
Abstract
Toll-like receptors (TLRs) represent an important part of the innate immune system. While human and murine TLRs have been intensively studied, little is known about TLRs in non-model species. The order Perissodactyla comprises a variety of free-living and domesticated species exposed to different pathogens in different habitats and is therefore suitable for analyzing the diversity and evolution of immunity-related genes. We analyzed TLR genes in the order Perissodactyla with a focus on the family Equidae. Twelve TLRs were identified by bioinformatic analyses of online genomic resources; their sequences were confirmed in equids by genomic DNA re-sequencing of a panel of nine species. The expression of TLR11 and TLR12 was confirmed in the domestic horse by cDNA sequencing. Phylogenetic reconstruction of the TLR gene family in Perissodactyla identified six sub-families. TLR4 clustered together with TLR5; the TLR1-6-10 subfamily showed a high degree of sequence identity. The average estimated evolutionary divergence of all twelve TLRs studied was 0.3% among the Equidae; the most divergent CDS were those of Equus caballus and Equus hemionus kulan (1.34%) in the TLR3, and Equus africanus somaliensis and Equus quagga antiquorum (2.1%) in the TLR1 protein. In each TLR gene, there were haplotypes shared between equid species, most extensively in TLR3 and TLR9 CDS, and TLR6 amino acid sequence. All twelve TLR genes were under strong negative overall selection. Signatures of diversifying selection in specific codon sites were detected in all TLRs except TLR8. Differences in the selection patterns between virus-sensing and non-viral TLRs were observed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The emergence and evolution of infectious diseases result from a permanent confrontation between pathogens and hosts. Each of them use different strategies to survive. Pathogens often rely on their short generation interval and can rapidly change their surface antigens or target receptors. In higher organisms, the immune system has evolved over millions of years to cope with these changes. While the adaptive immune system responds to variable epitopes and compensates for pathogen variability via MHC class II, T- and B-cell receptors and antigen-specific antibodies, the innate, non-adaptive part of the immune system relies on evolutionarily older strategies. One of them is the recognition of conserved molecular patterns by various receptors (pattern recognition receptors, PRRs).
Toll-like receptors (TLRs) are representative examples of PRRs. They recognize patterns that are either associated with pathogens themselves (pathogen-associated molecular patterns, PAMPs) or released by damaged or dying cells (damage-associated molecular patterns, DAMPs). A typical TLR molecule contains three domains: an N-terminal pattern-recognizing outer part with multiple leucine-rich repeat domains (LRRs), a transmembrane domain, and an intracytoplasmic part with the toll/interleukin-1 receptor (TIR) domain responsible for intracellular signaling (reviewed e.g. in Behzadi et al. 2021). When activated, TLR signaling pathways elicit the production of type I interferons and inflammatory cytokines (Kawai and Akira 2011). Besides their role in innate immunity, TLRs can also modulate adaptive immune responses (Kumar 2022). Innate immune cells such as dendritic cells, macrophages and/or NK cells as well as epithelial and endothelial cells, but also T and B cells, express various TLRs. Mammalian TLRs 1, 2, 4, 5 and 6, which detect microbial cell components, are localized on the outer plasma membrane, while viral nucleic acid-sensing TLRs 3, 7, 8 and 9, as well as TLRs 10, 11, 12 and 13, are found in endosomes (as reviewed in Vijay 2018; Fitzgerald and Kagan 2020).
The Toll gene was first discovered in Drosophila melanogaster (Anderson et al. 1985), but evolutionary TLR prototypes have been identified in organisms pre-dating bilaterians, such as Cnidaria (reviewed in Brennan and Gilmore 2018). To date, 28 different TLRs have been identified in vertebrates, including 13 in mammals (Behzadi et al. 2021). Purifying selection appears to dominate the evolution of all vertebrate TLRs, but patterns of diversifying selection can be detected in specific codons concentrated in the ligand-binding domains (Liu et al. 2020). While negative selection preserves the essential functions of TLRs, diversifying selection helps TLRs cope with changes in the pathogen pressure. The spectrum of TLRs present in each species and the sites under selection thus reflect the history of species-specific host-pathogen interactions.
The functional importance of TLR gene polymorphisms, especially of single nucleotide polymorphisms (SNPs), is reflected in their associations with various types of diseases observed in multiple mammalian species. In humans, immunity-related gene polymorphisms are associated with increased susceptibility or resistance to various infectious agents, such as Mycobacterium spp., Plasmodium spp., and herpes viruses, as well as with an increased risk of cancer and autoimmune diseases such as Crohn’s disease and asthma (Mukherjee et al. 2019). In a meta-analysis, the TLR4 896 A/G polymorphism was associated with a higher risk of viral infections (Silva et al. 2022). In domestic animals, polymorphisms in various TLR genes have been associated with mastitis and other economically important traits in cattle (reviewed in Novák 2014). In horses, an association between a SNP in TLR4 and West Nile virus infection has been reported (Stejskalova et al. 2019).
Compared to humans and model mammalian species, little is known about TLRs in non-model animals, such as domestic horses and the entire family Equidae. Despite their theoretical and practical importance, only fragmentary knowledge of their TLR genes consisting mostly of annotations in the current reference genome assemblies is available. As for their expression, TLR1-10 mRNA has been identified in domestic horses (Uddin et al. 2016); TLR 2 and TLR5 expression was reported for the Damara zebra (Dugovich et al. 2019). Only sporadic reports focusing on the characterization of TLR9 have been published (Manuja et al. 2019; Smith et al. 2020). Although the Equidae family consists of a single genus, Equus (Price and Bininda-Emonds 2009), it includes a variety of free-living and domesticated species exposed to different pathogens in different habitats and is therefore suitable for analyzing the diversity and evolution of immunity-related genes (Janova et al. 2009).
The general aim of the study was to provide comprehensive factual information on the set of TLR genes in the entire family Equidae in the context of the entire order Perissodactyla. The specific objectives of this study were (i) to perform a comparative analysis of available genomic resources in terms of the presence, functionality, copy number, localization and genomic organization of TLR loci in all equid species (ii) to determine TLR coding sequences on a panel of equid species for all TLR loci identified, and (iii) to carry out evolutionary (phylogenetic) and selection analyses of individual TLR genes in the family Equidae.
Materials and methods
Study design
The aim of this study was to perform comparative analysis of TLR loci in available genomic resources of equid species in the context of the order Perissodactyla, to determine TLR coding sequences on an experimental set of equid species for all TLR loci identified, and to carry out evolutionary (phylogenetic) and selection analyses of individual TLR genes in the family Equidae and in the entire order Perissodactyla.
Blood sample collection
Blood samples were collected from animals living in the ZOO Dvůr Králové and Prague ZOO, Czech Republic. Two (three where available) individuals representing the following nine species including four subspecies of the family Equidae were selected: Grevy’s zebra (Equus grevyi, EqGr); Mountain zebra (Equus hartmannae, EqHa); Plain zebras (Equus quagga antiquorum - EqQuAn, Equus quagga boehmi - EqQuBoe, Equus quagga chapmani - EqQuCh, Equus quagga borensis - EqQuBor); African wild ass (Equus africanus somaliensis, EqAfSo); donkey (Equus asinus, EqAs); Asian ass (Equus kiang - EqKi and Equus hemionus kulan - EqHeKu); domestic horse (Equus caballus, EqCa) and Przewalski’s horse (Equus przewalskii, EqPr). Since the taxonomic classification of zebras and donkeys is rather inconsistent, we followed the classification by ITIS (Integrated Taxonomic Information System; https://doi.org/10.5066/F7KH0KBK). Blood samples were stored at -20 °C until DNA extraction. All extracted DNAs were then used for sequencing of all TLR genes.
Genomic resources
Four currently available equid reference genome assemblies and two non-reference assemblies, together with tapirs (Tapirus indicus, TaIn; Tapirus terrestris, TaTe), rhinoceroses (Ceratotherium simum simum, CeSi; Diceros bicornis, DiBi; Rhinoceros unicornis, RhUn; Dicerorhinus sumatrensis, DiSu), bovine (Bos taurus, BoTa), mouse (Mus musculus, MuMu) and human (Homo sapiens sapiens, HoSa) assemblies were searched for TLR 1–13 (Table 1). In non-reference assemblies with no gene annotations available, the BLAST algorithm was used with Equus caballus TLR sequences as queries. Genomic sequences identified as TLR genes were then aligned for each individual gene. When multiple splice variants were present, the variant with a validated status was selected. In case all variants were only predicted, the one with coding sequence (CDS) length matching the length of the CDS of other species was chosen. Moreover, the NCBI nucleotide database was searched for equid TLR sequences using direct queries, BLASTn and tBLASTn algorithms. On the protein level, the UniProt database was searched. Data on domain structure and their localization within the gene and protein sequences were obtained from the NCBI GenBank and the UniProt database.
DNA extraction
Two hundred microliters of whole blood were used for DNA extraction according to the manufacturer´s instructions (NucleoSpin Blood kit, Macherey-Nagel, Düren, Germany).
Primer design and PCR
The genomic sequences of all equid TLR genes retrieved from the genomic resources were aligned in BioEdit (v7.2.5). Based on the Equus caballus EquCab3.0 sequence as a reference sequence, intron/exon boundaries and coding sequences (CDS) were determined for each TLR gene alignment. Conserved intronic regions surrounding the exon CDS were then identified and primer pairs for their amplification were designed by Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast). PCR reactions were performed using EliZyme HS Robust MIX Red (Elisabeth Pharmacon, Brno, Czech Republic), 2x PCR BIO Ultra Mix (PCR BioSystems, London, United Kingdom) and Expand LongRange, dNTPack polymerase (Roche, Mannheim, Germany) according to the manufacturer’s instructions. The PCR reaction volume was 12.5 µl. Primer sequences and PCR protocols are given in Online Resource 1.
TLR gene re-sequencing
A minority of PCR amplicons for TLR4 were originally sequenced by Sanger sequencing (Macrogen, Seoul, South Corea); all other sequences were then obtained by next-generation sequencing of the same sample set. The Roche GS Junior 454 platform (Roche, Mannheim, Germany) as described in Bayerova et al. (2016) was used originally. However, the vast majority of the next-generation sequencing (NGS) was performed on the MiSeqTM System (Illumina, San Diego, California, USA). Sequencing libraries were prepared using the Nextera XT DNA Library Preparation Kit (Illumina, San Diego, California, USA). Amplicons from the same individual were tagged with the same index if there was no significant sequence similarity; otherwise, amplicons were indexed separately. Raw reads were checked in FastQC (v0.11.9) for quality and processed using Trimmomatic (v0.39). Reads were mapped to reference sequences by the BWA-MEM (v0.7.15) software. Alignments were checked using SAMtools (v1.4.1), GATK (v3.5) and Picard (v2.20.4). No more than 5% soft-clipping and 10% mismatches was allowed, and and a minimum read length of 70 nucleotides was required, for the final alignments using NGSUtils (v0.5.9) and BBMap (v38.58). NGS alignments were inspected in IGV software (v2.3.94) and further edited in BioEdit (v7.2.5). Consensus sequences were generated for each animal and each TLR gene. When potential heterozygous positions were identified, variable positions were verified by the SAMtools or GATK software; visual inspection and confirmation of the variable site was then done in the IGV browser. The confirmed heterozygous positions were replaced with IUPAC ambiguity code letters.
cDNA sequencing
The expression of TLR11 and TLR12 in equine leukocytes was assessed by Sanger sequencing (Macrogen, Seoul, South Corea) and next-generation sequencing of equine cDNA amplified with primers designed based on the reference genome sequence and using Qiagen HotStarTaq Mix polymerase (Qiagen, Venlo, Netherlands) (see Online Resource 1 for primer sequence and reaction set up). Equine cDNA was prepared as described by Futas and Horin (2013).
Bioinformatic sequence analyses
Alignments of genomic NGS-generated sequences were made for each TLR gene using BioEdit’s ClustalW multiple alignment algorithm and then trimmed according to the reference sequence to generate CDS alignments. Haplotypes were inferred using the PHASE algorithm in DnaSP (v6.12.03) and assigned to samples. Final alignments were then merged with previously retrieved alignments of GenBank sequences.
Amino acid sequences were inferred by translation of CDS following the standard genetic code. Variable nucleotide and amino acid sites were identified in MEGA (v11.0.13). The impact of amino acid changes on protein function and structure was evaluated at the Sorting Intolerant From Tolerant (SIFT, https://sift.bii.a-star.edu.sg/index.html) website.
Estimates of the evolutionary divergence between sequences were calculated in MEGA using a maximum composite likelihood model for nucleotide sequences and a Poisson correction model for protein sequences. For this purpose, tapirs, rhinoceroses, bovine, human and/or mouse sequences were included in the alignments (see Online Resource 2 for sequence IDs and Online Resource 3 for aligned sequences).
Phylogenetic analysis
Combined alignments of both NGS-generated and GenBank retrieved sequences were evaluated for each TLR gene. The evolutionary history was reconstructed using maximum likelihood (ML) and the lowest BIC score nucleotide substitution model. The topology with the highest log-likelihood was always selected. Bovine and/or mouse sequences were used as outgroups to root the trees. Neighbor-joining trees (NJ) (uncorrected p-values, bootstrap consensus of 1000 replicates) were built as well. All calculations were performed in MEGA (v11.0.13).
Selection analyses
All analyses were performed first for all perissodactyls and then for the Equidae separately. Merged alignments of both NGS-generated and GenBank retrieved sequences were evaluated. The codon-based Z-test of selection in MEGA software was used for the analysis averaging over all sequence pairs. First, the probability of rejecting the null hypothesis of strict neutrality (non-synonymous mutations frequency equals synonymous; dN = dS) was tested, followed by the probability of rejecting the null hypothesis in favor of one of the alternative hypotheses (dN > dS for diversifying selection, dN < dS for purifying selection). The variance of the difference was calculated using the bootstrap method (1000 replicates). Analyses were performed using the Kumar method. Pervasive site-specific selection was evaluated by three methods (FEL, FUBAR, SLAC; performed by Datamonkey.org); selected amino acid sites (SAAS) where p was ≤ 0.05 or at p ≤ 0.1 but confirmed by two methods were considered to be under selection. Episodic site-specific diversifying selection was tested by MEME (Datamonkey.org); here, p values ≤ 0.05 were considered significant. Fisher’s exact probability test (two-tailed) was used to compare rates of SAAS in different groups of TLRs.
Results
In silico analysis of TLR genes in equids
Twelve functional TLR genes (TLR1-12) were identified in all the equid genomes analyzed. All coding sequences retrieved in silico are provided in Online Resource 3. The GenBank search did not yield any additional results for TLR sequences of equid origin beyond those derived by prediction from the assemblies. The major features of the genomic organization of TLRs in equids, including chromosomal locations, gene and protein length, and exon counts, are summarized in Table 2. Multiple splice variants were predicted for some of the TLR genes analyzed, with a maximum of 10 variants for TLR8 in Equus caballus and Equus asinus.
Although in general the TLR sequences were conserved across the species analyzed, some exceptions were observed. The computer-predicted, but not validated, CDS of TLR2 in Equus przewalskii and TLR9 in Equus quagga differed greatly in length from the rest of equids. Therefore, they were excluded from further bioinformatic analyses.
Twelve functional TLR genes (1–12) were also identified in available genomes of other odd-toed ungulates, namely tapirs and rhinoceroses. The predicted CDS of TLR2 based on BLAST hits in Tapirus terrestris as well as TLR2, 6 and 11 in Rhinoceros unicornis were excluded from further analyses due to incomplete sequences and/or to frameshifts with multiple stop codons.
All equid TLR genes and predicted proteins examined shared common features with their mammalian orthologues. The overview of TLR1-12 protein structure, i.e. the position of the signal peptide, of the LRR domain region and the LRR-C-terminal domain, and of the transmembrane region and the TIR domain, is shown in Fig. 1. Details including the exact locations of the respective domains are provided in Supplementary Table 1 in Online Resource 4.
In terms of TLR gene expression, mRNAs and derived CDS have so far been mostly predicted only in silico, and thus have a provisional status. A validated status has been assigned to the TLR1, 4, 8, 9 reference sequences of the domestic horse. According to the UniProt database, there is evidence of gene transcription for horse TLRs 2–4 and 7–10, and evidence for translation for TLR9 in domestic horses and donkeys. However, the expression of equine TLR1-10 has been confirmed as well (Astakhova et al. 2009; Uddin et al. 2016; Tarlinton et al. 2016). Dugovich et al. (2019), and Smith et al. (2020) confirmed the expression of TLR2, 5 and 9 in Equus quagga spp.
Re-sequencing of equid TLR genes
Coding sequences of all 12 TLR genes were obtained for all 12 species and subspecies included in the experimental panel. All sequences were submitted to GenBank; accession numbers are provided in Online Resource 5, while complete alignments are provided in Online Resource 3. On average, 1.69% of all CDS nucleotide positions and 2.27% of amino acid positions were variable between the species of the family. The least variable was the TLR8 sequence (0.96/0.29%); the highest variability was observed in TLR12 (2.46/3.85%). The numbers of unique haplotypes ranged from 18 in TLR7 and TLR8 to 32 in TLR12. See Table 3 for detailed numbers.
In silico inferred amino acid TLR sequences
Altogether, 208 variable amino acid sites were identified in TLR1-12 in the Equidae. Online Resource 4, Suppplementary Table 2, provides the complete list of variable sites detected and their localization within the respective protein; a brief summary is presented in Fig. 1. Eighteen variable amino acid sites were identified by SIFT as potentially affecting protein function (SIFT scores for all sites are provided in Online Resource 4). Six of them were identified in TLR10, four of them in the TIR domain. A rather high proportion of variable AA sites located in the TIR domain was observed in TLR1, 10 and 12.

A graphic overview of known and/or predicted domain organization of TLR1-12 in Equus caballus. Some domain positions were inferred from human and murine data (see Online Resource 4 for details). The LRR region, shown as a single block for simplicity, represents a region where numerous LRR domains (typically 10–30 in number) occur at different spacing. The number inside each domain indicates the sum of variable amino acid (AA) sites identified in translated alignments of NGS and GenBank retrieved sequences in the family Equidae. AA changes which may affect the protein function (according to SIFT) are in red. *This variation was only present in GenBank data; LRRCtd – Leucine-rich repeat C-terminal domain
cDNA sequencing
Sanger and NGS sequencing showed that equine TLR11 and TLR12 are transcribed genes and confirmed their expression in equine white blood cells. The sequences retrieved (provided in the Online Resource 3) matched the predicted coding sequences for these two genes. Both sequences were submitted to GenBank under accession numbers OQ971889 (TLR11) and OQ971888 (TLR12).
Interspecific comparisons and trans-species allele sharing
The average estimated CDS evolutionary divergence of all twelve TLRs studied was 0.3% among the Equidae, with the lowest value for TLR7 (0.13%), and the highest value for TLR3 (0.5%). The most divergent sequences were those of Equus caballus and Equus hemionus kulan (1.34%) in TLR3. On the other hand, in each TLR gene there were identical haplotypes shared between species (see Table 4; all shared haplotypes and species combinations can be found in Online Resource 6 and 7). While only one out of 26 haplotypes was shared in TLR2, five haplotype alleles were shared in TLR3 (out of 29) and in TLR9 (out of 24). At the amino acid level, the number of AA sequences shared by at least two species ranged from one (in TLR8) to five (in TLR3 and TLR6). The only AA sequence shared in TLR8 covered the widest range of species, as it was detected in all equids except Equus quagga and Equus hemionus kulan.
When comparing the nucleotide sequences of equids with other odd-toed ungulates, the average divergence between equids and tapirs was 6.69%, while it was 6.89% between equids and rhinoceroses. The least divergent were the sequences of TLR6, while the most different were TLR4 in tapirs (9.86%) and TLR12 in rhinoceroses (10.22%). No haplotype sharing was observed between equids and tapirs or rhinoceroses. The average divergence from bovine, human and murine sequences were 14.62%, 13.94% and 27.79%, respectively. In these species, the sequence of TLR8 was the most different from equids. Detailed interspecific comparison is provided in Online Resource 6.
At the protein level, the average estimated amino acid sequence divergence of all twelve TLRs was 0.44% among the Equidae; the divergence was lowest in TLR8 (0.08%) and highest in TLR1 (0.72%). The most different were Equus africanus somaliensis and Equus quagga antiquorum (2.1%) in TLR1. When comparing equids with other perissodactyls and bovine amino acid sequences, the most different were sequences of TLR4. Compared to humans, the most different was TLR8; compared to mice, the most different was TLR12. The estimated average divergence from equids to tapirs was 10.15%, while to rhinoceroses it was 10.53%. The average divergence from bovine, human and murine sequences were 19.69%, 19.95% and 32.6%, respectively. Divergence matrices for all species combinations on both the CDS and protein levels are provided in Online Resource 6 and 7, respectively.
Phylogenetic analyses
The inferred phylogenetic history represented by the constructed trees was generally consistent with the current taxonomy of the Perissodactyla order and the family Equidae, where three basic clades can be distinguished: zebras, asses and horses. However, occasional deviations from neutrality were observed in individual trees, although some lineages were only weakly supported. For the sake of better readability of the main text, all Neighbor-joining (NJ) and maximum-likelihood (ML) trees for TLR1-12 in the Perissodactyla are provided only as Supplementary Figs. 1–24 (Online Resource 8).
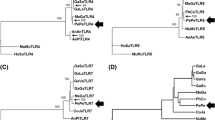
Figure 2 shows an unrooted ML tree with twelve TLR genes clustering into six families: the family of TLR1-6-10 with the single member family of TLR2 branching next to it; the two-member families of TLR4, 5 and TLR11,12; then TLR3 which clustered by itself but in proximity to the family comprising TLR 7, 8 and 9.

The phylogeny of TLR1-12 in the Perissodactyla inferred by the Maximum Likelihood method and the General Time Reversible model. The tree with the highest log likelihood (-63670.76) is shown. This analysis involved 177 nucleotide sequences, one representative sequence per species for each TLR gene. The percentage of trees (1000 bootstraps) in which the associated taxa clustered together is shown next to the branches. Eq – equids, Ta – tapirs, Rh – rhinoceroses
Selection analyses
The effects of selection acting on entire genes are summarized in Table 5. For the whole order Perissodactyla, all twelve TLR genes were under very strong negative selection. For the Equidae alone, six genes - TLR 3, 4, 7, 8, 9 and 11 - showed a deviation from neutral evolution at the p < 0.05 significance level. Strong negative selection was also confirmed in these six genes, most evidently in TLR9. Evidence of diversifying selection acting on whole genes was detected neither in perissodactyls nor in equids.
However, individual selected amino acid sites (SAAS) were observed both in the Perissodactyla and in the Equidae. The full list of SAAS, including p-values, along with a summary table (Supplementary Table 3), are provided in Online Resource 9. Purifying selection clearly predominated, as only 65 of the 615 SAAS detected were under diversifying selection in the Perissodactyla (0.61% of all sites analyzed), and 8 out of 103 in the Equidae (0.06% of all sites analyzed). An overview of the numbers of sites under episodic and pervasive site-specific positive selection identified in the Perissodactyla within each TLR is provided in Table 6. Figure 3 shows amino acid changes at all PSS, their distribution within the domains of respective proteins and SIFT prediction of the change impact. The majority of PSS (positively selected sites) were located in the LRRs and LRR-Ct domains (60% in the Perissodactyla, 50% in the Equidae), while 18.5% (16.7% in the Equidae) were located in the TIR domain. Interestingly, there were four PSS located in the TIR domain of TLR1 in the Perissodactyla, in contrast to just one in the LRR region.

Positively selected amino acid sites (PSS) in TLR1-12 in the Perissodactyla. Site positions correspond to the sequence of Equus caballus. AA changes which may affect protein function (according to SIFT) are in red
Comparisons of selected amino acid sites – interspecific differences
PSS identified in the Equidae and the Perissodactyla were compared to PSS previously described in vertebrates (Liu et al. 2020). Matches were found for the following PSS: TLR4-R342, A395; TLR5- V493; TLR9- V446, S699; TLR11- V287; TLR12- A78, A148, V675 (Equus caballus site positions). In comparison with the solely mammalian PSS identified by Areal et al. (2011), four shared PSS were found: TLR3-F712; TLR4-A395, L614; TLR10-V492.
Comparisons of selected amino acid sites – “Viral” vs. “Non-viral” TLRs
Comparisons between numbers of PSS and negatively selected sites (NSS) among non-viral (TLR1, 2, 4, 5, 6, 11, 12) and viral TLRs (TLR 3, 7, 8, 9) in the Perissodactyla showed more NSS and fewer PSS in the viral group than in the non-viral group (Fisher’s exact test, p = 2.5e-7). Separate comparisons of PSS and NSS rates within the LRR domains and the TIR domains also showed a significant difference between the viral and non-viral group (p = 0.00062 for LRR, p = 0.00061 for TIR domains). No significant difference between non-viral and viral TLR groups was found for the Equidae alone.
Discussion
TLR genes and their evolution have been studied in various vertebrate groups, including several mammalian families and model mammalian species, such as humans and mice (Liu et al. 2020). This study brings comprehensive information on the genomic organization and evolution of TLR genes in the family Equidae in the evolutionary context of the order Perissodactyla.
Both in silico analyses of WGS from GenBank and the resequencing of genomic DNA amplicons showed that equids as well as other perissodactyls (rhinoceroses and tapirs) possess 12 TLR genes. They are highly conservative in terms of their nucleotide sequences and the structure of their putative protein products. Based on the data obtained, it may be assumed that they play similar roles in immunity as in other mammalian species. In the mouse genome, TLRs 1–9 and TLRs 11–13 have been identified, while TLR10 is considered missing (Kawai and Akira 2011). In the human genome, ten TLR genes (1–10) and a TLR12 pseudogene have been annotated so far. In the current reference genome version of cattle (ARS-UCD1.2), only TLRs 2–10 have been annotated. However, the expression of the TLR1-6-10 family was confirmed and the genomic organization of the corresponding chromosome region was determined (Opsal et al. 2006). In agreement with this finding, the UniProt database contains data on bovine TLR1 identified at transcript level (ACH92575.1).
Unlike humans but similarly to mice, the genomes of all species analyzed here contained the TLR11 and TLR12 genes. Both of these genes can be found also in Rodentia, Lagomorpha, several Chiroptera species, and also in the Elephantidae. A TLR12, but not a TLR11 sequence has been annotated in most of the Cetaceans’ genomes as a pseudogene. Neither of the two genes can be found in genomes of other Cetartiodactyla members, such as Bovidae or Suidae. TLR11 recognizes bacterial flagellin (Mathur et al. 2012) and protozoan profilin (Yarovinski et al. 2005). The latter is also the main ligand of TLR12, and TLR11/12 heterodimers play important roles in the resistance of mice to Toxoplasma gondii (Koblansky et al. 2013; Andrade et al. 2013). As rodents are the major intermediate hosts for this parasite, the rodent immune system is adapted to better cope with T. gondii infection (Gazzineli et al., 2014). Possible role(s) of TLR11/12 in equine immune mechanisms involved in responses to T. gondii infection are yet to be elucidated. While clinical toxoplasmosis in horses is extremely rare (Kimble et al. 2021), the seroprevalence among equids is high, as many serological studies have detected antibodies against T. gondii in both domestic and wild equids (reviewed in Dubey et al. 2020). Here, we have shown that both TLR11 and TLR12 are transcribed in Equus caballus white blood cells. This is consistent with the expression pattern of these two genes described in mice (Koblansky et al. 2013). The presence of potentially functional genes TLR11 and TLR12 is a special feature of the Equidae that merits further attention in the context of their immune mechanisms and resilience/susceptibiliy to diseases.
According to the current annotation of the donkey genome (ASM1607732v2), both TLR7 and TLR8 are located on the Y chromosome in Equus asinus. This is in disagreement with the annotations of TLR7 and TLR8 genes in other mammalian species as well as with our findings. In most mammals – including humans, rodents, cattle and horses – these two genes are located on the X chromosome; in Equus quagga spp. (UCLA_HA_Equagga_1.0) they are currently annotated on chromosome 10. Both of the two Equus asinus samples we examined were heterozygous for two different TLR7 alleles, and both of the two African wild ass (Equus africanus somaliensis) samples were heterozygous for two different TLR8 alleles. We have checked the genes flanking TLR7 and TLR8 in this Equus asinus genome assembly, and they Blast-mapped in the vicinity of TLR7 and TLR8 on the X chromosome in the horse reference genome. Therefore, the localization of the donkey TLR7 and TLR8 genes on the X chromosome is likely to be an accurate assumption. The assembly of this part of the donkey genome thus seems to be incorrect due to the limitations of the short-read technique, but there is currently no WGS based on long reads that could confirm this assumption. Similarly, it seems that the localization of TLR7 and TLR8 on chromosome 10 in Equus quagga spp. might be due to an incorrect assembly of the genome of this species.
TLRs 7 and 8 show the lowest variability across the Equidae species. The low number of variable nucleotide positions combined with high rate of synonymous substitutions result in a very low number of variable amino acid sites. In TLR8, 90% of nucleotide substitutions were synonymous: 18 CDS haplotypes created only 4 amino acid alleles. Two of them were specific to Equus quagga spp., one to Equus hemionus kulan, and the remaining one was shared across the remaining equids. This is consistent with the high degree of conservation of the TLR7 and TLR8 genes previously described in mammals (Khan et al. 2019), except for lagomorphs (Neves et al. 2022). Interestingly, the highest variability was observed for TLR12, a gene missing in the genomes of several mammalian species (Behzadi et al. 2021), with a single currently known ligand, the T. gondii profilin. TLR11/12 heterodimers are required to elicit a response to profilin (Andrade et al. 2013); TLR11 showed a low degree of polymorphism in equids.
The phylogenetic analysis of TLR genes revealed six TLR gene clusters in the Perissodactyla. The clusters generally matched the TLR families recognized in vertebrates : TLR1-6-10,2; TLR3; TLR4; TLR5; TLR7-9; TLR11-23 (Roach et al. 2005; Liu et al. 2020). In our tree, TLR4, which clustered together with TLR5 with very high branch support (99%), represents an exception. According to Roach et al. (2005) and Liu et al. (2020), TLR4 and TLR5 are single members of separate TLR families. It is not clear whether the different position of the TLR4 in perissodactyls is due to the overall conservation and lesser differentiation of the entire Equidae family.
A conserved synteny of three paralogue genes, TLRs 1-6-10, was observed. This gene family arose by successive tandem duplications of an ancestral gene. In mammals, TLR10 emerged first, followed by TLR1 and TLR6 (Roach et al. 2005). In agreement with Kruithof et al. (2007), we observed a very high degree of sequence identity in a region of 300 amino acids (approx. 440–740) in the C-terminus of equid TLR1 and TLR6, which could be due to a gene conversion. There was no such a region of similarity found between TLR1 and TLR10, or TLR6 and TLR10. TLR10 diverged from the common ancestor much earlier than TLR1 and TLR6. Based on currently known TLR ligands (reviewed in Behzadi et al. (2021), the recognition of bacterial lipopeptides remained preserved in all three members of the TLR-1 gene family; TLR10 gained additionally the ability to recognize viral motifs, which may explain its lower sequence similarity to TLR1 and TLR6.
Phylogenetic trees reconstructed for individual TLRs in the Perissodactyla showed that in agreement with the current taxonomy, rhinoceroses, tapirs and equids always formed distinct groups. Within the Equidae group, the clustering of species was less distinct. According to the available mitochondrial and nuclear gene analysis, caballines, asian asses, african asses and zebra clades can be distinguished (Steiner and Ryder 2011). However, despite the general conservation of the TLR genes, a clear separation between zebras and asses was not always observed (Online Resource 8). As immune-related genes, TLR genes are subject to various selective pressures, reflecting the history of host-pathogen interactions. A dynamic balance between diversifying and balancing selection then drives allelic variation within and between species to cope with changes in PAMPs (Minias and Vinkler 2022). The deviations from the zoological taxonomy observed in the phylogenetic trees may be interpreted as deviations from neutrality, which is in agreement with the general findings of diversifying selection reported for TLR genes in different mammalian families (Ghosh et al. 2022; Darfour-Oduro et al. 2016).
The idea of the functional importance of some of the observed polymorphic variants is also supported by the findings of trans-species allele sharing and the presence of PSS in equids. In general, inferred TLR allelic haplotypes were shared mostly within equid clades (e.g. E. caballus-E. przewalskii), but for TLR 1, 4, 5, 6, 7, 8, 11, 12, at least one allele was shared across the clades. Although the allelic haplotypes were inferred based on short-read NGS using standard bioinformatic tools and have not been confirmed as physical haplotypes, they are the most likely existing combinations of the SNP sites. In fact, several of these inferred haplotypes were identical to the GenBank reference genome sequences, and some of those are shared between species. For example, the inferred haplotype 2 of TLR7 we identified in Equus hartmannae and Equus asinus was identical to Equus asinus XP_014724205.1; similarly, the TLR6 inferred haplotype 2 found in Equus grevyi was identical to Equus quagga XP_046512886.1. Nevertheless, all inferred haplotypes remain to be confirmed by long-read NGS.
Trans-species allele sharing due to polymorphism preceding speciation has been documented in immunity-related genes as well as in several other genes in humans and other species (Klumplerova et al. 2020; Azevedo et al. 2015; Halldórsdóttir and Árnason 2015). We observed trans-species allele sharing within the Equidae, while based on the few tapir and rhino sequences available, no alleles common to equids, tapirs and rhinoceroses, which diverged approximately 56 MY ago (Bai et al. 2018), were identified. Equids diverged into horses, zebras and asses approximately 4–5 MY ago (Librado and Orlando 2021). The existence of alleles shared across these clades after such a period of time may be explained by their adaptive value for the entire family, but also could be due to rapid speciation under strong negative selection. As a result, the branches of the constructed trees would not be well separated.
The two above explanations are not mutually exclusive. The adaptive value of TLR gene polymorphisms may also be estimated based on selection analyses of the sequences retrieved. Different types of selective pressures exerted on innate immunity genes may be reflected at the level of whole genes and/or at the level of selected amino acid sites. Liu et al. (2020) found evidence of purifying selection acting on entire vertebrate TLR genes, along with signatures of diversifying selection in specific codons. Here, we report that the same occurs in the Perissodactyla. All twelve TLR genes were under overall negative selection in perissodactyls. Significant negative selection was observed for TLR genes 3, 4, 7, 8, 9, and 11 even in the relatively small group of equids. On the other hand, signatures of diversifying, site-specific selection were detected in each of the TLR genes in the group of perissodactyls as a whole (Fig. 3 and Online Resource 9). Some of these PSS remained significant when assessed only within the Equidae. This was the case for TLR3, 10, 11, 12. Some PSS were not located in the LRR domains as is usually observed (Downing et al. 2010; Velová et al. 2018), but were instead in the transmembrane region (TLR1) and in the cytoplasmic TIR domains (TLR9-12), which are typically conserved regions (Xu et al. 2000). Although PSS outside the LRR domains are rare, they have been reported (Areal et al. 2011). Notably, PSS E704G in TLR10 located in the TIR domain has the potential to affect protein function (SIFT score < 0.01) if glutamic acid is substituted by glycine. With a single exception, all other PSS with significant SIFT scores were located in the LRR or LRR-Ctd domains. Considering the role of the variability in LRR domains in the recognition of various PAMPs, this is not a surprising finding. A comparison of PSS rates with the variability of the overall amino acid sequence within each TLR showed no clear relationships between greater variation in the sequence and the number of amino acid sites under diversifying selection.
Twelve PSS that we identified in perissodactyls could be matched to PSS previously identified across vertebrates (Liu et al. 2020) and mammals (Areal et al. 2011). According to the SIFT analysis, amino acid variations at these sites did not have direct influence on the function of the protein. Nevertheless, the TLR4-A395 PSS, identified both by us as well as by both of the aforementioned studies, is located in the LRR domain and shows extensive variation (alanine, serine, threonine and arginine were all detected in perissodactyls), which may be related to the variability of the PAMPs recognized. In contrast to the two prior studies, we did not detect any signs of episodic diversifying selection in any of the TLR8 codons.
Based on their ligands and cell localization, two major subgroups of TLRs may be recognized. Receptors expressed on the cell membrane recognize primarily bacterial components (TLR 1,2,4,5,6). Receptors expressed on intracellular membranes (endosomes) (TLR 3,7,8,9) recognize viral nucleic acids (Kawai and Akira 2011). For the purpose of this analysis, we have expanded the non-viral group to include also TLR11 and TLR12, as they bind bacterial and protozoan motifs. Since TLR10 molecules recognize both bacterial and viral motifs and are mostly involved in anti-inflammatory responses (Su et al. 2021; Oosting et al. 2014), TLR10 was not included in this analysis. Differences in the selection patterns of viral and non-viral TLRs have been reported in primates (Barreiro et al. 2009) and carnivores (Liu et al. 2017), and these observations were extended to vertebrates by Liu et al. (2020). The authors showed that diversifying selection acted more strongly on non-viral TLRs, while viral TLRs were under stronger evolutionary constraints. A possible explanation is the higher redundancy and therefore evolutionary flexibility of non-viral TLRs, as bacteria display each several different PAMPs, which are detected simultaneously by different non-viral TLRs. In contrast, non-redundant intracellular viral sensors have only a narrow choice of targets (viral nucleic acids) and changes are not tolerated easily (Barreiro et al. 2009). In our results, diversifying selection prevailed in the non-viral group in perissodactyls, which is consistent with findings by Liu et al. (2020) for all vertebrates.
The adaptive value of Toll-like receptor polymorphisms may also be reflected in their associations with the host’s susceptibility or resistance to infectious diseases (Mukherjee et al. 2019). Based on the overall sequence similarities of TLR gene sequences across mammalian species, it is possible to compare amino acid sites associated with disease in one species with the selection status of those sites in another species. In our study, a PSS identified in TLR1 in perissodactyls (Y309Q, H) corresponded to the H305L site associated with pulmonary tuberculosis in humans (Ma et al. 2007; Meyer et al. 2016). I602S polymorphism is associated with aspergillosis (Kesh et al. 2005) and leprosy (Johnson et al. 2007) in humans; in equids, this site was also variable (I606V), but it was not a PSS. For TLR2, a F227L amino-acid change was associated with Mycobacterium avium spp. paratuberculosis infection in cattle (Fisher et al. 2011). A corresponding variable site F227L was also found in equids (but not in tapirs or rhinos) but with no signs of diversifying selection. In humans and mice, genetic polymorphisms in TLR4 are involved in the LPS-induced signal transduction, but this was not confirmed for horses (Werners et al. 2006). The equine TLR4 site 420, participating in TLR4/MD2 binding, and sites 345, 364, 365, 367, 369, 385 and 414 that contribute to TLR4/TLR4 contact (Walsh et al. 2008), were variable in tapirs and rhinos, but not in equids. None of these sites were under positive selection. These comparisons show that amino acid sites of potential functional importance are often variable in equids or in perissodactyls. However, the significance of these variations in these species remains unknown.
Conclusions
Toll-like receptors are core components of innate immunity. While most studies on TLRs are focused on humans and mice, only limited data are available for non-model species. This study identified twelve TLR genes in perissodactyls both by bioinformatic analyses and re-sequencing. The expression of TLR11 and TLR12 was confirmed by cDNA sequencing. Phylogenic reconstruction of the TLR gene family identified six sub-families. TLR4 clustered together with TLR5; the TLR1-6-10 subfamily showed a high degree of sequence identity corresponding to its evolution through gene conversion. In general terms, TLR genes are rather conserved in the family Equidae, similarly to other immunity. However, distinction between zebras and asses was not always observed, which may be due to selective pressures acting on these genes. Although phylogenetic trees constructed for each TLR did not show any major deviations from neutrality, trans-species sharing of inferred haplotypes was identified across all equids, but not across other perissodactyls. All twelve TLR genes were under strong negative overall selection. Signatures of diversifying selection in specific codon sites were detected in all TLRs except TLR8. Differences in the selection patterns of viral and non-viral TLRs were observed.
Data Availability
All data generated or analysed during this study are included in this published article, its supplementary information files and/or the GenBank repository.
References
Anderson KV, Bokla L, Nüsslein-Volhard C (1985) Establishment of dorsal-ventral polarity in the Drosophila embryo: the induction of polarity by the toll gene product. Cell 42:791–798. https://doi.org/10.1016/0092-8674(85)90275-2
Andrade WA, Souza M, do C, Martinez ER et al (2013) Combined action of nucleic acid-sensing toll-like receptors (TLRs) and TLR11/TLR12 heterodimers imparts resistance to Toxoplasma Gondii in mice. Cell Host Microbe 13:42–53. https://doi.org/10.1016/j.chom.2012.12.003
Areal H, Abrantes J, Esteves PJ (2011) Signatures of positive selection in toll-like receptor (TLR) genes in mammals. BMC Evol Biol 11:368. https://doi.org/10.1186/1471-2148-11-368
Astakhova NM, Perelygin AA, Zharkikh AA et al (2009) Characterization of equine and other vertebrate TLR3, TLR7, and TLR8 genes. Immunogenetics 61:529–539. https://doi.org/10.1007/s00251-009-0381-z
Azevedo L, Serrano C, Amorim A, Cooper DN (2015) Trans-species polymorphism in humans and the great apes is generally maintained by balancing selection that modulates the host immune response. Hum Genomics 9:21. https://doi.org/10.1186/s40246-015-0043-1
Bai B, Wang Y-Q, Meng J (2018) The divergence and dispersal of early perissodactyls as evidenced by early Eocene equids from Asia. Commun Biol 1:115. https://doi.org/10.1038/s42003-018-0116-5
Barreiro LB, Ben-Ali M, Quach H et al (2009) Evolutionary dynamics of human toll-like receptors and their different contributions to host defense. PLoS Genet 5:e1000562. https://doi.org/10.1371/journal.pgen.1000562
Bayerova Z, Janova E, Matiasovic J et al (2016) Positive selection in the SLC11A1 gene in the family Equidae. Immunogenetics 68:353–364. https://doi.org/10.1007/s00251-016-0905-2
Behzadi P, García-Perdomo HA, Karpiński TM (2021) Toll-Like Receptors: General Molecular and Structural Biology. J Immunol Res 2021:9914854. https://doi.org/10.1155/2021/9914854
Brennan JJ, Gilmore TD (2018) Evolutionary origins of toll-like receptor signaling. Mol Biol Evol 35:1576–1587. https://doi.org/10.1093/molbev/msy050
Darfour-Oduro KA, Megens H-J, Roca AL et al (2016) Evolutionary patterns of toll-like receptor signaling pathway genes in the Suidae. BMC Evol Biol 16:33. https://doi.org/10.1186/s12862-016-0602-7
Downing T, Lloyd AT, O’Farrelly C, Bradley DG (2010) The differential evolutionary dynamics of avian cytokine and TLR gene classes. J Immunol 184:6993–7000. https://doi.org/10.4049/jimmunol.0903092
Dubey JP, Murata FHA, Cerqueira-Cézar CK, Kwok OCH (2020) Toxoplasma gondii Infections in horses, donkeys, and other equids: the last decade. Res Vet Sci 132:492–499. https://doi.org/10.1016/j.rvsc.2020.07.005
Dugovich BS, Crane LL, Alcantar BB et al (2019) Multiple innate antibacterial immune defense elements are correlated in diverse ungulate species. PLoS ONE 14:e0225579. https://doi.org/10.1371/journal.pone.0225579
Fisher CA, Bhattarai EK, Osterstock JB et al (2011) Evolution of the Bovine TLR Gene Family and Member associations with Mycobacterium avium subspecies paratuberculosis Infection. PLoS ONE 6:e27744. https://doi.org/10.1371/journal.pone.0027744
Fitzgerald KA, Kagan JC (2020) Toll-like receptors and the control of immunity. Cell 180:1044–1066. https://doi.org/10.1016/j.cell.2020.02.041
Futas J, Horin P (2013) Natural killer cell receptor genes in the family Equidae: not only Ly49. PLoS ONE 8:e64736. https://doi.org/10.1371/journal.pone.0064736
Gazzinelli RT, Mendonça-Neto R, Lilue J et al (2014) Innate resistance against Toxoplasma Gondii: an evolutionary tale of mice, cats and men. Cell Host Microbe 15:132–138. https://doi.org/10.1016/j.chom.2014.01.004
Ghosh M, Basak S, Dutta S (2022) Natural selection shaped the evolution of amino acid usage in mammalian toll like receptor genes. Comput Biol Chem 97:107637. https://doi.org/10.1016/j.compbiolchem.2022.107637
Halldórsdóttir K, Árnason E (2015) Trans-species polymorphism at antimicrobial innate immunity cathelicidin genes of Atlantic Cod and related species. PeerJ 3:e976. https://doi.org/10.7717/peerj.976
Hatai H, Lepelley A, Zeng W et al (2016) Toll-like receptor 11 (TLR11) interacts with Flagellin and Profilin through disparate mechanisms. PLoS ONE 11:e0148987. https://doi.org/10.1371/journal.pone.0148987
Janova E, Matiasovic J, Vahala J et al (2009) Polymorphism and selection in the major histocompatibility complex DRA and DQA genes in the family Equidae. Immunogenetics 61:513–527. https://doi.org/10.1007/s00251-009-0380-0
Johnson CM, Lyle EA, Omueti KO et al (2007) Cutting edge: a common polymorphism impairs cell surface trafficking and functional responses of TLR1 but protects against Leprosy. J Immunol 178:7520–7524. https://doi.org/10.4049/jimmunol.178.12.7520
Kawai T, Akira S (2011) Toll-like receptors and their crosstalk with other innate receptors in Infection and immunity. Immunity 34:637–650. https://doi.org/10.1016/j.immuni.2011.05.006
Kesh S, Mensah NY, Peterlongo P et al (2005) TLR1 and TLR6 polymorphisms are associated with susceptibility to invasive aspergillosis after allogeneic stem cell transplantation. Ann N Y Acad Sci 1062:95–103. https://doi.org/10.1196/annals.1358.012
Khan I, Maldonado E, Silva L et al (2019) The Vertebrate TLR Supergene Family Evolved dynamically by gene Gain/Loss and positive selection revealing a host–Pathogen Arms race in birds. Diversity 11:131. https://doi.org/10.3390/d11080131
Kimble KM, Gomez G, Szule JA et al (2021) Systemic toxoplasmosis in a horse. J Comp Pathol 182:27–31. https://doi.org/10.1016/j.jcpa.2020.11.004
Klumplerova M, Splichalova P, Oppelt J et al (2020) Genetic diversity, evolution and selection in the major histocompatibility complex DRB and DQB loci in the family Equidae. BMC Genomics 21:677. https://doi.org/10.1186/s12864-020-07089-6
Koblansky AA, Jankovic D, Oh H et al (2013) Recognition of Profilin by toll-like receptor 12 is critical for Host Resistance to Toxoplasma Gondii. Immunity 38:119–130. https://doi.org/10.1016/j.immuni.2012.09.016
Kruithof EK, Satta N, Liu JW et al (2007) Gene conversion limits divergence of mammalian TLR1 and TLR6. BMC Evol Biol 7:148. https://doi.org/10.1186/1471-2148-7-148
Kumar V (2022) Toll-like receptors in adaptive immunity. Handb Exp Pharmacol 276:95–131. https://doi.org/10.1007/164_2021_543
Librado P, Orlando L (2021) Genomics and the Evolutionary history of Equids. Annu Rev Anim Biosci 9:81–101. https://doi.org/10.1146/annurev-animal-061220-023118
Liu G, Zhang H, Sun G et al (2017) Characterization of the peripheral blood transcriptome and adaptive evolution of the MHC I and TLR gene families in the wolf (Canis lupus). BMC Genomics 18:584. https://doi.org/10.1186/s12864-017-3983-0
Liu G, Zhang H, Zhao C, Zhang H (2020) Evolutionary history of the toll-like receptor gene family across vertebrates. Genome Biol Evol 12:3615–3634. https://doi.org/10.1093/gbe/evz266
Ma X, Liu Y, Gowen BB et al (2007) Full-exon resequencing reveals toll-like receptor variants contribute to human susceptibility to Tuberculosis Disease. PLoS ONE 2:e1318. https://doi.org/10.1371/journal.pone.0001318
Manuja A, Manuja BK, Singha H (2019) Sequence and functional variability of toll-like receptor 9 gene in equines. Mol Immunol 105:276–282. https://doi.org/10.1016/j.molimm.2018.10.010
Mathur R, Oh H, Zhang D et al (2012) A mouse model of Salmonella typhi Infection. Cell 151:590–602. https://doi.org/10.1016/j.cell.2012.08.042
Meyer CG, Reiling N, Ehmen C et al (2016) TLR1 variant H305L Associated with Protection from Pulmonary Tuberculosis. PLoS ONE 11:e0156046. https://doi.org/10.1371/journal.pone.0156046
Minias P, Vinkler M (2022) Selection balancing at Innate Immune genes: adaptive polymorphism maintenance in toll-like receptors. Mol Biol Evol 39:msac102. https://doi.org/10.1093/molbev/msac102
Mukherjee S, Huda S, Sinha Babu SP (2019) Toll-like receptor polymorphism in host immune response to infectious Diseases: a review. Scand J Immunol 90:e12771. https://doi.org/10.1111/sji.12771
Neves F, Marques JP, Areal H et al (2022) TLR7 and TLR8 evolution in lagomorphs: different patterns in the different lineages. Immunogenetics 74:475–485. https://doi.org/10.1007/s00251-022-01262-9
Novák K (2014) Functional polymorphisms in toll-like receptor genes for innate immunity in farm animals. Vet Immunol Immunopathol 157:1–11. https://doi.org/10.1016/j.vetimm.2013.10.016
Oosting M, Cheng S-C, Bolscher JM et al (2014) Human TLR10 is an anti-inflammatory pattern-recognition receptor. Proc Natl Acad Sci U S A 111:E4478–E4484. https://doi.org/10.1073/pnas.1410293111
Opsal MAa, Våge DI, Hayes B et al (2006) Genomic organization and transcript profiling of the bovine toll-like receptor gene cluster TLR6-TLR1-TLR10. Gene 384:45–50. https://doi.org/10.1016/j.gene.2006.06.027
Price SA, Bininda-Emonds ORP (2009) A comprehensive phylogeny of extant horses, rhinos and tapirs (Perissodactyla) through data combination. Zoosyst Evol 85:277–292. https://doi.org/10.1002/zoos.200900005
Roach JC, Glusman G, Rowen L et al (2005) The evolution of vertebrate toll-like receptors. Proc Natl Acad Sci U S A 102:9577–9582. https://doi.org/10.1073/pnas.0502272102
Silva MJA, Santana DS, de Oliveira LG et al (2022) The relationship between 896A/G (rs4986790) polymorphism of TLR4 and infectious Diseases: a meta-analysis. Front Genet 13:1045725. https://doi.org/10.3389/fgene.2022.1045725
Smith RM, Kotzé A, Grobler JP, Dalton DL (2020) Molecular characterization in the toll-like receptor 9 gene of Cape Mountain Zebra (Equus zebra zebra) from three populations. Infect Genet Evol 78:104118. https://doi.org/10.1016/j.meegid.2019.104118
STEINER CC, RYDER OA (2011) Molecular phylogeny and evolution of the Perissodactyla. Zoo j Linn Soc-Lond 163:1289–1303. https://doi.org/10.1111/j.1096-3642.2011.00752.x
Stejskalova K, Janova E, Horecky C et al (2019) Associations between the presence of specific antibodies to the West Nile Virus Infection and candidate genes in Romanian horses from the Danube delta. Mol Biol Rep 46:4453–4461. https://doi.org/10.1007/s11033-019-04900-w
Su S-B, Tao L, Deng Z-P et al (2021) TLR10: insights, controversies and potential utility as a therapeutic target. Scand J Immunol 93:e12988. https://doi.org/10.1111/sji.12988
Tarlinton RE, Alder L, Moreton J et al (2016) RNA expression of TLR10 in normal equine tissues. BMC Res Notes 9:353. https://doi.org/10.1186/s13104-016-2161-9
Uddin MJ, Suen WW, Bosco-Lauth A et al (2016) Kinetics of the West Nile virus induced transcripts of selected cytokines and toll-like receptors in equine peripheral blood mononuclear cells. Vet Res 47:61. https://doi.org/10.1186/s13567-016-0347-8
Velová H, Gutowska-Ding MW, Burt DW, Vinkler M (2018) Toll-like receptor evolution in birds: gene duplication, pseudogenization, and diversifying selection. Mol Biol Evol 35:2170–2184. https://doi.org/10.1093/molbev/msy119
Vijay K (2018) Toll-like receptors in immunity and inflammatory Diseases: past, present, and future. Int Immunopharmacol 59:391–412. https://doi.org/10.1016/j.intimp.2018.03.002
Walsh C, Gangloff M, Monie T et al (2008) Elucidation of the MD-2/TLR4 interface required for signaling by lipid IVa1. J Immunol 181:1245–1254. https://doi.org/10.4049/jimmunol.181.2.1245
Werners AH, Bull S, Vendrig JC et al (2006) Genotyping of toll-like receptor 4, myeloid differentiation factor 2 and CD-14 in the horse: an investigation into the influence of genetic polymorphisms on the LPS induced TNF-alpha response in equine whole blood. Vet Immunol Immunopathol 111:165–173. https://doi.org/10.1016/j.vetimm.2005.12.003
Xu Y, Tao X, Shen B et al (2000) Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature 408:111–115. https://doi.org/10.1038/35040600
Yarovinsky F, Zhang D, Andersen JF et al (2005) TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 308:1626–1629. https://doi.org/10.1126/science.1109893
Acknowledgements
The authors appreciate the contribution of MVDr. Jiri Vahala (Zoo Dvur Kralove n.L.) and MVDr. Roman Vodicka, PhD. et PhD. (Zoo Prague) who collected and shared samples of captive perissodactyls for the original projects and allowed their archiving for follow-up studies. The authors thank Dr. Barbara Wallner from Vetmeduni Vienna, Austria for her consultations on equid Y chromosomes.
Funding
The project was funded by internal grant of VETUNI no. 2021ITA12.
Open access publishing supported by the National Technical Library in Prague.
Author information
Authors and Affiliations
Contributions
K.S. and P.S. performed the GenBank search, amplified and sequenced samples, analyzed data. E.J. and K.S. ran selection and phylogenetic analyses. J.F. amplified and sequenced cDNA, and consulted analyses. R.V. provided blood samples of equids and consulted phylogenetic analyses. K.S. and P.H. wrote the article. P.H. conceived and supervised the project. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All blood samples used in this study were archived samples originally collected for the purposes of other projects (Futas and Horin 2013; Bayerova et al. 2016; Klumplerova et al. 2020). Based on the legislation at the time, all samples were collected by licensed veterinarians in agreement with all ethical, welfare and professional standards.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stejskalova, K., Janova, E., Splichalova, P. et al. Twelve toll-like receptor (TLR) genes in the family Equidae – comparative genomics, selection and evolution. Vet Res Commun 48, 725–741 (2024). https://doi.org/10.1007/s11259-023-10245-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11259-023-10245-4