Fiorentino G, Hall A, Thomas-Oates J, Ashton PD et al (2010) A multidisciplinary study of archaeological grape seeds. Naturwissenschaften 97:205–217
CazzollaGatti R, Reich PB, Gamarra JGP, Crowther T, Hui C, Morera A, Bastin J-F et al (2022) The number of tree species on Earth. Proc Natl Acad Sci 119:1–11
Cheek M, NicLughadha E, Kirk P, Lindon H, Carretero J, Looney B, Douglas B et al (2020) New scientific discoveries: plants and fungi. Plants, People, Planet 2:371–388
Chessel D, Dufour A, Thioulouse J (2007) The ade4 Package - I: One-Table Methods. R News 7:47–52
Chybicki IJ, Burczyk J (2009) Simultaneous estimation of null alleles and inbreeding coefficients. J Heredity 100(1):106–113. https://doi.org/10.1093/jhered/esn088
Cornuet JM, Piry S, Luikart G, Estoup A, Solignac M (1999) New methods employing multilocus genotypes to select or exclude populations as origins of individuals. Genetics 153:1989–2000
Couvreur TLP, Dauby G, Blach-Overgaard A, Deblauwe V, Dessein S, Droissart V, Hardy OJ et al (2021) Tectonics, climate and the diversification of the tropical African terrestrial flora and fauna. Biol Rev 96:16–51
Daïnou K, Blanc-jolivet C, Degen B, Kimani P, Ndiade-bourobou D, Donkpegan ASL, Tosso F et al (2016) Revealing hidden species diversity in closely related species using nuclear SNPs, SSRs and DNA sequences – a case study in the tree genus Milicia. BMC Evol Biol 16:259
Daïnou K, Mahy G, Duminil J, Dick CW, Doucet J, Donkpégan ASL, Pluijgers M et al (2014) Speciation slowing down in widespread and long-living tree taxa: insights from the tropical timber tree genus Milicia (Moraceae). Heredity 113:74–85
Dauby G, Duminil J, Heuertz M, Koffi GK, Stévart T, Hardy OJ (2014) Congruent phylogeographical patterns of eight tree species in Atlantic Central Africa provide insights into the past dynamics of forest cover. Mol Ecol 23:2299–2312
Demenou BB, Migliore J, Heuertz M, Monthe FK, Ojeda DI, Wieringa JJ, Dauby G et al (2020) Plastome phylogeography in two African rain forest legume trees reveals that Dahomey Gap populations originate from the Cameroon volcanic line. Mol Phylogenet Evol 150:106854
De Quieroz K (2007) Species concepts and species delimitation. Syst Biol 56:879–886
Doucet JL (2003) L’alliance délicate de la gestion forestière et de la biodiversité dans les forêts du centre du Gabon. PhD thesis. Sciences agronomiques de Gembloux, Université de Liège
Doyle JJ (1995) The irrelevance of allele tree topologies for species delimitation, and a non-topological alternative. Syst Bot 20:574–588
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh plant tissue. Focus 12:13–15
Droissart V, Dauby G, Hardy OJ, Deblauwe V, Harris DJ, Janssens S, Mackinder BA et al (2018) Beyond trees: biogeographical regionalization of tropical Africa. J Biogeogr 45:1153–1167
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Ewédjè EEBK, Jansen S, Koffi GK, Staquet A, Piñeiro R, Essaba RA, Obiang NLE et al (2020) Species delimitation in the African tree genus Lophira (Ochnaceae) reveals cryptic genetic variation. Conserv Genet 21:501–514
Flot J-F, Couloux A, Tillier S (2010) Haplowebs as a graphical tool for delimiting species: a revival of Doyle’s ‘field for recombination’ approach and its application to the coral genus Pocillopora in Clipperton. BMC Evol Biol 10:372
Flot JF (2010) SeqPHASE: A web tool for interconverting phase input/output files and fasta sequence alignments. Mol Ecol Resour 10:162–166
Fouilloy R (1974) Myristicaceae. Flore du Cameroun. Volume 18. Muséum National d’Histoire Naturelle, Paris, pp 89–105
Gilbert G and Troupin G (1951) Flore du Congo Belge et du Ruanda-Urundi: Myristicaceae. vol. 2. Institut national pour l'étude agronomique du Congo belge. Bruxelles, Belgium
Hardy OJ, Born C, Budde K, Daïnou K, Dauby G, Duminil J, Ewédjé EEBK et al (2013) Comparative phylogeography of African rain forest trees: a review of genetic signatures of vegetation history in the Guineo-Congolian region. CR Geosci 345:284–296
Hardy OJ, Charbonnel N, Fréville H, Heuertz M (2003) Microsatellite allele sizes: a simple test to assess their significance on genetic differentiation. Genetics 163:1467–1482
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Helmstetter AJ, Amoussou BEN, Bethune K, Kamdem NG, GlèlèKakaï R, Sonké B, Couvreur TLP (2020a) Phylogenomic approaches reveal how climate shapes patterns of genetic diversity in an African rain forest tree species. Mol Ecol 29:3560–3573
Helmstetter AJ, Béthune K, Kamdem NG, Sonké B, Couvreur TLP (2020b) Individualistic evolutionary responses of Central African rain forest plants to Pleistocene climatic fluctuations. Proc Natl Acad Sci USA 117:32509–32518
Heuertz M, Duminil J, Dauby G, Savolainen V and Hardy OJ (2014) Comparative phylogeography in rainforest trees from lower Guinea, Africa. PLoS One 9:e84307. https://doi.org/10.1371/journal.pone.0084307
Hoban S, Bruford MW, Funk WC, Galbusera P, Griffith MP, Grueber CE, Heuertz M et al (2021) Global commitments to conserving and monitoring genetic diversity are now necessary and feasible. Bioscience 71:964–976
Hohenlohe PA, Funk WC, Rajora OP (2021) Population genomics for wildlife conservation and management. Mol Ecol 30:62–82
Holbrook KM, Smith TB (2000) Seed dispersal and movement patterns in two species of Ceratogymna hornbills in a West African tropical lowland forest. Oecologia 125:249–257
Ikabanga DU, Stévart T, Koffi KG, Monthé FK, Nzigoudoubindou EC, Dauby G, Souza A et al (2017) Combining morphology and population genetic analysis uncover species delimitation in the widespread African tree genus Santiria (Burseraceae). Phytotaxa 321:166–180
Janssens S, Geuten K, Yuan Y-M, Song Y, Küpfer P, Smets E (2006) Phylogenetics of Impatiens and Hydrocera (Balsaminaceae) using chloroplast atpB-rbcL spacer sequences. Syst Bot 31:171–180
Jombart T (2008) adegenet: a R package for multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S et al (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191
Laikre L, Hoban S, Bruford MW, Segelbacher G, Allendorf FW, Gajardo G, Rodríguez AG et al (2020) Post-2020 goals overlook genetic diversity. Science 367:1083–1085. https://doi.org/10.1126/science.abb2748
Ley AC, Dauby G, Köhler J, Wypior C, Röser M, Hardy OJ (2014) Comparative phylogeography of eight herbs and lianas (Marantaceae) in central African rainforests. Front Genet 5:1–14
Li M, Wunder J, Bissoli G, Scarponi E, Gazzani S, Barbaro E, Saedler H, Varotto C (2008) Development of COS genes as universally amplifiable markers for phylogenetic reconstructions of closely related plant species. Cladistics 24:727–745
Lischer HEL, Excoffier L (2012) PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28:298–299
Lissambou B-J, Couvreur TLP, Atteke C, Stévart T, Piñeiro R, Dauby G, Monthe FK et al (2019) Species delimitation in the genus Greenwayodendron based on morphological and genetic markers reveals new species. Taxon 68:442–454
Maley J (1996) The African rain forest main characteristics of changes in vegetation and climate from the Upper Cretaceous to the Quaternary. Proceed Royal Soc Edinburgh Sect B Biol Sci 104:31–73
Matvijev K, Dellicour S, Kaymak E and Hardy OJ (2022) Spatially explicit phylogeographical reconstruction sheds light on the history of the forest cover in the Congo Basin. J Biogeograph 49:2256–2268. https://doi.org/10.1111/jbi.14507
Migliore J, Kaymak E, Mariac C, Couvreur TLP, Lissambou BJ, Piñeiro R, Hardy OJ (2019) Pre-Pleistocene origin of phylogeographical breaks in African rain forest trees: new insights from Greenwayodendron (Annonaceae) phylogenomics. J Biogeogr 46:212–223
Monthe FK, Duminil J, Kasongo Yakusu E, Beeckman H, Bourland N, Doucet JL, Sosef MSM and Hardy OJ (2018) The African timber tree Entandrophragma congoense (Pierre ex De Wild.) A.Chev. is morphologically and genetically distinct from Entandrophragma angolense (Welw.) C.DC. Tree Genet Genomes 14:66. https://doi.org/10.1007/s11295-018-1277-6
Nielsen R, Tarpy DR, Reeve HK (2003) Estimating effective paternity number in social insects and the effective number of alleles in a population. Mol Ecol 12:3157–3164
Parmentier I, Duminil J, Kuzmina M, Philippe M, Thomas DW, Kenfack D, Chuyong GB et al (2013) How effective are DNA barcodes in the identification of African rainforest trees? PLoS ONE 8:e54921
Piñeiro R, Dauby G, Kaymak E, Hardy OJ (2017) Pleistocene population expansions of shade-tolerant trees indicate fragmentation of the African rainforest during the Ice Ages. Proceed Royal Soc b: Biol Sci 284:20171800
Piñeiro R, Hardy OJ, Tovar C, Gopalakrishnan S, Garrett F (2021) Contrasting genetic signal of recolonization after rainforest fragmentation in African trees with different dispersal abilities. Proc Natl Acad Sci 118:1–9
Piry S, Luikart G, Cornuet J-M (1999) Computer note. BOTTLENECK: a computer program for detecting recent reductions in the effective size using allele frequency data. J Hered 90:502–503
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Puechmaille SJ (2016) The program STRUCTURE does not reliably recover the correct population structure when sampling is uneven: subsampling and new estimators alleviate the problem. Mol Ecol Resour 16:608–627
QGIS.org. 2021. QGIS Geographic Information System. Open Source Geospatial Foundation. http://qgis.osgeo.org
RStudio Team (2019) RStudio: integrated development for R. RStudio, PBC, Boston, MA. http://www.rstudio.com/
Sang T, Crawford DJ, Stuessy TF (1997) Chloroplast DNA phylogeny, reticulate evolution, and biogeography of Paeonia (Paeoniaceae). Am J Bot 84:1120–1136
Scheffers BR, De Meester L, Bridge TCL, Hoffmann AA, Pandolfi JM, Corlett RT, Butchart SHM et al (2016) The broad footprint of climate change from genes to biomes to people. Science 354. https://doi.org/10.1126/science.aaf7671
Shapiro AC, Grantham HS, Aguilar-Amuchastegui N, Murray NJ, Gond V, Bonfils D, Rickenbach O (2021) Forest condition in the Congo Basin for the assessment of ecosystem conservation status. Ecol Ind 122:107268
Slatkin M (1995) Slatkin 1995 microsatellite interpretation using Rst. Genetics 139:457–462
Sosef MSM, Dauby G, Blach-Overgaard A, van der Burgt X, Catarino L, Damen T, Deblauwe V et al (2017) Exploring the floristic diversity of tropical Africa. BMC Biol 15:1–23
Spöri Y, Flot JF (2020) HaplowebMaker and CoMa: two web tools to delimit species using haplowebs and conspecificity matrices. Methods Ecol Evol 11:1434–1438
Stange M, Barrett RDH, Hendry AP (2021) The importance of genomic variation for biodiversity, ecosystems and people. Nat Rev Genet 22:89–105
Steeves R (2011) Anintrageneric and intraspecific study of morphological and genetic variation in the Neotropical Compsoneura and Virola (Myristicaceae). PhD thesis. The University of Guelph, Ontario
Stegemann S, Keuthe M, Greiner S, Bock R (2012) Horizontal transfer of chloroplast genomes between plant species. Proc Natl Acad Sci USA 109:2434–2438
Stephens M, Smith NJ, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68:978–989
Struck TH, Feder JL, Bendiksby M, Birkeland S, Cerca J, Gusarov VI, Kistenich S et al (2018) Finding evolutionary processes hidden in cryptic species. Trends Ecol Evol 33:153–163
Tate JA, Simpson BB (2003) Paraphyly of Tarasa (Malvaceae) and diverse origins of the polyploid species. Syst Bot 28:723–737
Thomas DW, Kenfack D, Chuyong GB, Moses SN, Losos EC, Condit RS and Songwe NC (2003) Tree species of Southwestern Cameroon: tree distribution maps, diameter tables, and species documentation of the 50-hectare Korup Forest Dynamics Plot. Thomas DW, Kenfack D, Chuyong GB, Moses SN, Losos EC, Condit RS, Songwe NC (eds) Center for Tropical Forest Science, Smithsonian Tropical Research Institute, Washington, D.C.
Vanden Abeele S, Hardy OJ and Janssens SB (2018) Isolation of microsatellite loci in the African tree species Staudtia kamerunensis (Myristicaceae) using high-throughput sequencing. Mole Biol Rep 45:1539–1544. https://doi.org/10.1007/s11033-018-4239-z
Vanden Abeele S, Janssens SB, Piñeiro R, Hardy OJ (2021) Evidence of past forest fragmentation in the Congo Basin from the phylogeography of a shade-tolerant tree with limited seed dispersal: Scorodophloeus zenkeri (Fabaceae, Detarioideae). BMC Ecology and Evolution 21:50
Vermoesen C (1923) Manuel des Essences forestières du Congo Belge
Wang J (2017) The computer program structure for assigning individuals to populations: easy to use but easier to misuse. Mol Ecol Resour 17:981–990
Warburg O (1897) Monographie der Myristicaceen. Nova Acta Academiae Caesareae Leopoldino-Carolinae Germanicae Naturae Curiosorum 68:1–680
Warburg O (1904) Myristicaceae africanae. In: Engler A (ed) Botanische Jahrbücher für Systematik, Pflanzengeschichte und Pflanzengeographie, 381–386. Schweizerbart, Stuttgart
Weir BS, Cockerham CC (1984) Estimating F-statitics for the analysis of population structure. Evolution 38:1358–1370
White F (1979) The Guineo-Congolian region and its relationships to other phytochoria. Bullet Du Jardin Botanique Natl De Belgique / Bullet Van De Nationale Plantentuin Van België 49:11–55
White F (1983) The vegetation of Africa. Natural Resources Research XX, UNESCO, Paris
Willi Y, Kristensen TN, Sgrò CM, Weeks AR, Ørsted M, Hoffmann AA (2022) Conservation genetics as a management tool: the five best-supported paradigms to assist the management of threatened species. Proc Natl Acad Sci 119:e2105076119
Acknowledgements
We are grateful to Esra Kaymak (ULB-EBE), Wim Baert (Meise Botanic Garden), and Pieter Asselman (UGhent) for their assistance in the laboratory. We would like to thank the staff from the herbaria which kindly provided us with the herbarium material, namely Tariq Stevart and Geoffrey Fadeur at the Université Libre de Bruxelles (BRLU), David Harris at RBG Edinburgh (E), and Nina Davies and Isabel Larridon at the Royal Botanic Gardens, Kew (K). The samples collected in DR Congo by SVA were exported with a permit from the DRC ministère de l’Environnement et Développement Durable and are a part of the research agreement between Meise Botanic Garden and Université de Kisangani (UNIKIS).
Data archiving statement
Voucher information, sampling locations, and GenBank accession numbers for the Staudtia kamerunensis accessions included in this study are given in Online Resource 1 of the Supporting Information. The dataset with the corresponding genotypes is available from the Zenodo repository (https://doi.org/10.5281/zenodo.7767340).
Funding
This study is part of the HERBAXYLAREDD and AFRIFORD projects (BR/143/A3/HERBAXYLAREDD, BR/132/A1/AFRIFORD), funded by the Belgian Belspo-BRAIN program axis 4. This study has also received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement N° 765000, and the Fund for Scientific Research F.R.S.-FNRS (grant J.0292.17F). SVA is currently supported by a Postdoctoral Fellowship of the Wiener-Anspach Foundation. KM and OJH are supported by the Fonds de la Recherche Scientifique (F.R.S.-FNRS).
Author information
Authors and Affiliations
Contributions
SVA, SBJ, and OJH conceived and designed the study. SVA wrote the manuscript, with contributions from KM, SBJ, and OJH. SVA and KM did the molecular work, and SVA, KM, OJH, VD, and J-FF analyzed the data. SVA, KM, OJH, D-MA, BIA, HB, GB, CAB, J-FG, NGK, JL, FM, and BS contributed data and samples. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Communicated by C. Rellstab.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vanden Abeele, S., Matvijev, K., Hardy, O.J. et al. Genetic breaks caused by ancient forest fragmentation: phylogeography of Staudtia kamerunensis (Myristicaceae) reveals distinct clusters in the Congo Basin. Tree Genetics & Genomes 19, 27 (2023). https://doi.org/10.1007/s11295-023-01601-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-023-01601-1