
Abstract
Canine parvovirus type 2 (CPV-2) is one of the most relevant pathogens associated with enteritis in dogs and is frequently reported in association with the detection of other pathogens in faeces. In this study the concomitant presence of Canine circovirus (CanineCV) and Canine adenovirus (CAdV) DNA in faecal or intestine samples of 95 dogs with parvovirus enteritis sampled in Italy (1995–2017) was investigated and the viruses identified were genetically characterised. Potential correlations with the antigenic variant of CPV-2 and with signalment data and outcome were evaluated. Twenty-eight of 95 (29.5%) CPV-2 infected dogs tested positive to other viruses: 7/28 were also positive to CanineCV, 1/28 to CAdV-1, 18/28 to CAdV-2, 1/28 to CanineCV and CAdV-2, and 1/28 to CAdV-1 and CAdV-2. The frequency of CAdV DNA detection and coinfections was significantly higher in purebred dogs compared to mixed breed ones (P = 0.002 and 0.009, respectively). The presence of coinfection was not associated with any other relevant data available, including CPV-2 variant and final outcome. The detection of CanineCV in a dog sampled in 2009 allowed to backdating its circulation in dogs. The eight CanineCV completely sequenced were phylogenetically related to the CanineCV identified in dogs, wolves and a badger from Europe, USA, Argentina and China. Nine CAdV were partially sequenced and phylogenetic analysis showed a separate branch for the oldest CAdV-2 identified (1995). From the results obtained in this study population, CanineCV and CAdV coinfections in dogs with parvoviral enteritis did not result in more severe disease.
Similar content being viewed by others


Introduction
Coinfections involving viral diseases represent an increasingly reported problem in veterinary medicine, as they could worsen illness severity and interfere with diagnostic and therapeutic protocols (Kumar et al. 2018a). Among viral diseases, viral enteritis are widespread causes of morbidity and mortality in dogs (Cardillo et al. 2020), and are commonly sustained by multiple pathogens of different virulence (da Rocha Gizzi et al. 2014; Deng et al. 2018; Ortega et al. 2017). Canine parvovirus type 2 (CPV-2) is one of the most relevant pathogens causing severe acute haemorrhagic enteritis in dogs, being highly contagious and lethal especially in non-vaccinated puppies (Decaro and Buonavoglia 2012; Mylonakis et al. 2016). CPV-2 infection is frequently associated with the detection of other pathogens in faecal samples (Alves et al. 2018; da Rocha Gizzi et al. 2014; Ortega et al. 2017; Qi et al. 2020; Zobba et al. 2021). Specifically, several studies reported the coexistence of CPV-2 with Canine circovirus (CanineCV) and Canine adenovirus (CAdV) type 1 and 2 (Dowgier et al.2017; Headley et al. 2013, 2018, 2019). CanineCV are non-enveloped single-stranded and circular DNA viruses, belonging to the genus Circovirus of the family Circoviridae, associated with several disease entities (Bexton et al. 2015; Kapoor et al. 2012; Piewbang et al. 2018; Zaccaria et al. 2016). CAdV are non-enveloped double-stranded and linear DNA viruses belonging to the genus Mastadenovirus of the family Adenoviridae, responsible of a serious multisystem disease, the infectious canine hepatitis (CAdV-1), or implicated in the aetiopathogenesis of infectious tracheobronchitis (CAdV-2) (Decaro et al. 2008; Buonavoglia and Martella 2007). CAdV-2, although associated with respiratory disease, has also been frequently identified in the stool or internal organs of dogs and wildlife (Balboni et al. 2013, 2014; Chaturvedi et al.2008; Dowgier et al. 2018). To date, although the reported frequency of these coinfections is high in dogs with diarrhoea, the effects of multiple pathogens on the disease outcomes remain unclear (da Rocha Gizzi et al. 2014; Dowgier et al. 2017). Furthermore, no information is available on the frequency of multiple infections in association with the different antigenic variants of CPV-2 (2a, 2b and 2c) in dogs with enteritis.
Aims of this study were i) to investigate the presence of coinfections sustained by CanineCV and CAdV (type 1 and type 2) in dogs with parvoviral enteritis; ii) to evaluate whether coinfections were related with the antigenic variant of CPV-2, signalment data and outcome, ii) to genetically characterise the viruses identified.
Materials and methods
Study design, inclusion criteria and samples
For the purposes of the study, dogs with a diagnosis of parvoviral enteritis recorded from 1994 to 2017 were retrospectively selected. The final diagnosis of parvoviral enteritis was achieved if dogs had clinical signs consistent with enteritis including anorexia or lethargy, foul-smelling diarrhoea which could range from mucoid to purely haemorrhagic, vomiting, dehydration and fever (Mylonakis et al. 2016), and if they tested positive to CPV-2 DNA using a qualitative PCR assay (Mochizuki et al. 1993) carried out on faecal or intestine samples. Samples were tested and DNA extracts were stored after routine diagnostic activity at the Service of Clinical Pathology (Department of Veterinary Medical Sciences – DIMEVET, University of Bologna, Italy). Only dogs for which the VP2 gene of CPV-2 was partially or completely sequenced [569 nucleotides (nts) (Balboni et al. 2018) or 1745 nts (Battilani et al. 2019), respectively] and the antigenic variant of CPV-2 deduced from the predicted amino acid residue 426 were included in this study. Signalment data (year of sampling, sex, age, breed and geographical origin), vaccination status and outcome of enrolled dogs were retrieved from medical records.
Detection of Canine circovirus and Canine adenovirus type 1 and 2 DNA
Viral DNA extraction from faecal or intestine samples was performed using the NucleoSpin Tissue Kit (Macherey–Nagel, Germany) according to the manufacturer’s instructions. The extracted DNA was eluted in 100 µL of elution buffer and stored at -20 °C until use. Canine circovirus DNA screening was carried out using a SYBR Green real-time PCR (qPCR) assay according to De Arcangeli and collaborators (De Arcangeli et al. 2020). Canine AdV-1 and 2 DNA screening was carried out using a SYBR Green qPCR assay able to discriminate the two viral types on the basis of melting curve analysis developed and validated by Balboni and collaborators (Balboni et al. 2015). The two qPCR assays were performed using the PowerUp SYBR Green Master Mix (Thermo Fisher Scientific, Life Technologies, USA) and the StepOnePlus Real-Time PCR System (Thermo Fisher Scientific, Life Technologies, USA) following the manufacturer’s instructions. Reactions were carried out testing in duplicate eight tenfold dilutions of a standard plasmid (pCR4 plasmid, Invitrogen, USA) containing one copy of the target sequence used as external positive standard controls for the construction of the assay standard curve, the DNA extracts and a no template negative control. Samples were considered positive when the fluorescence curve in the amplification plot showed an exponential increase and a specific melting peak was observed.
Genetic characterisation of the viruses identified
The complete genome of CanineCV was amplified in positive samples integrating rolling circle amplification (RCA) and endpoint PCR methods (De Arcangeli et al. 2020). The RCA was performed on the positive samples to increase the amount of circular DNA using the TempliPhi 100 amplification kit (GE Healthcare, USA) following the manufacturer’s instructions. Subsequently, viral DNA was amplified by end-point PCR using two couples of primers and a proofreading DNA polymerase (Phusion Hot Start II High-Fidelity DNA Polymerase, Thermo Fisher Scientific, USA) (De Arcangeli et al. 2020).
For the CAdV identified, amplification of partial E3 gene and flanking regions, and of hexon and fiber genes was carried out according to Hu et al. (Hu et al. 2001) and Balboni et al. (Balboni et al. 2017), respectively. Reactions were performed using a proofreading DNA polymerase (Phusion Hot Start II High-Fidelity DNA Polymerase, Thermo Fisher Scientific, USA).
The amplicons obtained were directly sequenced, assembled, aligned with reference sequences from GenBank (see Online Resource 1 for CanineCV and Online Resource 2 for CAdV) and translated into amino acid sequences using the ClustalW method implemented in the BioEdit 7.2.5 software. The variability of the different nucleotide residues of replicase (Rep) and capsid (Cap) genes of CanineCV and of hexon and fiber genes of CAdV-2 was evidenced using entropy (H(x)) plot function implemented in BioEdit 7.2.5; only the reference strains, from which both Rep and Cap or hexon and fiber genes sequences were available, were used for the analysis (Online Resource 1 and 2). The total number of polymorphic sites, the total number of mutations, nucleotide diversity, average number of nucleotide differences, and the number of haplotypes were calculated on the viral genes using DnaSP package version 5.10.01 (Librado and Rozas 2009) and compared from the two viruses.
Phylogeny was carried out on complete genome nucleotide sequences of CanineCV and on partial E3 gene and flanking regions sequences and multiple gene sequences (concatenated hexon and fiber genes sequences) of CAdV using MEGA X version 10.1.7 (Kumar et al. 2018b).
Statistical analysis
The data were evaluated using descriptive statistics and reported as median and range. Categorical data were analysed using the Chi-squared test, while continuous data (age) were analysed by the Mann–Whitney U test. Statistical significance was set at P < 0.05. Statistical analysis was performed using a commercially available statistical software (MedCalc Statistical Software version 16.8.4).
Results
Study population
A total of 95 dogs with clinical signs indicative of parvoviral enteritis tested positive to CPV-2 DNA (90 from faecal samples and five from intestine samples), sampled between 1995 and 2017, were included in this study (Table 1). 48/95 (50.5%) dogs were males, 30/95 (31.6%) were females and for 17/95 (17.9%) this data was unknown. The median age of all dogs was 3 months (range 1 month—10 years). 65/95 (68.4%) dogs were purebred, 24/95 (25.3%) were mixed breed and for 6/95 (6.3%) dogs this data was not available. On the basis of the amino acid residue 426 of the predicted VP2 protein, the canine parvovirus identified belonged to CPV-2a variant in 49/95 (51.6%) dogs, CPV-2b in 21/95 (22.1%) dogs and CPV-2c in 25/95 (26.3%) dogs. 32/95 (33.7%) dogs had undergone at least one administration of a trivalent modified live vaccine against canine parvovirosis (original CPV-2 or CPV-2b), infectious canine hepatitis (CAdV-2) and canine distemper (canine distemper virus, CDV). This group was composed by dogs that undergone a full vaccination scheme and dogs that undergone an incomplete vaccination scheme because they showed gastrointestinal signs and were sampled when they were too young to complete the vaccination protocol. 54/95 (56.8%) dogs did not received any dose of the vaccine and for 9/95 (9.5%) dogs this data was not available. 36/95 dogs (37.9%) recovered from the disease, 22/95 (23.2%) died and for 37/95 (38.9%) dogs the outcome was not available. Based on the availability of DNA extracts, 85/95 (89.5%) dogs were tested for the presence of both CanineCV and CAdV DNA, whereas 4/95 (4.2%) and 6/95 (6.3%) dogs were tested only for the presence of CanineCV and CAdV DNA, respectively.
Detection of Canine circovirus and Canine adenovirus type 1 and 2 DNA
From the 89 dogs tested for CanineCV DNA, 8 (8.9%) were positive (Table 1). The median amount of viral DNA detected in tested samples was 1.8 × 103 copies of target DNA / μL of extracted DNA (range 5.1—1.9 × 105). No significant association was found between the positivity to CanineCV DNA and the variables analysed (Table 1). The oldest sample in which CanineCV DNA was identified was collected in 2009 (lab ID 800/2009).
From the 91 dogs tested to CAdV DNA, 21 (23.1%) were positive (Table 1): 1/21 was positive to CAdV-1, 19/21 to CAdV-2 and 1/21 was positive to both CAdV types. The median amounts of viral DNA detected in tested samples were 6.1 (range 4.5—7.6) and 1.1 × 102 (range 5.7—8.5 × 105) copies of target DNA / μL of extracted DNA for CAdV-1 and CAdV-2, respectively. The frequency of CAdV DNA positivity was significantly higher in purebred dogs (all positive dogs were purebred) compared to mixed breed ones. (P = 0.002, Table 1). No other significant association was found between the positivity to CAdV DNA and the variables analysed (Table 1).
From the 95 dogs included in the study, 28 (29.5%) were coinfected. In 26/28 coinfected dogs, one other viral DNA were detected in addition to CPV-2: 7/28 were positive to CanineCV, 1/28 was positive to CAdV-1 and 18/28 were positive to CAdV-2. The remaining 2/28 coinfected dogs showed triple infection: one was positive for CPV-2, CanineCV and CAdV-2, and one was positive for CPV-2, CAdV-1 and CAdV-2. From 1994 to 2008, before the first detection of CanineCV DNA, 5/44 (11.4%) dogs included in the study were coinfected by CPV-2 and CAdV. From 2009 (year in which the first CanineCV was detected) to 2017, 23/51 (45.1%) dogs included in the study were coinfected. Among the coinfected dogs, 13/28 (46.4%) were infected by CPV-2a variant, 4/28 (14.3%) by CPV-2b and 11/28 (39.3%) by CPV-2c. No significant association was found between the CPV-2 variant and the presence of other viruses. From the 28 coinfected dogs, 26 were purebred and 2 were mixed breed. A significant association was found between the presence of a coinfection and the purebred status (P = 0.009, Table 1). No other significant association was found between the state of coinfection and the variables analysed (Table 1).
Sequence data
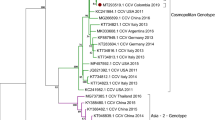
The complete genome sequences of eight CanineCV were obtained and were 2063 nts in length. The genome structure was the same described elsewhere (Decaro et al. 2014; Kotsias et al. 2019; Li et al. 2013; Piewbang et al. 2018; Urbani et al. 2021). Nucleotide alignment between the CanineCV sequences obtained and 110 reference sequences showed an overall nucleotide identity ranging from 80.8 to 100%. Entropy plot analysis showed that nucleotide variation was equally distributed throughout the Rep and Cap genes (Online Resource 3). DnaSP analysis showed a very high and comparable nucleotide variability between the CanineCV Rep and Cap genes, which are approximately the same length (Table 2). Furthermore, the values of nucleotide diversity, average number of nucleotide differences and number of haplotypes calculated for CanineCV genes were clearly higher than those calculated for CAdV-2 genes, regardless of the different number of sequences analysed. The phylogenetic tree constructed with complete genome nucleotide sequences showed a well distinguishable clustering of the CanineCV nucleotide sequences into five groups (Fig. 1), as previously reported by Urbani and colleagues (Urbani et al. 2021). The CanineCV identified in this study were included in the group 1 together with other CanineCV identified in dogs, wolves and a badger from Europe, USA, Argentina and China.

Unrooted phylogenetic tree based on the complete genome of canine circovirus (CanineCV) obtained in this study and 110 reference strains retrieved from the GenBank database (Online Resource 1). Phylogeny was carried out using the software MEGA X version 10.1.7 (Kumar et al. 2018b) and the Maximum Likelihood method. The best-fit model of nucleotide substitution was determined using the Find Best DNA/Protein Model function implemented in MEGA X. General Time Reversible (GTR) model with gamma distribution and invariable sites resulted optimal for the sequence data. Statistical support was provided by bootstrapping with 1000 replicates. Bootstrap values greater than 70% are indicated on the respective branches. The scale bars indicate the estimated numbers of nucleotide substitutions. Highlighted in black: Sequences generated in this study. Numbers in grey are the groups evidenced in this study and from 1 to 4 correspond to genotypes proposed by Niu et al. (Niu et al. 2020)
Nucleotide sequences of partial E3 gene and flanking regions were obtained for one CAdV-1 and eight CAdV-2. The only one CAdV-1 E3 nucleotide sequence was 462 nts in length, while the eight CAdV-2 E3 were 870 nts in length. The unrooted phylogenetic tree constructed with these nucleotide sequences and 71 reference sequences identified in dogs, foxes and wolves showed a clear subdivision of CAdV sequences into two main clusters: the CAdV-1 clade, including the only one CAdV-1 nucleotide sequence obtained in this study, and the CAdV-2 clade, including all the CAdV-2 sequenced in this study (Online Resource 4). The CAdV-2 618/1995 formed a separated branch, while other CAdV-2 sequences obtained in this study clustered together. PCR products specific for the hexon gene (2718 nts in length, corresponding to 905 amino acid residues) and the fiber gene (1629 nts in length, corresponding to 542 amino acid residues) were generated from eight and seven CAdV-2, respectively (GenBank ID: MT193135-MT193149). For both hexon and fiber genes, all the nucleotide sequences obtained in this study showed a complete identity between themselves, except for CAdV-2 618/1995 that showed for the two genes an identity of 99.7% and 99.4% with other viruses sequenced in this study. Entropy plot analysis showed that nucleotide variation was equally distributed throughout the hexon gene, whereas greater nucleotide variability was present in the 3’ portion of the fiber gene (Online Resource 5). DnaSP analysis showed greater nucleotide variability in the hexon gene than the fiber gene (Table 2), with a higher number of polymorphic sites (12 and 6, respectively) and haplotypes (3 and 2, respectively). In the rooted phylogenetic tree constructed from the concatenated nucleotide sequences of hexon and fiber genes obtained in this study and 15 reference sequences, the CAdV-1 and CAdV-2 sequences formed two distinct clusters (Fig. 2). The CAdV-2 618/1995 formed a separated branch while other CAdV-2 sequences obtained in this study grouped together with the vaccine strain Toronto A26/61.

Rooted phylogenetic tree constructed with nucleotide sequences of concatenated nucleotide sequences of hexon and fiber genes obtained in this study and 15 reference strains retrieved from the GenBank database (Online Resource 2). Phylogeny was carried out using the software MEGA X version 10.1.7 (Kumar et al. 2018b) and the Maximum Likelihood method. The best-fit model of nucleotide substitution was determined using the Find Best DNA/Protein Model function implemented in MEGA X. The Hasegawa-Kishino-Yano (HKY) model with gamma distribution and invariable sites resulted optimal for the sequence data. Statistical support was provided by bootstrapping with 1000 replicates. Bootstrap values greater than 70% are indicated on the respective branches. The scale bars indicate the estimated numbers of nucleotide substitutions. Highlighted in black: Sequences generated in this study. On the bottom of the figure, a portion of the obtained tree is enlarged to better visualise the phylogenetic relationships existing between the CAdV-2 nucleotide sequences
Discussion
In this study, 28 out of 95 (29.5%) dogs infected by CPV-2 (1995–2017) were found also positive to at least a different virus of those examined. In particular, 7/28 were also positive to CanineCV, 1/28 to CAdV-1, 18/28 to CAdV-2, 1/28 to CanineCV and CAdV-2, and 1/28 to CAdV-1 and CAdV-2. From 2009 onwards there was an increase in the frequency of viral coinfections detected: passing from 5/44 (11.4%) in 1994–2008 to 23/51 (45.1%) in 2009–2017. Since no CanineCV DNA was detected prior to 2009 in this study, the low frequency of coinfection found can be explained by a genuine limited spread of the CanineCV in the dogs sampled before 2009 or by a degradation of the DNA due to prolonged storage of samples over time with reduced detection of small amounts of viral DNA. No significant association was found between the CPV-2 variant and the presence of the other viruses examined. Thus, it seems that the antigenic CPV-2 variant causing parvoviral enteritis is not a predisposing factor for the onset of coinfections. Furthermore, no significant association was found between the state of coinfection and all the variables analysed, with the exception of purebred status (P = 0.009). Indeed, 26/28 coinfected dogs were purebred, with all dogs testing positive to CAdV DNA that were purebred (P = 0.002). This result suggests a possible predisposition of purebred dogs infected by CPV-2 to be coinfected with CAdV. A previous study that investigated the presence of CAdV DNA in dogs referred to a veterinary hospital in Italy did not detect this association (Balboni et al. 2014), but the study did not investigate the presence of other infectious agents and the dogs were not enrolled on the basis of clinical signs related to gastroenteritis. In the absence of epidemiological data to support this finding, the potential association found between purebred dogs infected by CPV-2 and co-infection with CAdV should be considered with caution. Indeed, this result could be a mere representation of the dogs included in the study, since the origin s of most of the included dogs were from one geographical location. In light of this, further studies are needed to confirm this possible predisposition and clarify which factors can determine it. The mortality of CPV-2 infected dogs appeared not increased if they were coinfected with CanineCV or CAdV. Our findings agree with da Rocha Gizzi and colleagues (da Rocha Gizzi et al. 2014) who did not report increased mortality in dogs coinfected by CPV-2 and other pathogens. In contrast, Anderson and colleagues (Anderson et al. 2017) reported a significantly higher mortality rate of dogs coinfected by CPV-2 and CanineCV. Different case series compositions and variable epidemiological features of the considered viruses might explain such different results.
CanineCV DNA was detected in faecal samples of 8/89 (8.9%) dogs with parvoviral enteritis. Several studies reported a higher frequency of CanineCV infection in diarrhoeic dogs (Dowgier et al. 2017; Hsu et al. 2016; Niu et al. 2020). The low frequency of CanineCV infection found in our study might recognise a genuine limited spread of the CanineCV in the dogs sampled before 2009 or a degradation of the DNA due to prolonged storage of samples over time ass discussed above for coinfections. CanineCV DNA was detected in a dog sampled in 2009 (lab ID 800/2009), two years before the first report of CanineCV infection in dogs (Kapoor et al. 2012). This data, together with the identification of CanineCV DNA in arctic foxes (Vulpes lagopus) in 1996–2001 from Svalbard archipelago (Urbani et al. 2021), supports the hypothesis that CanineCV has been circulating in canids for much longer than previously assumed. All the complete CanineCV nucleotide sequences analysed showed an overall identity ≥ 80.8%. According to the species demarcation threshold of 80% genome-wide nucleotide sequence identity for members of the family Circoviridae (Breitbart et al. 2017; Rosario et al. 2017), this result confirms the existence of a unique canine circovirus species, including the viruses detected in this study. Phylogeny reconstruction evidenced that the CanineCV identified in this study were included in the group 1 of five clusters (Niu et al. 2020; Urbani et al. 2021), together with other viruses identified in dogs, wolves and a badger from Europe, USA, Argentina and China, supporting the hypothesis of a possible transmission of CanineCV from dogs to wild carnivores and vice versa (Balboni et al. 2021; De Arcangeli et al. 2020).
CAdV DNA was detected in faecal samples of 21/91 (23.1%) dogs with parvoviral enteritis, the majority of which belonged to type 2 (20/21, one of which tested positive to both CAdV-1 and CAdV-2). Ji and collaborators (Ji et al. 2020) reported 19/224 (8.5%) dogs with diarrhoea tested positive for CAdV-2, five of which were concomitantly infected with CPV-2, but no association between the infection with CAdV-2 and clinical signs was demonstrated. A frequent detection of CAdV-2 DNA in faecal samples (58.8%) has already been reported in a study performed on dogs not enrolled in relation to gastrointestinal signs and showing various pathological conditions or no clinical symptoms (Balboni et al. 2014).. Further studies should be carried out to investigate whether CAdV-2 may actually play a pathogenic role in gastrointestinal diseases. CAdV-2 sequences analysis evidenced a clear distinction of the oldest virus detected in this study, identified in a dog sampled in 1995 (lab ID 618/1995), showing that CAdV-2 accumulated mutations in the following years. The other CAdV-2 identified in this study grouped phylogenetically with the vaccine strain Toronto A26/61, suggesting that the vaccine currently adopted should not exhibit reductions in efficacy.
From the comparison between CanineCV and CAdV genes, a greater nucleotide variability for CanineCV than CAdV-2 emerged. Since viruses with small and circular genomes tend to mutate faster than viruses with large and linear genome (Sanjuán and Domingo-Calap 2016; Shackelton et al. 2005), this result was expected from the genome characteristics of the two viruses analysed: small single-stranded and circular DNA for CanineCV and medium-large double-stranded and linear DNA for CAdV-2. In particular, CanineCV showed higher nucleotide variability than the one reported for CPV-2, which has a similarly sized but linear genome and for which genomic substitution rate comparable to those of RNA viruses was reported (Battilani et al. 2019; Shackelton et al. 2005). This result highlights a greater propensity of viruses with circular genomes to accumulate mutations. From the analysis of hexon and fiber genes sequences of CAdV-2, it was shown the presence of some nucleotide mutations equally distributed throughout the hexon gene, and preferentially localized in the 3’ portion of the fiber gene. The 3’ portion of the fiber gene codify for the head region (also known as the knob) of the fiber protein, that is responsible to receptor binding and antigenic property (King et al. 2011). A large number of mutations in this region of the fiber gene have already been reported for CAdV-2 in China by Ji and colleagues (Ji et al. 2020) and for CAdV-1 in Italy by Balboni and colleagues (Balboni et al. 2019), highlighting that this genetic region undergoes rapid evolution in all canine mastadenoviruses. Contrary to CAdV-1 (Balboni et al. 2019), a higher nucleotide variability was found in the CAdV-2 hexon gene compared to the fiber gene.
The present study has some limitations. First of all, a small number of dogs were included in a very long period of time, limiting the representativeness of the results obtained. Secondly, only DNA viruses were screened in our population due to the availability of stored DNA extracts in our lab. Thus, coinfections with RNA viruses (such as canine coronavirus, canine distemper virus, canine calicivirus, canine astrovirus, etc.) were not investigated. Moreover, for the purposes of the study, the frequency of coinfections was evaluated in dogs with parvoviral enteritis; the inclusion of dogs with gastroenteritis of different origin as well as of healthy controls would have better clarified the pathogenetic role of such coinfections. Finally, due to the retrospective nature of the study, clinical data indicative of disease severity and days of hospital stay were not available, and data regarding outcome were lacking for some patients. Hence, prospective studies focused on the clinical course of dogs with parvoviral enteritis and eventually coinfected by these viruses are needed to better understand the impact of such coinfections in clinical practice.
Conclusions
In this study, we report new data on the concomitant presence of CanineCV and CAdV in dogs with parvoviral enteritis. From the results obtained, the presence of CanineCV and CAdV coinfection was not associated with the antigenic variant of canine parvovirus causing enteritis and coinfections do not seem to worsen the outcome of enrolled dogs. The genetic characterisation of the identified viruses allowed clarifying new aspects concerning spread and evolution of CanineCV and CAdV in the canine population. The detection of CanineCV in a dog sampled in 2009 allowed to backdating its circulation in the domestic dog population and supports the hypothesis that CanineCV circulates in canids for much longer than previously assumed.
Data availability
The datasets generated and analysed during the current study are available in the International Nucleotide Sequence Database Collaboration (INSDC) repository (http://www.insdc.org/; ID: MT193135-MT193166).
Code availability
Not applicable.
References
Alves CDBT, Granados OFO, Budaszewski RDF, Streck AF, Weber MN, Cibulski SP, Pinto LD, Ikuta N, Canal CW (2018) Identification of enteric viruses circulating in a dog population with low vaccine coverage. Braz J Microbiol 49:790–794. https://doi.org/10.1016/j.bjm.2018.02.006
Anderson A, Hartmann K, Leutenegger CM, Proksch AL, Mueller RS, Unterer S (2017) Role of canine circovirus in dogs with acute haemorrhagic diarrhoea. Vet Rec 180:542. https://doi.org/10.1136/vr.103926
Balboni A, Verin R, Morandi F, Poli A, Prosperi S, Battilani M (2013) Molecular epidemiology of canine adenovirus type 1 and type 2 in free-ranging red foxes (Vulpes vulpes) in Italy. Vet Microbiol 162:551–557. https://doi.org/10.1016/j.vetmic.2012.11.015
Balboni A, Mollace C, Giunti M, Dondi F, Prosperi S, Battilani M (2014) Investigation of the presence of canine adenovirus (CAdV) in owned dogs in Northern Italy. Res Vet Sci 97:631–636. https://doi.org/10.1016/j.rvsc.2014.10.010
Balboni A, Dondi F, Prosperi S, Battilani M (2015) Development of a SYBR green real-time PCR assay with melting curve analysis for simultaneous detection and differentiation of canine adenovirus type 1 and type 2. J Virol Methods 222:34–40. https://doi.org/10.1016/j.jviromet.2015.05.009
Balboni A, Dondi F, Agnoli C, Verin R, Gruarin M, Morini M, Battilani M (2017) Novel sequence variants of viral hexon and fibre genes in two dogs with canine adenovirus type 1-associated disease. Vet J 223:73–75. https://doi.org/10.1016/j.tvjl.2017.05.011
Balboni A, Bassi F, De Arcangeli S, Zobba R, Dedola C, Alberti A, Battilani M (2018) Molecular analysis of carnivore Protoparvovirus detected in white blood cells of naturally infected cats. BMC Vet Res 14:41. https://doi.org/10.1186/s12917-018-1356-9
Balboni A, Tryland M, Mørk T, Killengreen ST, Fuglei E, Battilani M (2019) Unique genetic features of canine adenovirus type 1 (CAdV-1) infecting red foxes (Vulpes vulpes) in northern Norway and arctic foxes (Vulpes lagopus) in Svalbard. Vet Res Commun 43:67–76. https://doi.org/10.1007/s11259-019-09746-y
Balboni A, Urbani L, Delogu M, Musto C, Fontana MC, Merialdi G, Lucifora G, Terrusi A, Dondi F, Battilani M (2021) Integrated use of molecular techniques to detect and genetically characterise dna viruses in italian wolves (Canis lupus italicus). Animals 11:2198. https://doi.org/10.3390/ani11082198
Battilani M, Modugno F, Mira F, Purpari G, Di Bella S, Guercio A, Balboni A (2019) Molecular epidemiology of canine parvovirus type 2 in Italy from 1994 to 2017: recurrence of the CPV-2b variant. BMC Vet Res 15:393. https://doi.org/10.1186/s12917-019-2096-1
Bexton S, Wiersma LC, Getu S, van Run PR, Verjans GMGM, Schipper D, Schapendonk CME, Bodewes R, Oldroyd L, Haagmans BL, Koopmans MMP, Smits SL (2015) Detection of Circovirus in foxes with Meningoencephalitis, United Kingdom, 2009–2013. Emerg Infect Dis 21:1205–1208. https://doi.org/10.3201/eid2107.150228
Breitbart M, Delwart E, Rosario K, Segalés J, Varsani A, Ictv Report Consortium (2017) ICTV virus taxonomy profile: circoviridae. J Gen Virol 98:1997–1998. https://doi.org/10.1099/jgv.0.000871
Buonavoglia C, Martella V (2007) Canine respiratory viruses. Vet Res 38:355–373. https://doi.org/10.1051/vetres:2006058
Cardillo L, Piegari G, Iovane V, Viscardi M, Alfano F, Cerrone A, Pagnini U, Montagnaro S, Galiero G, Pisanelli G, Fusco G (2020) lifestyle as risk factor for infectious causes of death in young dogs: a retrospective study in Southern Italy (2015–2017). Vet Med Int 2020:6207297. https://doi.org/10.1155/2020/6207297
Chaturvedi U, Tiwari AK, Ratta B, Ravindra PV, Rajawat YS, Palia SK, Rai A (2008) Detection of canine adenoviral infections in urine and faeces by the polymerase chain reaction. J Virol Methods 149:260–263. https://doi.org/10.1016/j.jviromet.2008.01.024
da Rocha Gizzi AB, Oliveira ST, Leutenegger CM, Estrada M, Kozemjakin DA, Stedile R, Marcondes M, Biondo AW (2014) Presence of infectious agents and co-infections in diarrheic dogs determined with a real-time polymerase chain reaction-based panel. BMC Vet Res 10:23. https://doi.org/10.1186/1746-6148-10-23
De Arcangeli S, Balboni A, Kaehler E, Urbani L, Verin R, Battilani M (2020) Genomic characterization of Canine Circovirus detected in red foxes (Vulpes vulpes) from Italy using a new real-time PCR assay. J Wildl Dis 56:239–242
Decaro N, Buonavoglia C (2012) Canine parvovirus–a review of epidemiological and diagnostic aspects, with emphasis on type 2c. Vet Microbiol 155:1–12. https://doi.org/10.1016/j.vetmic.2011.09.007
Decaro N, Martella V, Buonavoglia C (2008) Canine adenoviruses and herpesvirus. Vet Clin North Am Small Anim Pract 38:799–814. https://doi.org/10.1016/j.cvsm.2008.02.006
Decaro N, Martella V, Desario C, Lanave G, Circella E, Cavalli A, Elia G, Camero M, Buonavoglia C (2014) Genomic characterization of a circovirus associated with fatal hemorrhagic enteritis in dog. Italy Plos One 9:e105909. https://doi.org/10.1371/journal.pone.0105909
Deng X, Zhang J, Su J, Liu H, Cong Y, Zhang L, Zhang K, Shi N, Lu R, Yan X (2018) A multiplex PCR method for the simultaneous detection of three viruses associated with canine viral enteric infections. Arch Virol 163:2133–2138. https://doi.org/10.1007/s00705-018-3828-4
Dowgier G, Lorusso E, Decaro N, Desario C, Mari V, Lucente MS, Lanave G, Buonavoglia C, Elia G (2017) A molecular survey for selected viral enteropathogens revealed a limited role of Canine circovirus in the development of canine acute gastroenteritis. Vet Microbiol 204:54–58. https://doi.org/10.1016/j.vetmic.2017.04.007
Dowgier G, Lahoreau J, Lanave G, Losurdo M, Varello K, Lucente MS, Ventriglia G, Bozzetta E, Martella V, Buonavoglia C, Decaro N (2018) Sequential circulation of canine adenoviruses 1 and 2 in captive wild carnivores, France. Vet Microbiol 221:67–73. https://doi.org/10.1016/j.vetmic.2018.05.025
Headley SA, Alfieri AA, Fritzen JT, Garcia JL, Weissenböck H, da Silva AP, Bodnar L, Okano W, Alfieri AF (2013) Concomitant canine distemper, infectious canine hepatitis, canine parvoviral enteritis, canine infectious tracheobronchitis, and toxoplasmosis in a puppy. J Vet Diagn Invest 25:129–135. https://doi.org/10.1177/1040638712471344
Headley SA, Oliveira TES, Pereira AHT, Moreira JR, Michelazzo MMZ, Pires BG, Marutani VHB, Xavier AAC, Di Santis GW, Garcia JL, Alfieri AA (2018) Canine morbillivirus (canine distemper virus) with concomitant canine adenovirus, canine parvovirus-2, and Neospora caninum in puppies: a retrospective immunohistochemical study. Sci Rep 8:13477. https://doi.org/10.1038/s41598-018-31540-0
Headley SA, de Mello Zanim Michelazzo M, Elias B, Viana NE, Pereira YL, Pretto-Giordano LG, da Silva JF, da Silva FES, Vilas-Boas LA, da Costa Flaiban KKM, Alfieri AA, Gomes LA (2019) Disseminated melanized fungal infection due to Cladosporium halotolerans in a dog coinfected with canine adenovirus-1 and canine parvovirus-2. Braz J Microbiol 50:859–870. https://doi.org/10.1007/s42770-019-00082-6
Hsu HS, Lin TH, Wu HY, Lin LS, Chung CS, Chiou MT, Lin CN (2016) High detection rate of dog circovirus in diarrheal dogs. BMC Vet Res 12:116. https://doi.org/10.1186/s12917-016-0722-8
Hu RL, Huang G, Qiu W, Zhong ZH, Xia XZ, Yin Z (2001) Detection and differentiation of CAV-1 and CAV-2 by polymerase chain reaction. Vet Res Commun 25:77–84. https://doi.org/10.1023/a:1006417203856
Ji J, Li W, Hu W, Xu X, Kan Y, Yao L, Bi Y, Xie Q (2020) Novel genotype definition and the first epidemiological investigation of Canine Adenovirus type 2 in dogs in Central China. Front Vet Sci 7:534. https://doi.org/10.3389/fvets.2020.00534
Kapoor A, Dubovi EJ, Henriquez-Rivera JA, Lipkin WI (2012) Complete genome sequence of the first canine circovirus. J Virol 86:7018. https://doi.org/10.1128/JVI.00791-12
King AMQ, Lefkowitz E, Adams MJ, Carstens EB (2011) Family Adenoviridae. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz E (eds) Virus taxonomy: IXth Report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press, London, pp 125–141
Kotsias F, Bucafusco D, Nuñez DA, Lago Borisovsky LA, Rodriguez M, Bratanich AC (2019) Genomic characterization of canine circovirus associated with fatal disease in dogs in South America. PLoS ONE 14:e0218735. https://doi.org/10.1371/journal.pone.0218735
Kumar N, Sharma S, Barua S, Tripathi BN, Rouse BT (2018a) Virological and immunological outcomes of coinfections. Clin Microbiol Rev 31:e00111-e117. https://doi.org/10.1128/CMR.00111-17
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018b) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Li L, McGraw S, Zhu K, Leutenegger CM, Marks SL, Kubiski S, Gaffney P, Dela Cruz FN Jr, Wang C, Delwart E, Pesavento PA (2013) Circovirus in tissues of dogs with vasculitis and hemorrhage. Emerg Infect Dis 19:534–541. https://doi.org/10.3201/eid1904.121390
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. https://doi.org/10.1093/bioinformatics/btp187
Mochizuki M, San Gabriel MC, Nakatani H, Yoshida M, Harasawa R (1993) Comparison of polymerase chain reaction with virus isolation and haemagglutination assays for the detection of canine parvoviruses in faecal specimens. Res Vet Sci 55:60–63. https://doi.org/10.1016/0034-5288(93)90035-e
Mylonakis ME, Kalli I, Rallis TS (2016) Canine parvoviral enteritis: an update on the clinical diagnosis, treatment, and prevention. Vet Med (auckl) 7:91–100. https://doi.org/10.2147/VMRR.S80971
Niu L, Wang Z, Zhao L, Wang Y, Cui X, Shi Y, Chen H, Ge J (2020) Detection and molecular characterization of canine circovirus circulating in northeastern China during 2014–2016. Arch Virol 165:137–143. https://doi.org/10.1007/s00705-019-04433-4
Ortega AF, Martínez-Castañeda JS, Bautista-Gómez LG, Muñoz RF, Hernández IQ (2017) Identification of co-infection by rotavirus and parvovirus in dogs with gastroenteritis in Mexico. Braz J Microbiol 48:769–773. https://doi.org/10.1016/j.bjm.2017.03.008
Piewbang C, Jo WK, Puff C, van der Vries E, Kesdangsakonwut S, Rungsipipat A, Kruppa J, Jung K, Baumgärtner W, Techangamsuwan S, Ludlow M, Osterhaus ADME (2018) Novel canine circovirus strains from Thailand: evidence for genetic recombination. Sci Rep 8:7524. https://doi.org/10.1038/s41598-018-25936-1
Qi S, Zhao J, Guo D, Sun D (2020) A mini-review on the epidemiology of canine parvovirus in China. Front Vet Sci 7:5. https://doi.org/10.3389/fvets.2020.00005
Rosario K, Breitbart M, Harrach B, Segalés J, Delwart E, Biagini P, Varsani A (2017) Revisiting the taxonomy of the family Circoviridae: establishment of the genus Cyclovirus and removal of the genus Gyrovirus. Arch Virol 162:1447–1463. https://doi.org/10.1007/s00705-017-3247-y
Sanjuán R, Domingo-Calap P (2016) Mechanisms of viral mutation. Cell Mol Life Sci 73:4433–4448. https://doi.org/10.1007/s00018-016-2299-6
Shackelton LA, Parrish CR, Truyen U, Holmes EC (2005) High rate of viral evolution associated with the emergence of carnivore parvovirus. Proc Natl Acad Sci U S A 102:379–384. https://doi.org/10.1073/pnas.0406765102
Urbani L, Tryland M, Ehrich D, Fuglei E, Battilani M, Balboni A (2021) Ancient origin and genetic segregation of canine circovirus infecting arctic foxes (Vulpes lagopus) in Svalbard and red foxes (Vulpes vulpes) in Northern Norway. Transbound Emerg Dis 68:1283–1293. https://doi.org/10.1111/tbed.13783
Zaccaria G, Malatesta D, Scipioni G, Di Felice E, Campolo M, Casaccia C, Savini G, Di Sabatino D, Lorusso A (2016) Circovirus in domestic and wild carnivores: an important opportunistic agent? Virology 490:69–74. https://doi.org/10.1016/j.virol.2016.01.007
Zobba R, Visco S, Sotgiu F, Pinna Parpaglia ML, Pittau M, Alberti A (2021) Molecular survey of parvovirus, astrovirus, coronavirus, and calicivirus in symptomatic dogs. Vet Res Commun 45:31–40. https://doi.org/10.1007/s11259-020-09785-w
Author information
Authors and Affiliations
Contributions
Conceptualization: [Andrea Balboni, Massimo Giunti, Mara Battilani]; Methodology: [Andrea Balboni, Alessia Terrusi, Lorenza Urbani, Silvia A M Stefanelli, Roberta Troia]; Formal analysis and investigation: [Andrea Balboni, Alessia Terrusi, Lorenza Urbani, Silvia A M Stefanelli, Roberta Troia]; Writing—original draft preparation: [Andrea Balboni, Alessia Terrusi, Lorenza Urbani]; Writing—review and editing: [Massimo Giunti, Mara Battilani]; Resources: [Massimo Giunti, Mara Battilani]; Supervision: [Mara Battilani].
Corresponding author
Ethics declarations
Ethics approval
The study was carried out using stored DNA extracts of faecal samples which had been collected with the agreement of the dog owners for clinical and diagnostic purposes independent of the study. As stored DNA extracts of faecal samples were used, no separate ethical approval was required for the study. All efforts were made to minimise the discomfort of the animals during sampling.
Consent to participate
All authors participated voluntarily in the research.
Consent for publication
All authors read and approved the final manuscript.
Conflicts of interest/Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Balboni, A., Terrusi, A., Urbani, L. et al. Canine circovirus and Canine adenovirus type 1 and 2 in dogs with parvoviral enteritis. Vet Res Commun 46, 223–232 (2022). https://doi.org/10.1007/s11259-021-09850-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11259-021-09850-y