
Abstract
The taxonomical classification among fungi kingdom in the last decades was evolved. In this work the targeted metabolomics study based on 1H NMR spectroscopy combined with chemometrics tools was reported to be useful for differentiation of three model of fungal strains, which represent various genus of Ascomycota (Aspergillus pallidofulvus, Fusarium oxysporum, Geotrichum candidum) were selected in order to perform metabolomics studies. Each tested species, revealed specific metabolic profile of primary endo-metabolites. The species of A. pallidofulvus is represented by the highest concentration of glycerol, glucitol and Unk5. While, F. oxysporum species is characterised by increased level of propylene glycol, ethanol, 4-aminobutyrate, succinate, xylose, Unk1 and Unk4. In G. candidum, 3-methyl-2-oxovalerate, glutamate, pyruvate, glutamine and citrate were elevated. Additionally, a detailed analysis of metabolic changes among A. pallidofulvus, F. oxysporum and G. candidum showed that A. pallidofulvus seems to be the most pathogenic fungi. The obtained results demonstrated that targeted metabolomics analysis could be utilized in the future as a supporting taxonomical tool for currently methods.
Graphical Abstract

Similar content being viewed by others


Introduction
The old taxonomic approach for fungi was generally based on phenotype (Guarro et al. 1999). This approach included comparative studies of the morphological structures, cell wall composition, cytological testing, ultrastructure, cellular metabolism, fossil record, and sexual cycle (Bartnicki-Garcia 1970, 1987; Fuller 1976; Hawksworth et al. 1995; Heath 1980, 1986; Le´John 1974; Taylor 1978; Vogel 1964). According to this method, the fungi kingdom was divided into the following four main phyla: Zygomycota, Ascomycota, Basidomycota and Fungi Imperfecti, namely Deuteromycota (Guarro et al. 1999). This division has changed during the last two decades, especially due to the development of new cladistics and molecular approaches (phylogenetic), such as a PCR method in which universal oligonucleotide primers specific to fungi were selected and the 18S subunits of the rRNA sequences were compared (Bruns et al. 1992; Golenberg et al. 1990; Hendriks 1992; Hausner et al. 1992; Haase et al. 1995; Spatafora and Blackwell 1993; Swann and Taylor 1995). Currently, the classification system of fungi contains the following seven phyla: Chytridiomycota, Blastocladiomycota, Neocallimastigomycota, Microsporidia, Glomeromycota, Ascomycota and Basidiomycota (Kirk et al. 2001, 2008; Hibbett et al. 2007; Blackwell et al. 2006; David et al. 2011). It can be imagined that this systematic scheme might further evolve in the future because of the huge biodiversity of fungal species and techniques that are still being developed.
The procedure of microbial identification can also be based on the analysis of the chemical compositions of cells (Ivanisˇevic´ et al. 2011; Semmar et al. 2007; Cevallos-Cevallosa et al. 2009). This approach is successfully used to screen for metabolic differences of various living systems. For example, Zieliński et al. reported that chemometrics studies are useful for determination of the origin of polish monofloral and multifloral honeys, whereas Deja et al. showed that multivariate analysis of primary endo-metabolites can exhibit correlation between fruit bodies and the topsoil type as well as differences in the chemical compositions of the stem and cap of Amanita muscaria (Zieliński et al. 2014; Deja et al. 2014). Apart from a few papers showing the possibility of organism classification according to their intracellular metabolite compositions, most of literature data demonstrated differentiation power based on secondary endo-metabolites (Frisvad et al. 2008; Andersen et al. 2008; Frisvad 1992; Jennessen et al. 2005). Generally, this trend could be observed in studies on fungal biodiversity, where chemotaxonomy is referred to as being successful for its classification. Metabolomics approaches have proven to be useful to distinguish among genera such as Penicilium, Aspergillus and Fusarium (Larsen et al. 2005; Smedsgaard and Nielsen 2005). Kadlec et al. exhibited differentiation between Tolypocladium, Beauceria and Paecilomyces by using gas chromatography combined with mass spectrometric analysis (Kadlec et al. 1994). Moreover, chemotaxonomic diversity among Saccharomyces cerevisiae mutant groups was observed (Smedsgaard and Nielsen 2004; Smedsgaard et al. 2004; Mas et al. 2007). Although chemotaxonomic studies associated with a metabolomics approach have been conducted based on secondary endo-metabolites, they have rarely been conducted on primary endo-metabolites. A few reports could be found in which components of the grow medium (metabolic footprint) were used for classification purposes (Junka et al. 2013; Zheng et al. 2011).
The main goal of this study was to examine a potential metabolomics-based approach for the supporting currently taxonomy of filamentous fungal pathogens by applying 1H NMR spectroscopy in association with chemometric analysis. The common fungal human pathogens Aspergillus pallidofulvus (A. pal), Fusarium oxysporum (F. oxy) and Geotrichum candidum (G. can) were chosen as model microorganisms. These fungal species belong to three different classes (Eurotiomycetes, Sordariomycetes, and Saccharomycetes) of the Ascomycota phylum. It should be noted that Eurotiomycetes and Sordariomycetes come from the same subphylum Pezizomycotina, whereas Saccharomycetes come from Saccharomycotina. With regard to previous reports, this is the first study in which the simple distinguish of filamentous fungi based on primary not secondary metabolites was observed. Moreover, the second objective was to determine characteristic metabolites related to the occurrence of the tested fungal pathogens. Finally, the specific biochemical pathways were discussed.
Materials and methods
Fungal strains and culture conditions
In this study, three genus of filamentous fungi including Aspergillus pallidofulvus (ZK0431), Fusarium oxysporum (DSM 12646) and Geotrichum candidum (DSM 6593) were tested. Fungal strains were routinely maintained on potato dextrose agar (PDA, Difco), which provided profuse sporulation suitable for collection of the inoculum.
To evaluate the differences and biodiversity between Aspergillus pallidofulvus, Fusarium oxysporum and Geotrichum candidum based on intracellular metabolites, fungi were cultured on potato dextrose agar (PDA, Difco) in 10-cm Petri dishes for 5 days at 28 °C. The inoculums of fungi were prepared by washing with 5 mL of 0.1% Tween 20. The spore suspensions were adjusted to a final concentration of 106 conidia/mL in 100 mL of potato dextrose broth (PDB, Difco) and incubated with shaking for 48 h at 28 °C.
Extraction of the fungal metabolites
The fungal biomasses were filtered, washed with saline solution and then weighed in aliquots of 100 mg of wet weight cells. In the next step, fungal cells were frozen for 10 min at −80 °C. The procedure of fungal cell disintegration included two separate consecutive steps. In the first one, frozen samples were disrupted using QIAGEN–TissueLyser (50 Hz, 5 min), followed by the addition of 650 µL of PBS buffer (10% D2O, 0.05 M, pH = 7.0, 0.15 mM TSP) to each sample. The second step based on ice bath ultrasonic cell disruption was performed for 30 min. After the disintegration procedure was completed, samples were centrifuged (10 min, 17 500 rpm, 4 °C), and 550 µL of clarified homogenate was transferred into a 5 mm NMR tube. Ten repetitions for each strain were performed in this study.
1H NMR spectroscopy analysis of the fungal metabolites
Standard NMR experiments were performed on a Bruker AVANCE II 600.58 MHz spectrometer equipped with a 5 mm TBO probe at 300 K. All one-dimensional 1H NMR spectra were carried out using the zgpr1d (in Bruker notation) pulse sequence by suppression of water resonance by presaturation. Acquisition parameters were as follows: spectral width, 20 ppm; acquisition time, 1.36 s per scan; time domain points, 32 K; relaxation delay, 3.5 s; and number of scans, 256. Prior to Fourier transformation, the FIDs were multiplied by an exponential function equivalent to that of a 0.3 Hz line-broadening factor. The spectra were manually corrected for phase and baseline (by a fifth order polynomial baseline fitting) and referenced to the TSP resonance at 0.0 ppm. Additionally, the metabolite conformations were assigned through two-dimensional NMR experiments 1H-1H-TOCSY (Total Correlation Spectroscopy) and 1H-13C-HSQC (Heteronuclear Single Quantum Coherence).
Data processing and multivariate statistical data analysis
All spectra were exported to Matlab (Matlab v. 8.1, Mathwork Inc.) for preprocessing. Regions affected by solvent suppression were excluded (4.500–5.100 ppm) and alignment procedures involving the correlation of optimized warping (COW) and interval correlation shifting (icoshift) algorithms were applied (Tomasi et al. 2004; Savorani et al. 2010). The spectra consisted of 30,811 data points and were normalized using the probabilistic quotient method to overcome the issue of dilution (Dieterle et al. 2006).
The multivariate and statistical data analysis were performed on a set of the 51 assigned metabolites. The metabolite concentration measured by NMR was obtained as the sum of the intensities of the non-overlapping resonances (or a cluster of partly overlapping resonances). Such a transformed data matrix was the input for SIMCA-P software (v 13.0, Umetrics, Umeå, Sweden) and Matlab for follow-up analysis.
Prior to the chemometric analysis, the data sets were unit variance scaled. For classification of the fungal strains, principal component analysis (PCA) was carried out. The fungal strains were assigned to groups according to the results of the homologous metabolite profiling that coincided with those of PCA-hierarchical cluster analysis (PCA-HCA). The graphical representation of the metabolite and fungal strains biplot was constructed from the first two components. The scores and loadings in the biplot were expressed using correlation scaling. A heat map with dendrograms to show dynamic changes in metabolites in the hierarchical clustering of data was created.
Metabolites responsible for the separation in models were tested using STATISTICA 10 with the Mann–Whitney–Wilcoxon test (MWW). A 0.01 level of probability was used as the criterion for statistical significance. Correlation coefficients (r) were also calculated.
Results
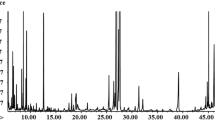
Representative 1H NMR-spectra for fungal strains are shown in Fig. 1. In total, 45 metabolites were identified based on typical 2D NMR experiments, namely 1H-1H-TOCSY and 1H-13C-HSQC. All assignments were verified using the Biological Magnetic Resonance Data Bank (BMRDB) and the Human Metabolome Data Base (HMDB) (Andersen et al. 2001, 2003). Six metabolites were not assigned (Unk1, Unk2, Unk3, Unk4, Unk5, Unk6).

Median 600 MHz 1H NMR zgpr spectra of filamentous fungal pathogen strains obtained from cell-free crude extracts of the following: a Aspergillus pallidofulvus; b Fusarium oxysporum; c Geotrichum candidum. 1 leucine; 2 valine; 3 isoleucine; 4 isobutyrate; 5 3-methyl-2-oxovalerate; 6 propylene glycol; 7 ethanol; 8 lactate; 9 alanine; 10 arginine; 11 lysine; 12 acetate; 13 proline; 14 methionine; 15 4-aminobutyrate; 16 glutamate; 17 pyruvate; 18 succinate; 19 glutamine; 20 citrate; 21 2-oxoisocaproate; 22 malate; 23 aspartate; 24 asparagine; 25 creatine; 26 malonate; 27 CHOLINE; 28 sn-glycero-3-phosphocholine; 29 myo-inositol; 30 glycerol; 31 glucitol; 32 mannitol; 33 betaine; 34 threonine; 35 xylose; 36 glucose; 37 glucose-1-phosphate; 38 uracil; 39 guanosine; 40 fumarate; 41 tyrosine; 42 histidine; 43 phenylalanine; 44 tryptophan; 45 xanthine; 46 Unk1; 47 Unk2; 48 Unk3; 49 Unk4; 50 Unk5; 51 Unk6
Multivariate analysis of the metabolite fingerprinting in A. pallidofulvus, F. oxysporum and G. candidum
In general, PCA was calculated with four principal components (PC) and revealed the natural grouping of the various fungal strains based on concentrations of the assigned endo-metabolites (Fig. 2a).

a PCA PC1/PC2 score plots of 1H NMR of the 51 total assigned metabolites and b Clusters in a two-dimensional biplot for A. pallidofulvus (orange circles), F. oxysporum (yellow boxes) and G. candidum (red triangles). (Color figure online)
In agreement with PCA, PCA-HCA applied on the same dataset revealed consistent resolution of the fungal strains (Fig. 3). The hierarchical clustering of the metabolite data showed three major groups of samples that were similar (A. pallidofulvus, F. oxysporum and G. candidum) to those of PCA. As shown in Fig. 2a, G. can is clearly separated from the other fungal genus. Among the other genus, A. pal and F. oxy were found to be the most similar to themselves.

Dendrogram of HCA results obtained from PCA based on 4 PCs
The first and second PC (PC1 and PC2, respectively) accounted for 41.7% and 37.1% of the variance in the data, respectively. The data points of A. pal and F. oxy were separated from G. can in PC1, whereas differences between A. pal and F. oxy were in PC2 (Fig. 2a).
The dynamic changes in the metabolite concentrations in the three clusters were explored using a heat map with hierarchical clustering (Fig. 4). The cluster of A. pal was characterized by increased levels of malonate, choline, sn-glycero-3-phosphocholine, glycerol, glucitol, betaine, glucose and Unk5. The group of F. oxy showed higher levels of leucine, isoleucine, isobutyrate, propylene glycol, ethanol, acetate, 4-aminobutyrate, succinate, aspartate, xylose, guanosine, phenylalanine, Unk1, Unk4 and Unk6.

The heat map with hierarchical clustering of the metabolites. The metabolites assigned in columns are labeled with the same numbering scheme as in Fig. 1
In G. can the concentrations of 3-methyl-2-oxovalerate, alanine, proline, glutamate, pyruvate, glutamine, citrate, 2-oxoisocaproate, creatine, fumarate, histidine and xanthine were significantly higher.
The statistical analysis performed by using the MWW exhibited that in every possible comparison (A. pal vs. F. oxy, A. pal vs. G. can, F. oxy vs. G. can), almost every metabolite was statistically significant (Table 1).
The most important metabolites correlated with the tested fungal genus were demonstrated in a biplot (Fig. 2b). Additionally, the correlation coefficient (r) was calculated for each comparison (Table 2). Taking into account the variables and their correlation coefficients, in positive correlation r > 0.9, and for each genus the characteristic metabolites were selected. The species of A. pal is represented by the highest concentrations of glycerol, glucitol and Unk5 (Fig. 5; bold italics metabolites in Table 2).

The general scheme of the metabolic pathways of A. pallidofulvus (orange boxes), F. oxysporum (yellow boxes) and G. candidum (red boxes). (Color figure online)
The positively correlated metabolites in F. oxy species were propylene glycol, ethanol, 4-aminobutyrate, succinate, xylose, Unk1 and Unk4 (Fig. 2b; italics metabolites in Table 2). In G. can, 3-methyl-2-oxovalerate, glutamate, pyruvate, glutamine and citrate exhibited positive correlation (Fig. 2b; bold metabolites in Table 2).
Discussion
There is no evidence that previously published papers have comparatively analyzed the metabolite profiles of filamentous fungal pathogens including A. pallidofulvus, F. oxysporum and G. candidum. Our results show that a simple comparison of primary endo-metabolites using 1H NMR-based metabolomics can clearly separate these tested fungi. This is contradictory to most previous reports that postulated the lower power of primary endo-metabolites in chemotaxonomy and the differentiation of microorganisms (Frisvad et al. 2008; Andersen et al. 2008; Frisvad 1992; Jennessen et al. 2005). It is well-known that satisfactory growth of fungi and hence the production of metabolites is strongly dependent on the medium type and growth conditions (Frisvad et al. 2008; Andersen et al. 2001, 2003; Thrane 1993). Therefore, four types of media are routinely used in metabolic studies of fungi including the following: dichloran Rose Bengal yeast extract sucrose agar, malt extract agar, yeast extract sucrose (YES) agar, and potato dextrose agar (PBA) (Andersen et al. 2001, 2003; Thrane 1993). In this study, a medium based on potato-dextrose was used.
Biodiversity in fungal primary endo-metabolites showed that the three pathogenic strains, A. pal, F. oxy and G. can, are characterized by some particular variations in their fungal primary endo-metabolites despite the same growth conditions and general similarities in metabolism. The main differences are assigned in Fig. 5, where on a simple map of the biochemical pathways, increased concentrations of various metabolites can be seen for A. pal (orange boxes), F. oxy (yellow boxes) and G. can (red boxes).
Biodiversity of the metabolic pathways
In comparison to F. oxy and G. can, the pathogenic strains of A. pal are characterized by some elevated metabolite levels included in galactose metabolism, namely glucitol (d-sorbitol) and d-glucose. Sorbitol is a sugar alcohol that arises from sucrose or glucose and fructose but rarely from glucose or fructose alone (Baek et al. 2010). The formation of sorbitol is often related to protecting cells against osmotic stress (Yoo and Lee 1993; Shen et al. 1999). Thus, sorbitol acts as high-osmotic pressure metabolite as well as protecting against protein denaturation. However, it is well-known that sorbitol is a component of potato-dextrose medium. Therefore, this finding might also be associated with the better uptake of sorbitol by A. pal cells from the growth medium than with its production in biochemical pathways.
The second group of metabolic pathways where strains of A. pal revealed higher concentrations of some metabolites is glycerophospholipid metabolism. Choline, sn-glycero-3-phosphocholine, glycerol and malonate are involved in the lipid biosynthesis necessary to form cellular membranes. Glycerol can be utilized for the backbone of different lipids as well as playing a crucial role in osmoregulation and maintaining a proper anabolic reduction charge (Shen et al. 1999; Vries et al. 2003; Clark et al. 2003). Sn-glycero-3-phosphocholine and choline as a source of methyl groups can take part in the elongation of glycerol- and glycerophospholipid chains. Additionally, betaine is a precursor for choline synthesis.
Malonate is a simple three-carbon dicarboxylic acid well-known to be a competitive inhibitor of succinate dehydrogenase (Kim 2002). The role of malonate is unclear, but its main objectives are related to nitrogen metabolism (Kim 2002). In plants, malonate has been suggested as a defensive metabolite when under stress.
Therefore, A. pal malonate might play a different protective role, and in addition, malonate might be included in the fatty acid biosynthetic pathways (components of cell wall and/or membrane).
Analysis of the F. oxy primary endo-metabolites resulted in the observation that the main differences are associated with a check-point of the respiration process. In oxidative respiration, pyruvate is integrated via acetyl-CoA into the citric acid cycle. When the oxygen level becomes insufficient, pyruvate is metabolized to ethanol via acetate. Thus, the elevated levels of ethanol and acetate in the cells of F. oxy suggest that this fungus can switch its metabolism to direct anaerobic respiration. However, the level of succinate (main intermediate of oxidative respiration) was also elevated. Therefore, it could be assumed that F. oxysporum metabolizes pyruvate both via the citric acid cycle and ethanol production.
In F. oxy higher concentrations of aspartate, leucine, isoleucine and isobutyrate, the primary metabolites for protein metabolism, anabolism (biosynthesis) and catabolism (degradation), could be noted. Additionally, phenylalanine is increased in F. oxy and can be synthesized by the interconversion of d-xylose (up-regulated in F. oxy) via the pentose phosphate pathway. Propylene glycol is known as a competitive inhibitor of glycerol transport (Castro and Loureiro-Dias 1991). Thus, it seems that F. oxy accumulated their metabolites more from direct protein synthesis than lipids and lipid components. However, the regulation of cell wall growth might be controlled by an elevated concentration of guanosine.
Among all of the discussed pathogenic filamentous fungi, G. can were characterized by highly increased levels of metabolites involved in oxidative respiration. Higher concentrations of metabolites of the citric acid cycle (citrate and fumarate) were noted. The excess of citrate can be utilized in the synthesis of glutamate and lysine (both in higher concentrations in the case of G. can) (Kanehisa et al. 2017). Glutamate can then be metabolized towards glutamine (up-regulated in G. can), which might be used to produce purine, aminosugars and proline (up-regulated in G. can). Glutamate can also be utilized in the urea cycle that plays a pivotal role in the N-metabolism of fungi, which was observed as the level of creatine was up-regulated (Kanehisa et al. 2017).
Generally, fungi are capable of producing different secondary metabolites such as antibiotics, volatile compounds and others, which is related to the activation of different metabolic pathways involving the biosynthetic precursors of these reactions (Zhai et al. 2017; Alberti et al. 2017). In G. can, the levels of histidine and xanthine, the main precursor metabolites in the biosynthesis of secondary metabolites, were elevated. Thus, it seems that G. can switch their metabolism towards the production secondary metabolites more so than A. pal and F. oxy.
In G. can, the levels of two α-keto acids, 3-methyl-2-oxovalerate and 2-osoisocaproate, were elevated. These metabolites, unique for fungal metabolism, are derived from amino acid degradation (Kanehisa et al. 2017). However, detailed analysis and their role in comparing the tested species is still unclear.
Filamentous fungal virulence
The comparative primary endo-metabolome analysis of the three common filamentous fungi A. pal, F. oxy and G. can showed the main differences, but on this basis, it could also indicate the most dangerous fungal pathogens. These results suggest that A. pal is the most dangerous of the three, which is in agreement with previous reports. What makes A. pal a successful pathogen? In its metabolism can be observed the targeting metabolism on the protection of cell wall. Most antifungal agents act against the integrity of the cell wall. A. pal, by the synthesis of various components of lipids and the cell wall, might be able to resist these compounds. Additionally, the level of mannitol in A. pal is interesting. The concentration of mannitol is similar to that in F. oxy, and the HCA plot showed closer to grouping these two fungi. However, mannitol plays a very important role in the fungi as its presence allow cells to increase resistance and their virulence factors (Krahulec et al. 2011; Calmes et al. 2013; Ruijter et al. 2003).
Conclusion
In this study the 1H NMR-based metabolomics approach was applied for the analysis of the biodiversity of filamentous fungal pathogens. According to our preliminary results, each of the tested strain (A. pallidofulvus, F. oxysporum and G. candidum) cultured in the same growth conditions revealed a specific metabolite profile. Moreover, we demonstrated that targeted metabolomics analysis could be utilized after careful optimization by other omics as well as biochemical assays in the future as a supporting taxonomical tool for currently methods.
References
Alberti F, Foster GD, Bailey AM (2017) Natural products from filamentous fungi and production by heterologous expression. Appl Microbiol Biotechnol 101(2):493–500
Andersen B, Krøger E, Roberts RG (2001) Chemical and morphological segregation of Alternaria alternata, A. gaisen and A. longipes. Mycol Res 105:291–299
Andersen B, Nielsen KF, Thrane U, Szaro T, Taylor JW, Jarvis BB (2003) Molecular and phenotypic descriptions of Stachybotrys chlorohalonata sp. nov. and two new chemotypes of Stachybotrys chartarum found in water-damaged buildings. Mycologia 95:1227–1238
Andersen B, Dongo A, Pryor BM (2008) Secondary metabolite profiling of Alternaria dauci, A. porri, A. solani and A. tomatophila. Mycol Res 112:241–250
Baek JG, Shim S-M, Kwon DJ, Choi H-K, Lee CH, Kim YS (2010) Metabolite profiling of cheonggukjang, a fermented soybean paste, inoculated with various Bacillus strains during fermentation. Biosci Biotechnol Biochem 74(9):1860–1868
Bartnicki-Garcia S (1970) Cell wall composition and other biochemical markers in fungal phylogeny. In: Harbone JB (ed) Phytochemical phylogeny. Academic press, Ltd., London, pp 81–103
Bartnicki-Garcia S (1987) The cell wall in fungal evolution. In: Rayner ADM, Brasier CM, Moore D (eds) Evolutionary biology of the fungi. Cambridge University Press, New York, pp 389–403
Blackwell M, Hibbett DS, Taylor JW, Spatafora JW (2006) Research coordination networks: a phylogeny for kingdom fungi (Deep Hypha). Mycologia 98:829–837. doi:10.3852/mycologia.98.6.829
Bruns TD, Vilgalys R, Barns SM, Gonzalez D, Hibbett DS, Lane DJ, Simon L, Stickel S, Szaro TM, Weisburg WG, Sogin ML (1992) Evolutionary relationships within the fungi: analyses of small subunit ribosomal DNA sequences. Appl Environ Microbiol 61:681–689
Calmes B, Guillemette T, Teyssier L, Siegler B, Pigné S, Landreau A, Iacomi B, Lemoine R, Richomme P, Simoneau P (2013) Role of mannitol metabolism in the pathogenicity of the necrotrophic fungus Alternaria brassicicola. Front. Plant Sci. Front Plant Sci 4:131. doi:10.3389/fpls.2013.00131
Castro IM, Loureiro-Dias MC (1991) Glycerol utilization in Fusarium oxysporum var. lini: regulation of transport and metabolism. J Gen Microbiol 137(7):1497–1502
Cevallos-Cevallosa JM, Reyes-De-Corcueraa JI, Etxeberriaa E, Danyluka MD, Rodrickb GE (2009) Metabolomic analysis in food science: a review. Trends Food Sci Technol 20(11):557–566
Clark AJ, Blissett KJ, Oliver RP (2003) Investigating the role of polyols in Cladosporium fulvum during growth under hyperosmotic stress and in planta. Planta 216:614–619
David M, Geoffrey DR, Anthony PJ (2011) 21st century guidebook to fungi © Trinci. Cambridge University Press, New York
De Vries RP, Flitter SJ, van de Vondervoort PJI, Chaveroche MK, Fontaine T, Fillinger S, Ruijter GJG, d’Enfert C, Visser J (2003) Glycerol dehydrogenase, encoded by gldB is essential for osmotolerance in Aspergillus nidulans. Mol Microbiol 49:131–141
Deja S, Wieczorek PP, Halama M, Jasicka-Misiak I, Kafarski P, Poliwoda A, Młynarz P (2014) Do differences in chemical composition of stem and cap of Amanita muscaria fruiting bodies correlate withtopsoil type? PLoS ONE 9(12):e104084. doi:10.1371/journal.pone.0104084
Dieterle F, Ross A, Schlotterbeck G, Senn H, 2006. Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Anal Chem 78:4281–4290
Frisvad JC (1992) Chemometrics and chemotaxonomy: a comparison of multivariate statistical methods for the evaluation of binary fungal secondary metabolite data. Chemom Intell Lab Syst 14:253–269
Frisvad JC, Andersen B, Thrane U, Andersen B, Dongo A, Pryor BM (2008) The use of secondary metabolite profiling in chemotaxonomy of filamentous fungi. Mycol Res 112:231–240. doi:10.1016/j.mycres.2007.08.018
Fuller MS (1976) Mitosis in fungi. Int Rev Cytol 45:113–153
Golenberg EM, Giannasi DE, Clegg MT, Smiley CJ, Durbin M, Henderson D, Zurawski G (1990) Chloroplast DNA sequence from a miocene Magnolia species. Nature 344:656–658
Guarro J, Gené J, Stchigel AM (1999) Developments in fungal taxonomy. Clin Microbiol Rev 12(3):454–500
Haase G, Sonntag L, van de Peer Y, Uijthof JMJ, Podbielski A, Melzer-Krick B (1995) Phylogenetic analysis of ten black yeast species using nuclear small subunit rRNA gene sequences. Antonie Leeuwenhoek Int J Genet 68:19–33
Hausner G, Reid J, Klassen GR (1992) Do galeate-ascospore members of the Cephaloascaceae, Endomycetaceae and Ophiostomataceae share a common phylogeny? Mycologia 84:870–881
Hawksworth DL, Kirk PM, Sutton BC, Pegler DN, 1995. Ainsworth and Bisby’s dictionary of the fungi, 8th edn. International Mycological Institute, Egham
Heath IB (1980) Variant mitoses in lower eukaryotes: indicators of the evolution of mitosis? Int Rev Cytol 64:1–80
Heath IB (1986) Nuclear division: a marker for protist phylogeny. Prog Protistol 1:115–162
Hendriks L, Goris A, van de Peer Y, Neefs JM, Vancanneyt M, Kersters K, Berny JF, Hennebert GL, De Wachter R (1992) Phylogenetic relationships among ascomycetes and ascomycete-like yeasts as deduced from small ribosomal subunit RNA sequences. Syst Appl Microbiol 15:98–104
Hibbett DS, Bindera M, Bischoff JF, Blackwell M, Cannon PF, Eriksson OE, Huhndorf S, James T, Kirk PM, Lücking R et al (2007) A higher-level phylogenetic classification of the Fungi. Mycol Res 111:509–547. doi:10.1016/j.mycres.2007.03.004
Ivanisˇevic´ J, Thomas OP, Lejeusne C, Chevaldonne´ P, Pe´rez T, 2011. Metabolic fingerprinting as an indicator of biodiversity: towards understanding inter-specific relationships among Homoscleromorpha sponges. Metabolomics 7:289–304. doi:10.1007/s11306-010-0239-2.
Jennessen J, Nielsen KF, Houbraken J, Lyhne EK, Schnurer J, Frisvad JC, Samson RA (2005) Secondary metabolite and mycotoxin production by the Rhizopus microsporus group. J Agric Food Chem 53:1833–1840
Junka AF, Deja S, Smutnicka D, Szymczyk P, Ziółkowski G, Bartoszewicz M, Młynarz P (2013) Differences in metabolic profiles of planktonic and biofilm cells in Staphylococcus aureus—1H nuclear magnetic resonance search for candidate biomarkers. Acta Biochim Pol 60:701–706
Kadlec Z, Šimek P, Heydová A, Jegorov’ A, Maťha V, Landa Z, Eyal J (1994) Chemotaxonomic discrimination among the fungal genera Tolypocladium, beauveria and Paecilomyces. Biochem Syst Ecol 22:803–806
Kanehisa FM, Tanabe M, Sato Y, Morishima K (2017) KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 45:D353–D361
Kim YS (2002) Malonate metabolism: biochemistry, molecular biology, physiology, and industrial application. J Biochem Mol Biol 35:443–451
Kirk PM, Cannon PF, David JC, Stalpers JA (2001) Dictionary of the fungi, 9th edn. CAB International, Wallingford. ISBN 085199377X
Kirk PM, Cannon, PF, Minter DW, Stalpers JA (2008) Dictionary of the fungi, 10th edn. CAB International, Wallingford. ISBN 9780851998268
Krahulec S, Armao GC, Klimacek M, Nidetzky B (2011) Enzymes of mannitol metabolism in the human pathogenic fungus Aspergillus fumigatus—kinetic properties of mannitol-1-phosphate 5-dehydrogenase and mannitol 2-dehydrogenase, and their physiological implications. FEBS J 278:1264–1276
Larsen TO, Smedsgaard J, Nielsen KF, Hansen ME, Frisvad JC (2005) Phenotypic taxonomy and metabolite profiling in microbial drug discovery. Nat Prod Rep 22:672–695
Le´John HB (1974) Biochemical parameters of fungal phylogenetics. Evolutionary biology of the fungi. Cambridge University Press, New York. pp 79–115
Mas S, Villas-Boas SG, Hansen ME, Akesson M, Nielsen J (2007) A comparison of direct infusion MS and GC-MS for metabolic footprinting of yeast mutants. Biotechnol Bioeng 96:1014–1022
Ruijter GJ, Bax M, Patel H, Flitter SJ, Vondervoort van de PJ, de Vries RP, vanKuyk PA, Visser J (2003) Mannitol is required for stress tolerance in Aspergillus niger conidiospores. Eukaryot Cell 2:690–698
Savorani F, Tomasi G, Engelsen SB (2010) icoshift: A versatile tool for the rapid alignment of 1D NMR spectra. J Magn Reson 202:190–202, doi:10.1016/j.jmr.2009.11.012
Semmar N, Jay M, Nouira S (2007) A new approach to graphical and numerical analysis of links between plant chemotaxonomy and secondary metabolism from HPLC data smoothed by a simplex mixture design. Chemoecology 17:139–156
Shen B, Hohmann S, Jensen RG, Bohnert H (1999) Roles of sugar alcohols in osmotic stress adaptation. Replacement of glycerol by mannitol and sorbitol in yeast. Plant Physiol 121(1):45–52
Smedsgaard J, Nielsen J (2004) Metabolite profiling of fungi and yeast: from phenotype to metabolome by MS and informatics. J Exp Bot 56:273–286
Smedsgaard J, Nielsen J (2005) Metabolite profiling of fungi and yeast: from phenotype to metabolome by MS and informatics. J Exp Bot 56(410):273–286
Smedsgaard J, Hansen ME, Frisvad JC (2004) Classification of terverticillate Penicillia by electrospray mass spectrometric profiling. Stud Mycol 49:243–251
Spatafora JW, Blackwell M (1993) Molecular systematics of unitu- nicate perithecial ascomycetes: the Clavicipitales-Hypocreales connection. Mycologia 85:912–922
Swann EC, Taylor JW (1995) Toward a phylogenetic systematics of the Basidiomycota: integrating yeasts and filamentous Basidiomycetes using 18 rRNA gene sequences. Stud Mycol 38:147–161
Taylor FJR (1978) Problems in the development of an explicit hypothet- ical phylogeny of the lower eukaryotes. Biosystems 10:67–89
Thrane U (1993) Fusarium species and their specific profiles of secondary metabolites. In: Chelkowski J (ed) Fusarium: mycotoxins, taxonomy and pathogenicity. Elsevier, Amsterdam, pp. 199–225
Tomasi G, van den Berg F, Andersson C (2004) Correlation optimized warping and dynamic time warping as preprocessing methods for chromatographic data. J Chemometr 18:231–241. doi:10.1002/cem.859
Vogel HJ (1964) Distribution of lysine pathways among fungi: evolutionary implications. Am Nat 98:435–446
Yoo B, Lee CM (1993) Thermoprotective effect of sorbitol on proteins during dehydration. J Agric Food Chem 41:190–192
Zhai X, Jia M, Chen L, Zheng CJ, Rahman K, Han T, Qin LP (2017) The regulatory mechanism of fungal elicitor-induced secondary metabolite biosynthesis in medical plants. Crit Rev Microbiol 43(2):238–261
Zheng X, Xie G, Zhao A, Zhao L, Yao C, Chiu NHL, Zhou Z, Bao Y, Jia W, Nicholson JK, Jia W (2011) The footprints of gut microbial mammalian co-metabolism. J Proteome Res 10:5512–5522. doi:10.1021/pr2007945.
Zieliński L, Deja S, Jasicka-Misiak I, Kafarski P (2014) Chemometrics as a tool of origin determination of Polish monofloral and multifloral honeys. J Agric Food Chem 62(13):2973–2981. doi:10.1021/jf4056715
Acknowledgements
This project was supported by Wroclaw University of Technology S40531/Z0303 and WCB KNOW 2014-2018.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ząbek, A., Klimek-Ochab, M., Jawień, E. et al. Biodiversity in targeted metabolomics analysis of filamentous fungal pathogens by 1H NMR-based studies. World J Microbiol Biotechnol 33, 132 (2017). https://doi.org/10.1007/s11274-017-2285-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-017-2285-7