
Abstract
Current antimicrobial drug discovery and advancement attempts are aimed to identify new complexes that can work effectively against the infections caused by microbes. The aim of present study was to synthesize new diorganotin(IV) complexes of N'-((8-hydroxy-1,2,3,5,6,7-hexahydropyrido[3,2,1-ij]quinolin-9-yl)methylene)-2-nitrobenzohydrazide (H2L1), N'-((8-hydroxy-1,2,3,5,6,7-hexahydropyrido[3,2,1-ij]quinolin-9-yl)methylene)-3-methoxybenzohydrazide (H2L2) and N'-((8-hydroxy-1,2,3,5,6,7-hexahydropyrido[3,2,1-ij]quinolin-9-yl)methylene)-4-methylbenzohydrazide (H2L3) Schiff base ligands with formula R2SnL1−3 (where R = Me, Et, Bu and Ph). Various spectral and physico-analytical techniques such as elemental analysis, FT-IR, multinuclear (1H, 13C, 119Sn) NMR, HRMS and XRD were utilized to structurally elucidate the prepared compounds. Based on the spectral data, it was concluded that ligands behave in a tridentate manner and coordinates to the central tin atom through ONO donor atoms. Furthermore, the synthesized compounds were examined for in vitro antimicrobial activity against four bacterial and two fungal strains. Among the examined compounds, Ph2SnL1 complex (MIC = 0.0095 µmol/mL) was found to be the most active. To rationalize the preferred mode of interactions of the most active compound, a molecular docking study of compound Ph2SnL1 was executed in the binding site of 3-oxoacyl-[acyl-carrier-protein] synthase 2 (FabF) of E. coli and sterol 14-alpha demethylase of C. albicans. Compound Ph2SnL1 inhibits the microbes effectively and it could be an effective and new candidate for the modulation of various infections.
Graphical Abstract
New diorganotin(IV) complexes (4–15) were prepared from tridentate Schiff base hydrazones (1–3). The compounds were examined for in vitro antimicrobial activity. Further, molecular docking study of the most potent compound Ph2SnL1 (7) was performed against the enzyme 3-oxoacyl-[acyl-carrier-protein] synthase 2 (FabF) of E. coli and sterol 14-alpha demethylase of C. albicans.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Despite the accessibility of numerous antimicrobial drugs in the market, microbial infections are still the leading cause of death globally [1]. Similar to COVID-19, microbial resistance poses a threat to health security and if appropriate measures are not taken to stop it, might develop into the next major global health emergency [2]. The annual report of GARDP estimates the death of approximately 1.2 million people every year due to infections initiated by drug resistant microbes. The problem is made much worse by the irresponsible use of the drugs that are already available in the market, which in turn leads to reduction in the efficacy of the drugs [3]. This has resulted in global endeavours to develop innovative and more effective antimicrobial drugs. Antimicrobial agents with enhanced efficacy have been the subject of extensive research by scientists.
Schiff base ligands are the most frequently utilized organic compounds in the chemistry and bio-chemistry fields. This is due to their ease of synthesis, stability and potential pharmacological and catalytic applications [4, 5]. Additionally, these sorts of ligands have been engaged as biological models to recognize the bioactivity and structures of various biomolecules [6, 7]. Schiff bases are versatile ligands which act as building blocks for various organic substrates. The structural integrity of these ligands allows for the inoculation of different acceptors or donors which can readily form a variety of molecular structures with different coordination numbers and geometries around the metallic ions [8,9,10]. Tridentate ligands, specially tridentate ONO ligands, have been extensively utilized in coordination chemistry throughout the former few decades [11,12,13,14,15,16]. These ligands are usually flexible giving structurally versatile complexes. Furthermore, the opportunity of producing low-symmetry six- or five-coordinate chelated compounds, which are so exciting from an electronic standpoint, especially if main steric ligand limitations are present, is one major reason for the increasing attention towards these systems [17, 18].
Diorganotin(IV) compounds, as a subclass of organometallic compounds, have gained a lot of interest not only due to their immense structural diversity but also due to their potential usage in a variety of domains including organic synthesis, catalysis, anti-fouling coating materials and so forth [19, 20]. Diorganotin(IV) compounds have potent antimicrobial, anti-leishmanial, antitumour, cardiovascular, antioxidant, antiherpes, trypanocidal and antituberculosis activities [21,22,23,24,25,26,27,28]. Furthermore, the activity of diorganotin(IV) compounds can be easily altered by the choice of ligand attached to the central tin atom. Diorganotin(IV) compounds coordinated especially through oxygen, sulphur and nitrogen donor atoms have been investigated with a particular emphasis on their structure–activity relationships [29]. Diorganotin(IV) complexes of Schiff base ligands containing hydrazide motifs have been reported to be biologically important.
Motivated by the aforementioned observations and in continuation of our ongoing interest [30] in hydrazone ligands and their diorganotin(IV) complexes, we have synthesized a series of diorganotin(IV) complexes with hydrazone Schiff bases. The synthesized compounds were structurally elucidated with the help of various spectroscopic techniques and further evaluated for in vitro antimicrobial activity. Additionally, a molecular docking study of most potent compound was executed against the enzyme 3-oxoacyl-[acyl-carrier-protein] synthase 2 (FabF) of E. coli and sterol 14-alpha demethylase of C. albicans to determine the binding conformations.
Experimental
Materials
All the reagents used in this research work, such as 9-Formyl-8-hydroxyjulolidine (97%), 2-Nitrobenzhydrazide (98%), m-Anisic hydrazide (98%), p-Toluic hydrazide (99%), Dimethytin dichloride (97%), Diethyltin dichloride (97%), Dibutyltin dichloride (97%) and Diphenyltin dichloride (96%) were purchased from Sigma-Aldrich and Alfa Aesar. All solvents used were of analytical grade and were dried in as per the standard literature protocols [31].
Instrumentation
The Bruker Avance III 400 MHz NMR spectrometer was used to record the NMR spectra of the compounds. 1H, 13C, and 119Sn NMR spectra were obtained in DMSO-d6 and CDCl3 solvents using tetramethylsilane and tetramethyltin as internal standards. The FTIR spectra were recorded using the KBr matrix in the wavenumber range of 4000–400 cm−1 by utilizing Perkin Elmer BX II spectrometer. Using an electrical heating coil apparatus, the melting points were measured and reported uncorrected. The SCIEX TripleTOF 5600 and 5600 + /SCIEX instrument was used to record the mass spectra in acetonitrile solvent. Using a Perkin-Elmer 2400 sequence analyser, elemental analysis was performed. The X-ray diffraction study was performed using a Rigaku table top X-ray diffractometer with a scan rate of 2◦ min−1 in the 2θ range of 10–80◦.
Synthesis of Schiff base ligands
In a round bottom flask, 9-Formyl-8-hydroxyjulolidine (1.0863 g, 5 mmol) with 2-Nitrobenzhydrazide (0.90575 g, 5 mmol) for H2L1/ m-Anisic hydrazide (0.8309 g, 5 mmol) for H2L2/ p-Toluic hydrazide (0.7509 g, 5 mmol) for H2L3 were dissolved in dry methanol in a 1:1 molar ratio. The reaction mixtures were refluxed for about 4–5 h with the addition of a few drops of glacial acetic acid. After completion of the reactions, products were filtered off, washed, and dried to obtain pure products.
N'-((8-hydroxy-1,2,3,5,6,7-hexahydropyrido[3,2,1-ij]quinolin-9-yl)methylene)-2-nitrobenzohydrazide H 2 L.1 , (1)
Yield: 84%; Red solid; m.p.: 232–234 °C; Anal. calcd. for C20H20N4O4: C, 63.15; H, 5.30; N, 14.73, found: C, 63.13; H, 5.27; N, 14.67. FTIR (v, cm−1): 3450 (N–H), 2943 (O–H), 1632 (C = O), 1594 (C = N), 1226 (C–OH). 1H NMR (400 MHz, DMSO-d6) δ: 12.02 (s, 1H, NH), 11.54 (s, 1H, OH), 8.14 (s, 1H, -N = CH), 7.95–7.85 (m, 2H, Ar–H), 7.78 (d, J = 7.9 Hz, 2H, Ar–H), 6.74 (s, 1H, Ar–H), 3.21–3.17 (m, 4H), 2.64–2.59 (m, 4H), 1.90–1.82 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ: 161.13 (NH–C = O), 155.16 (HC = N), 153.82, 151.79, 147.61, 145.95, 134.37, 131.80, 130.09, 128.93, 124.82, 113.04, 106.69, 105.87 (Ar–C), 49.73, 49.33, 26.98, 21.93, 21.11, 20.65. HRMS: m/z [M + H]+ calcd. for C20H20N4O4: 381.15, found: 381.15.
N'-((8-hydroxy-1,2,3,5,6,7-hexahydropyrido[3,2,1-ij]quinolin-9-yl)methylene)-3-methoxybenzohydrazide H 2 L.2 , (2 )
Yield: 82%; Lemon yellow solid; m.p.: 221–224 °C; Anal. calcd. for C21H23N3O3: C, 69.02; H, 6.34; N, 11.50, found: C, 69.05; H, 6.27; N, 11.47. FTIR (v, cm−1): 3445 (N–H), 3056 (O–H), 1628 (C = O), 1592 (C = N), 1221 (C–OH). 1H NMR (400 MHz, DMSO-d6) δ: 11.79 (s, 1H, NH), 11.76 (s, 1H, OH), 8.32 (s, 1H, –N = CH), 7.49 (d, J = 8.9 Hz, 1H, Ar–H), 7.45 (dd, J = 9.1, 6.4 Hz, 2H, Ar–H), 7.16 (dd, J = 8.5, 3.2 Hz, 1H, Ar–H), 6.72 (s, 1H, Ar–H), 3.84 (s, 3H, –OCH3), 3.19–3.16 (m, 4H), 2.63–2.59 (m, 4H), 1.88–1.84 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ: 162.32 (NH–C = O), 159.71 (HC = N), 155.17, 151.48, 145.73, 135.03, 130.12, 128.75, 120.13, 117.82, 113.24, 112.91, 106.76, 106.23 (Ar–C), 55.83 (–OCH3), 49.80, 49.34, 27.01, 21.99, 21.16, 20.69. HRMS: m/z [M + H]+ calcd. for C21H23N3O3: 366.18, found: 366.18.
N'-((8-hydroxy-1,2,3,5,6,7-hexahydropyrido[3,2,1-ij]quinolin-9-yl)methylene)-4-methylbenzohydrazide H 2 L.3 , (3)
Yield: 87%; Yellow solid; m.p.: 213–215 °C; Anal. calcd. for C21H23N3O2: C, 72.18; H, 6.63; N, 12.03, found: C, 72.15; H, 6.67; N, 12.05. FTIR (v, cm−1): 3453 (N–H), 2938 (O–H), 1632 (C = O), 1594 (C = N), 1209 (C–OH). 1H NMR (400 MHz, DMSO-d6) δ: 11.83 (s, 1H, NH), 11.73 (s, 1H, OH), 8.32 (s, 1H, –N = CH), 7.83 (d, J = 8.1 Hz, 2H, Ar–H), 7.34 (d, J = 8.2 Hz, 2H, Ar–H), 6.72 (s, 1H, Ar–H), 3.20–3.16 (m, 4H), 2.65–2.60 (m, 4H), 2.39 (s, 3H, –CH3), 1.87–1.86 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ: 162.43 (NH–C = O), 155.14 (HC = N), 151.14, 145.66, 142.13, 130.74, 129.47, 128.69, 127.95, 112.87, 106.78, 106.31 (Ar–C), 49.80, 49.34, 27.00, 22.00, 21.49, 21.18, 20.70. HRMS: m/z [M + H]+ calcd. for C21H23N3O2: 350.18, found: 350.18.
Synthesis of diorganotin(IV) complexes
Diorganotin(IV) complexes have been synthesized by dissolving Schiff base ligand H2L1 (0.760 g, 2 mmol)/ H2L2 (0.730 g, 2 mmol)/ H2L3 (0.698 g, 2 mmol) in appropriate amount of THF solvent and refluxed for 30 min with the insertion of 2–3 drops of triethylamine. After that, dimethyltin dichloride (0.439 g, 2 mmol)/ diethyltin dichloride (0.495 g, 2 mmol)/ dibutyltin dichloride (0.607 g, 2 mmol)/ diphenyltin dichloride (0.687 g, 2 mmol) was separately dissolved to the above resulting mixtures with continuous stirring. The reaction mixtures were refluxed for 7–8 h. After completion of the reactions, Et3N.HCl was filtered off and the solvent was evaporated under reduced pressure on vacuum. The different coloured solid products obtained were washed with dry hexane and dried.
Me 2 SnL 1 (4)
Yield: 77%; Yellow solid; m.p.: 187–189 °C; Anal. calcd. for C22H24N4O4Sn: C, 50.12; H, 4.59; N, 10.63, found: C, 50.15; H, 4.57; N, 10.65. FTIR (v, cm−1): 1586 (C = N), 1233 (C–O), 561 (Sn–O), 473 (Sn–N). 1H NMR (400 MHz, CDCl3) δ: 8.34 (s, 1H, –N = CH), 7.83 (dd, J = 7.7, 1.4 Hz, 1H, Ar–H), 7.79 (dd, J = 8.0, 1.1 Hz, 1H, Ar–H), 7.62–7.58 (m, 1H, Ar–H), 7.53–7.49 (m, 1H, Ar–H), 6.55 (s, 1H, Ar–H), 3.29–3.23 (m, 4H), 2.68–2.60 (m, 4H), 1.95–1.91 (m, 4H), 0.78 (s, 6H, Me). 13C NMR (100 MHz, CDCl3) δ: 165.03 (NH = C–O), 163.73 (HC = N), 161.03, 149.23, 132.08, 131.72, 130.05, 129.86, 123.73, 113.16, 108.58, 105.96 (Ar–C), 50.25, 49.92, 30.33, 22.01, 21.05, 20.66, 1.24 (Me–C). 119Sn NMR (400 MHz, CDCl3) δ: − 143.2. HRMS: m/z [M + H]+ calcd. for C22H24N4O4Sn: 529.08, found: 529.08.
Et 2 SnL 1 (5)
Yield: 72%; Light yellow solid; m.p.: 179–181 °C; Anal. calcd. for C24H28N4O4Sn: C, 51.92; H, 5.08; N, 10.09, found: C, 51.95; H, 5.07; N, 10.05. FTIR (v, cm−1): 1587 (C = N), 1234 (C–O), 535 (Sn–O), 475 (Sn–N). 1H NMR (400 MHz, CDCl3) δ: 8.36 (s, 1H, –N = CH), 7.84 (dd, J = 7.7, 1.3 Hz, 1H, Ar–H), 7.76 (dd, J = 8.0, 1.0 Hz, 1H, Ar–H), 7.61–7.57 (m, 1H, Ar–H), 7.52–7.48 (m, 1H, Ar–H), 6.54 (s, 1H, Ar–H), 3.28–3.25 (m, 4H), 2.68–2.63 (m, 4H), 1.97–1.92 (m, 4H), 1.46–1.42 (m, 4H, Et-H), 1.31 (t, J = 7.8 Hz, 6H, Et-H). 13C NMR (100 MHz, CDCl3) δ: 165.22 (NH = C–O), 164.42 (HC = N), 161.00, 149.21, 149.13, 131.89, 131.69, 130.04, 129.97, 129.75, 123.60, 112.92, 108.58, 106.08 (Ar–C), 50.23, 45.81, 30.33, 27.18, 22.05, 21.13, 13.72 (Et-C), 9.23 (Et-C). 119Sn NMR (400 MHz, CDCl3) δ: − 182.1. HRMS: m/z [M + H]+ calcd. for C24H28N4O4Sn: 557.12, found: 557.12.
Bu 2 SnL 1 (6)
Yield: 71%; Light yellow solid; m.p.: 180–183 °C; Anal. calcd. for C28H36N4O4Sn: C, 55.01; H, 5.94; N, 9.16, found: C, 55.05; H, 5.97; N, 9.14. FTIR (v, cm−1): 1588 (C = N), 1234 (C–O), 537 (Sn–O), 475 (Sn–N). 1H NMR (400 MHz, CDCl3) δ: 8.33 (s, 1H, –N = CH), 7.83 (d, J = 8.8 Hz, 1H, Ar–H), 7.77 (d, J = 8.7 Hz, 1H, Ar–H), 7.59 (t, J = 8.1 Hz, 1H, Ar–H), 7.50 (t, J = 7.1 Hz, 1H, Ar–H), 6.54 (s, 1H, Ar–H), 3.28–3.23 (m, 4H), 2.68–2.61 (m, 4H), 1.95–1.91 (m, 4H), 1.71–1.67 (m, 4H, Bu–H), 1.50–1.38 (m, 8H, Bu–H), 0.91 (t, J = 8.0 Hz 6H, Bu–H). 13C NMR (100 MHz, CDCl3) δ: 165.18 (NH = C–O), 164.23 (HC = N), 160.83, 149.20, 149.08, 131.91, 131.69, 130.03, 130.00, 129.76, 123.61, 112.87, 108.57, 106.06 (Ar–C), 50.23, 49.93, 30.33, 29.71, 27.19, 26.83, 26.51, 22.05, 21.84, 21.14, 20.59, 13.68. 119Sn NMR (400 MHz, CDCl3) δ: − 190.3. HRMS: m/z [M + H]+ calcd. for C28H36N4O4Sn: 613.18, found: 613.18.
Ph 2 SnL 1 (7)
Yield: 79%; Yellow solid; m.p.: 169–172 °C; Anal. calcd. for C32H28N4O4Sn: C, 59.01; H, 4.33; N, 8.60, found: C, 59.03; H, 4.37; N, 8.57. FTIR (v, cm−1): 1587 (C = N), 1234 (C–O), 535 (Sn–O), 476 (Sn-N). 1H NMR (400 MHz, CDCl3) δ: 8.34 (s, 1H, –N = CH), 8.02 (dd, J = 7.7, 1.4 Hz, 1H, Ar–H), 7.88–7.86 (m, 4H, Ar–H), 7.73 (dd, J = 7.9, 1.2 Hz, 1H, Ar–H), 7.60 (dd, J = 7.6, 1.4 Hz, 1H, Ar–H), 7.55 (dd, J = 7.8, 1.5 Hz, 1H, Ar–H), 7.45–7.40 (m, 6H, Ar–H), 6.58 (s, 1H, Ar–H), 3.35–3.30 (m, 4H), 2.95 (t, J = 6.4 Hz, 2H), 2.67 (t, J = 6.2 Hz, 2H), 2.08–2.03 (m, 2H), 1.98–1.94 (m, 2H). 13C NMR (100 MHz, CDCl3) δ: 164.26 (NH = C-O), 164.12 (HC = N), 160.89, 149.56, 139.98, 136.25, 131.44, 130.20, 130.12, 128.65, 123.34, 113.61, 108.95, 106.21 (Ar–C), 50.31, 50.00, 27.17, 21.95, 21.15, 20.80. 119Sn NMR (400 MHz, CDCl3) δ: -329.0. HRMS: m/z [M + H]+ calcd. for C32H28N4O4Sn: 653.12, found: 653.12.
Me 2 SnL 2 (8)
Yield: 73%; Pale yellow solid; m.p.: 165–168 °C; Anal. calcd. for C23H27N3O3Sn: C, 53.93; H, 5.31; N, 8.20, found: C, 53.91; H, 5.33; N, 8.18. FTIR (v, cm−1): 1583 (C = N), 1234 (C–O), 553 (Sn–O), 473 (Sn-N). 1H NMR (400 MHz, CDCl3) δ: 8.45 (s, 1H, –N = CH), 7.65 (d, J = 7.7 Hz, 1H, Ar–H), 7.59 (s, 1H, Ar–H), 7.33 (d, J = 8.0 Hz, 1H, Ar–H), 6.98 (d, J = 7.5 Hz, 1H, Ar–H), 6.59 (s, 1H, Ar–H), 3.89 (s, 3H, –OCH3), 3.28–3.22 (m, 4H), 2.69–2.63 (m, 4H), 1.96–1.91 (m, 4H), 0.76 (s, 6H, Me-H). 13C NMR (100 MHz, CDCl3) δ: 166.44 (NH = C–O), 163.53 (HC = N), 160.14, 159.48, 148.81, 135.59, 131.50, 129.10, 119.68, 116.85, 112.85, 111.57, 108.66 (Ar–C), 55.39 (–OCH3), 50.23, 49.90, 30.33, 27.21, 22.07, 21.14, 1.27 (Me–C). 119Sn NMR (400 MHz, CDCl3) δ: − 145.6. HRMS: m/z [M + H]+ calcd. for C23H27N3O3Sn: 514.11, found: 514.51.
Et 2 SnL 2 (9)
Yield: 75%; Yellow solid; m.p.: 170–174 °C; Anal. calcd. for C25H31N3O3Sn: C, 55.58; H, 5.78; N, 7.78, found: C, 55.61; H, 5.79; N, 7.81. FTIR (v, cm−1): 1587 (C = N), 1237 (C–O), 554 (Sn–O), 475 (Sn–N). 1H NMR (400 MHz, CDCl3) δ: 8.46 (s, 1H, –N = CH), 7.67–7.64 (m, 1H, Ar–H), 7.60 (dd, J = 2.5, 1.4 Hz, 1H, Ar–H), 7.32 (d, J = 8.0 Hz, 1H, Ar–H), 6.99–6.96 (m, 1H, Ar–H), 6.58 (s, 1H, Ar–H), 3.89 (s, 3H, –OCH3), 3.27–3.22 (m, 4H), 2.69–2.64 (m, 4H), 1.95–1.92 (m, 4H), 1.45–1.40 (m, 4H, Et-H), 1.31 (t, J = 7.8 Hz, 6H, Et–H). 13C NMR (100 MHz, CDCl3) δ: 166.75 (NH = C–O), 164.15 (HC = N), 160.11, 159.47, 148.73, 135.73, 131.50, 129.08, 125.54, 119.70, 116.66, 112.63, 111.66, 108.63, 106.32 (Ar–C), 55.36 (–OCH3), 50.05 (–OCH3), 50.20, 45.81, 30.33, 27.20, 22.09, 21.19, 13.61 (Et–C), 9.34 (Et-C). 119Sn NMR (400 MHz, CDCl3) δ: − 183.6. HRMS: m/z [M + H]+ calcd. for C25H31N3O3Sn: 542.14, found: 542.14.
Bu 2 SnL 2 (10)
Yield: 69%; Yellow solid; m.p.: 188–191 °C; Anal. calcd. for C29H39N3O3Sn: C, 58.41; H, 6.59; N, 7.05, found: C, 58.43; H, 6.57; N, 7.01. FTIR (v, cm−1): 1587 (C = N), 1234 (C–O), 555 (Sn–O), 473 (Sn–N). 1H NMR (400 MHz, CDCl3) δ: 8.44 (s, 1H, –N = CH), 7.65 (d, J = 7.7 Hz, 1H, Ar–H), 7.60 (s, 1H, Ar–H), 7.32 (t, J = 7.9 Hz, 1H, Ar–H), 6.98 (dd, J = 7.8, 2.9 Hz, 1H, Ar–H), 6.58 (s, 1H, Ar–H), 3.89 (s, 3H, –OCH3), 3.27–3.22 (m, 4H), 2.69–2.63 (m, 4H), 1.96–1.92 (m, 4H), 1.72–1.64 (m, 4H, Bu–H), 1.47–1.39 (m, 8H, Bu–H), 0.90 (t, J = 8.0 Hz, 6H, Bu-H). 13C NMR (100 MHz, CDCl3) δ: 166.71 (NH = C–O), 164.00 (HC = N), 159.94, 159.46, 148.68, 135.80, 131.49, 129.07, 119.70, 116.63, 112.56, 111.67, 108.65, 106.34 (Ar–C), 55.35 (–OCH3), 50.20, 49.90, 45.83, 30.33, 27.20, 26.92, 26.51, 22.10, 21.66, 21.21, 20.67, 13.69. 119Sn NMR (400 MHz, CDCl3) δ: − 191.2. HRMS: m/z [M + H]+ calcd. for C29H39N3O3Sn: 598.20, found: 598.20.
Ph 2 SnL 2 (11)
Yield: 74%; Light yellow solid; m.p.: 164–166 °C; Anal. calcd. for C33H31N3O3Sn: C, 62.29; H, 4.91; N, 6.60, found: C, 62.30; H, 4.88; N, 6.61. FTIR (v, cm−1): 1583 (C = N), 1228 (C–O), 566 (Sn–O), 471 (Sn–N). 1H NMR (400 MHz, CDCl3) δ: 8.45 (s, 1H, –N = CH), 7.93–7.90 (m, 4H, Ar–H), 7.85–7.78 (m, 3H, Ar–H), 7.41–7.39 (m, 6H, Ar–H), 7.03 (ddd, J = 8.2, 2.7, 0.9 Hz, 1H, Ar–H), 6.60 (s, 1H, Ar–H), 3.93 (s, 3H, -OCH3), 3.34–3.29 (m, 4H), 2.97 (t, J = 6.4 Hz, 2H), 2.68 (t, J = 6.2 Hz, 2H), 2.09–2.04 (m, 2H), 1.97–1.94 (m, 2H). 13C NMR (100 MHz, CDCl3) δ: 166.15 (NH = C–O), 163.98 (HC = N), 159.99, 159.55, 149.10, 140.37, 136.28, 131.78, 130.02, 129.15, 128.59, 119.86, 116.49, 113.27, 112.21, 109.08, 106.42 (Ar–C), 55.43, 50.28, 49.97, 45.80, 27.19, 22.02, 21.25, 20.89, 8.61. 119Sn NMR (400 MHz, CDCl3) δ: − 332.9. HRMS: m/z calcd. for C33H31N3O3Sn: 638.14, found: 638.14.
Me 2 SnL 3 (12)
Yield: 76%; Light yellow solid; m.p.: 158–161 °C; Anal. calcd. for C23H27N3O2Sn: C, 55.67; H, 5.48; N, 8.47, found: C, 55.65; H, 5.46; N, 8.48. FTIR (v, cm−1): 1576 (C = N), 1232 (C–O), 554 (Sn–O), 465 (Sn–N). 1H NMR (400 MHz, CDCl3) δ: 8.44 (s, 1H, –N = CH), 7.93 (d, J = 8.2 Hz, 2H, Ar–H), 7.21 (d, J = 7.9 Hz, 2H, Ar–H), 6.58 (s, 1H, Ar–H), 3.26–3.22 (m, 4H), 3.15–3.10 (m, 4H), 2.40 (s, 3H, –CH3), 1.95–1.91 (m, 4H), 0.75 (s, 6H, Me–H). 13C NMR (100 MHz, CDCl3) δ: 166.75 (NH = C–O), 159.82 (HC = N), 148.68, 140.28, 131.41, 128.79, 127.09, 112.74, 108.72, 106.31 (Ar–C), 50.22, 49.89, 45.85, 27.20, 22.10, 21.17, 20.76, 12.41, 1.12 (Me–C). 119Sn NMR (400 MHz, CDCl3) δ: − 146.8. HRMS: m/z [M + H]+ calcd. for C23H27N3O2Sn: 498.12, found: 498.12.
Et 2 SnL 3 (13)
Yield: 72%; Yellow solid; m.p.: 161–164 °C; Anal. calcd. for C25H31N3O2Sn: C, 57.28; H, 5.96; N, 8.02, found: C, 57.25; H, 5.94; N, 8.03. FTIR (v, cm−1): 1577 (C = N), 1232 (C–O), 552 (Sn–O), 464 (Sn–N). 1H NMR (400 MHz, CDCl3) δ: 8.46 (s, 1H, –N = CH), 7.94 (d, J = 8.1 Hz, 2H, Ar–H), 7.21 (d, J = 8.0 Hz, 2H, Ar–H), 6.57 (s, 1H, Ar–H), 3.27–3.22 (m, 4H), 2.69–2.64 (m, 4H), 2.40 (s, 3H, –CH3), 1.96–1.92 (m, 4H), 1.44–1.38 (m, 4H, Et-H), 1.30 (t, J = 7.4 Hz, 6H, Et–H). 13C NMR (100 MHz, CDCl3) δ: 167.09 (NH = C–O), 164.07 (HC = N), 159.79, 148.58, 140.20, 131.41, 128.78, 127.10, 112.50, 108.69, 106.40 (Ar–C), 50.20, 49.89, 45.87, 27.20, 22.12, 21.48, 21.24, 13.52 (Et–C), 9.30 (Et–C). 119Sn NMR (400 MHz, CDCl3) δ: − 182.9. HRMS: m/z [M + H]+ calcd. for C25H31N3O2Sn: 526.15, found: 526.20.
Bu 2 SnL 3 (14)
Yield: 69%; Light yellow solid; m.p.: 181–184 °C; Anal. calcd. for C29H39N3O2Sn: C, 60.02; H, 6.77; N, 7.24, found: C, 60.05; H, 6.74; N, 7.23. FTIR (v, cm−1): 1577 (C = N), 1231 (C–O), 553 (Sn–O), 463 (Sn–N). 1H NMR (400 MHz, CDCl3) δ: 8.45 (s, 1H, –N = CH), 7.94 (d, J = 8.1 Hz, 2H, Ar–H), 7.21 (d, J = 8.0 Hz, 2H, Ar–H), 6.59 (s, 1H, Ar–H), 3.25–3.20 (m, 4H), 2.70–2.64 (m, 4H), 2.41 (s, 3H, –CH3), 1.97–1.91 (m, 4H), 1.74–1.67 (m, 4H, Bu–H), 1.46–1.37 (m, 8H, Bu–H), 0.90 (t, J = 8.0 Hz, 2H, 6H, Bu-H). 13C NMR (100 MHz, CDCl3) δ: 167.73 (NH = C-O), 163.15 (HC = N), 159.66, 148.64, 140.18, 132.34, 127.77, 118.59, 113.53, 110.78, 108.61, 106.35 (Ar–C), 50.25, 49.76, 45.83, 30.31, 27.24, 26.87, 26.51, 22.45, 21.87, 21.05, 20.65, 13.69. 119Sn NMR (400 MHz, CDCl3) δ: − 194.4. HRMS: m/z [M + H]+ calcd. for C29H39N3O2Sn: 582.21, found: 582.21.
Ph 2 SnL 3 (15)
Yield: 71%; Light yellow solid; m.p.: 157–160 °C; Anal. calcd. for C33H31N3O2Sn: C, 63.89; H, 5.04; N, 6.77, found: C, 63.87; H, 5.06; N, 6.73. FTIR (v, cm−1): 1575 (C = N), 1233 (C–O), 553 (Sn–O), 462 (Sn–N). 1H NMR (400 MHz, CDCl3) δ: 8.44 (s, 1H, –N = CH), 8.12 (d, J = 8.1 Hz, 2H, Ar–H), 7.93–7.90 (m, 4H, Ar–H), 7.41–7.37 (m, 6H, Ar–H), 7.29 (s, 1H, Ar–H), 7.27 (s, 1H, Ar–H), 6.60 (s, 1H, Ar–H), 3.33–3.29 (m, 4H), 2.97 (t, J = 6.5 Hz, 2H), 2.68 (t, J = 6.2 Hz, 2H), 2.44 (s, 3H, –CH3), 2.08–2.03 (m, 2H), 1.99–1.93 (m, 2H). 13C NMR (100 MHz, CDCl3) δ: 166.51 (NH = C–O), 163.87 (HC = N), 159.68, 148.97, 140.43, 136.29, 131.72, 129.98, 128.89, 128.56, 127.26, 113.16, 109.10, 106.44 (Ar–C), 50.27, 49.96, 27.18, 22.03, 21.55, 21.27, 20.91. 119Sn NMR (400 MHz, CDCl3) δ: − 329.6. HRMS: m/z [M + H]+ calcd. for C33H31N3O2Sn: 622.15, found: 622.15.
Antimicrobial activity
The in vitro antimicrobial activity of the synthesized Schiff base ligands (1–3) and their corresponding diorganotin(IV) complexes (4–15) was evaluated against two Gram-positive (Staphylococcus aureus MTCC 2901 and Bacillus subtilis MTCC 441), two Gram-negative (Escherichia coli MTCC 732, Pseudomonas aeruginosa MTCC 424) bacterial and two fungal (Candida albicans MTCC 227, Aspergillus niger MTCC 9933) strains using serial dilution method [32,33,34]. The results were reported in terms of MIC (µmol/mL). Potato dextrose broth and nutrient broth were utilized as the growth media for fungal and bacterial species, respectively. Initially, the stock solutions of synthesized compounds with a concentration of 100 µg/mL were prepared in dimethylsulphoxide, and the solutions were again serially diluted to obtain concentrations of 50, 25, 12.5, 6.25, 3.12, 1.56 µg/mL [35]. 1 mL of the above prepared solutions was poured into each test tube containing 1 mL of nutrient broth and potato dextrose broth for antibacterial and antifungal activity, respectively. The test tubes containing fungal strains and bacterial strains were kept for incubation (all the bacteria were incubated at 37 °C for 24 h and A. niger was incubated at 25 °C for 7 days, while C. albicans was kept at 25 °C for 2 days). The experimental procedure was repeated to get concordant readings, and their MIC values were calculated. The obtained results were compared with standard drugs, viz. Ciprofloxacin for antibacterial and Fluconazole for antifungal activity.
Molecular docking study
Ligand and protein preparation
The structure of the metal complex was sketched and optimized in 2D and 3D with MarvinSketch [36]. The X-ray crystallographic structures of enzymes (pdb id: 2gfx and 5tz1) were retrieved from the protein data bank [https://www.rcsb.org]. The protein was prepared by the removal of water molecules, and the binding site was defined by bound ligand coordinates. The binding site center was chosen by the bound ligand, and a radius of 10 Å was used for extraction of the binding site. The later was used for docking studies. The whole process of binding site preparation was accomplished with iGemdock2.1 [37].
Docking analysis
Computational simulations for docking were carried out using iGemdock2.1, and the suitable binding conformation was searched with a genetic algorithm comprising of population size 300, generations 80 and number of solutions 10 in docking accuracy settings. The conformation which was most overlapping with the co-crystallized ligand was chosen for study. The Discovery studio visualizer and Chimera-X [38, 39] tools were used for the visualization of the outcomes. The procedures of docking were attuned by the correct re-docking of co-crystallized ligands.
Results and discussion
Synthetic aspects
Schiff base ligands (1–3) were obtained by the condensation reaction of 9-Formyl-8-hydroxyjulolidine with Benzohydrazide derivatives in a 1:1 molar ratio. Diorganotin(IV) derivatives were prepared by the reaction of Schiff base ligands with R2SnCl2 (R = Me, Et, Bu and Ph groups). The schematic pathway for the synthesis of ligands and their diorganotin(IV) complexes is depicted in Scheme 1. The synthesized compounds were obtained in good yields and soluble in tetrahydrofuran, dimethylsulphoxide, acetonitrile, etc., but were insoluble in water. The molar conductivity with 10−3 M solutions was found to be in the range of 8–14 Ω−1cm2mol−1 which is very low, suggesting the non-electrolytic nature of the metal complexes. No suitable single crystals for the synthesized compounds could be obtained in any solution.
IR spectra
The FTIR measurements embodied the ubiquity of all the functionalities present in the synthesized compounds. The obtained results are depicted in Table 1. The azomethine band in the ligands was observed in the range of 1594–1592 cm−1, which shifts to a region of lower frequency in complexes, i.e. 1588–1575 cm−1, thereby suggesting the coordination of azomethine nitrogen to the central tin atom [40]. In IR spectra of Schiff base ligands, ν(N–N) stretching band appeared in the range of 968–902 cm−1, which got shifted to slightly lower frequency region upon complexation [41]. In ligands, the absorption bands appearing in the range of 3453–3445 and 3056–2938 cm−1 are due to ν(N–H) and ν(O–H) groups, respectively, which upon coordination with the tin metal disappears in the spectra of complexes. The data obtained strongly manifested the deprotonation of N–H and O–H groups and their subsequent coordination with the tin metal [42]. Another prominent band which helps in determining the binding mode is the ν(C = O) band which was observed at 1632–1628 cm−1 in the free ligands and its disappearance was noticed during complexation [43]. M-L vibration bands for ν(Sn–O) and ν(Sn–N) were detected in far infrared region of 566–535 cm−1 and 476–462 cm−1, respectively [44, 45]. The IR studies reveal that the ligands acted in a tridentate manner and the metal complexes have pentacoordinated geometry [46].
1 H NMR
The proton NMR spectra of the Schiff base ligands (H2L1−3) and their respective diorganotin complexes (R2SnL1−3) were recorded in DMSO-d6 and CDCl3 solvents. In 1H NMR spectra of the ligands, -NH and -OH protons gave singlets at δ 12.02–11.79 ppm and δ 11.76–11.54 ppm, respectively, which disappeared upon complexation, thereby showing the deprotonation and coordination of phenolic and enolic oxygen atoms with the tin metal [42]. The binding mode of the ligands with the tin metal was also explained by –CH = N proton signal at δ 8.32–8.14 ppm, which shifts downfield on complexation at 8.46–8.33 ppm and gives the tin satellite peak with 3 J(119Sn−1H) coupling constant in the range of 48–52 Hz, which further specify the coordination of azomethine nitrogen to the tin metal and formation of Sn–N bond [47]. The aromatic protons in ligands resonated at δ 7.96–6.72 ppm, which were unaffected or very less affected on complexation.
In 1H NMR spectra of diorganotin(IV) complexes, additional peaks were observed due to the presence of alkyl/aryl groups directly attached to the central Sn(IV) atom. Dimethytin derivatives (4, 8, 12) gave singlet in the range of δ 0.78–0.75 ppm. In diethyltin complexes (5, 9, 13), the methylene proton appears as a triplet in the range of 1.31–1.30 ppm while methyl proton appeared as multiplet at δ 1.46–1.38 ppm. Dibutyltin complexes (6, 10, 14) have multiplet and triplet in the range of δ 1.72–1.39 ppm and δ 0.91–0.90 ppm, respectively. Signals obtained in the range of δ 8.12–6.58 ppm are due to phenyl protons of diphenyltin complexes (7, 11, 15).
According to the literature, the coordination patterns of tin(IV) in dimethyltin(IV) derivatives with 2 J(119Sn-1H) coupling constant values are as follows: in tetra-coordinated tin complexes, 2 J values were less than 60 Hz; in pentacoordinated tin complexes, the values fall in the range of 65–80 Hz. Another parameter which helps to confirm the geometry around the tin atom is the C–Sn–C angle. Its values for tetra- and penta-coordinated complexes are θ ≤ 112◦ and θ = 115–130◦, respectively. For dimethyltin(IV) complexes, the observed coupling constant 2 J(119Sn, 1H) values lie in the range of 72–78 Hz and θ (C–Sn–C angle) was 121.82–128.39◦. The obtained values suggest five coordinated geometry for the complexes [48].
13 C NMR
In 13C NMR spectra of ligands, the signal for azomethine carbon resonate at δ 159.71–155.14 ppm and after complex formation its downfield shift was observed at δ 164.42–159.82 ppm, which effectively justify the participation of nitrogen in bonding with the tin metal [49]. Furthermore, the signal for NH–C = O carbon appeared at δ 162.43–161.13 ppm, which have been shifted downfield in the range of δ 167.73–164.26 ppm upon complexation. The signal in ligands at δ 155.17–151.14 ppm is due to the carbon attached to the phenolic oxygen, which shifts to δ 161.03–159.82 ppm in complexes thereby providing another evidence of complex formation.
The signal due to Sn–CH3 of dimethyltin complexes (4, 8, 12) appeared at δ 1.27–1.12 ppm, while diethyltin complexes (5, 9, 13) gave signals at δ 13.72–13.64 ppm and δ 9.34–9.23 ppm due to carbons of diethyl group. Dibutyl complexes (6, 10, 14) gave signals at δ 30.33–30.31 ppm, δ 27.19–26.87 ppm, δ 22.45–22.05 ppm and δ 13.69–13.68 ppm due to carbons of dibutyl group. The signals due to carbon atoms of diphenyltin complexes (7, 11, 15) obtained at δ 161.03–105.96 ppm. These additional signals confirm the existence of the Sn–C bond in complexes.
119 Sn NMR
To study the bonding and molecular structure in organotin compounds, 119Sn NMR spectroscopy proves to be a useful tool. The chemical shift values for dimethyltin (4, 8 and 12), diethyltin (5, 9 and 13), dibutyltin (6, 10 and 14), and diphenyltin (7, 11 and 15) complexes were found to be in the range of δ − 146.83 to − 143.24 ppm, δ − 183.56 to − 182.13 ppm, δ − 194.39 to − 190.34 ppm and δ − 332.89 to − 329.00 ppm, respectively. In complexes, the observed 119Sn chemical shift values specify a penta-coordinated environment around the tin atom [50,51,52].
Mass spectra
The mass spectra of the synthesized ligands (1–3) and their corresponding diorganotin(IV) complexes (4–15) were recorded in acetonitrile solvent and the obtained data was consistent with the proposed structures. The molecular ion peak of hydrazone ligand (3) was observed at m/z 349.1790, which was same as the calculated value. The molecular ion peak for its dimethyltin (12), diethyltin (13), dibutyltin (14) and diphenyltin (15) complexes were observed at m/z 498.1200, 526.2005, 582.2144 and 622.1564, respectively, which correspond to the predicted molecular weight and ensure the formation of complexes. The general possible mass fragmentation pattern for ligand (3) and its dimethyltin(IV) complex (12) is presented in Scheme 2, 3.

Synthesis of Schiff base ligands (1–3) and their diorganotin complexes (4–15)

General possible mass fragmentation for the Schiff base ligand (3)

General possible mass fragmentation for the dimethyltin(IV) complex (12)
X-ray diffraction analysis
The X-ray powder diffraction analysis was carried out to get information about the crystalline or amorphous nature of the synthesized compounds. The Schiff base ligand (1) and its complexes (4–7) were examined by the use of this technique over the 2θ range 10–80◦ at a wavelength of 1.5406 Å. The micrograph of ligand shows sharp peaks specifying its crystalline nature, while the micrographs of metal complexes show semi-crystalline nature. The obtained pattern of ligand is completely different from those of complexes, thereby further supporting the coordination of the ligands to the metal ion. The average crystallite size of the examined compounds was calculated according to Debye–Scherrer equation:
where λ is wavelength, θ is diffraction angle, constant 0.9 is the shape factor, and β is full width at half maxima (FWHM). The average crystallite size dXRD of the compounds H2L1 (1), Me2SnL1 (4), Et2SnL1 (5), Bu2SnL1 (6), and Ph2SnL1 (7) was found to be 42.45, 31.45, 33.19, 28.56 and 26.38 nm, respectively. The values of dislocation density of compounds 1 and 4–7 were found to be in the range of 0.00143–0.00055 nm−2.
Antimicrobial assay
The synthesized compounds (1–15) were tested for antimicrobial activity against four bacterial and two fungal strains. The serial dilution method was utilized in these assays and each experiment was repeated thrice [53]. Ciprofloxacin and Fluconazole were used as standard drugs for the assay. The activity results were reported in terms of MIC (µmol/mL), and the obtained results are summarized in Fig. 1 and Table 2.

Graphical representation of antimicrobial activity of the synthesized compounds
By comparing the activity of ligands (1–3), it was established that H2L1 (MIC = 0.0656–0.0328 µmol/mL) was more active as compared to other ligands. The greater activity of H2L1 ligand was due to the companionship of electron-withdrawing nitro group present on the salicylaldehydic ring, while H2L3 (MIC = 0.0714–0.0357 µmol/mL) was least active due to the presence of electron-donating methyl group [54]. The ligand H2L2 (MIC = 0.0684–0.0342 µmol/mL) having –OCH3 group attached to the salicylaldehyde ring was found to show moderate inhibition. Among the ligands, the antimicrobial activity follows the order H2L1 > H2L2 > H2L3, which suggests that compounds having electron withdrawing groups attached were found to be more active than compounds having electron donating groups attached [55].
Among the metal complexes, especially diphenyl complexes 7, 11 and 15 (MIC = 0.0200–0.0095 µmol/mL) exhibit better antimicrobial potential than the other substituted complexes. The inclusion of two phenyl groups enhances the solubility in lipids and they can easily cross through the cell walls with more efficiency [56]. Dibutyl complexes 6, 10 and 14 (MIC = 0.0214–0.0102 µmol/mL) showed better activity as compared to diethyl complexes 5, 9 and 13 (MIC = 0.0238–0.0112 µmol/mL) and dimethyl complexes 4, 8 and 12 (MIC = 0.0251–0.0118 µmol/mL). The general trend of activity of the complexes follows the order: Ph2SnL1−3 > Bu2SnL1−3 > Et2SnL1−3 > Me2SnL1−3 [57].
From the activity results, it was found that the studied diorganotin(IV) complexes (4–15) exhibit enhanced inhibitory activity against the tested strains as compared to hydrazone ligands (1–3). The increased activity of the complexes might be attributed to increased lipophilicity and efficient diffusion of the metal complexes into bacterial and fungal cells after coordination of ligand to the tin metal [58]. Furthermore, upon complexation the positive charge of the metal in the complexes is partially shared with the donor atoms of the ligands, so that there is an electron delocalization over the whole chelate ring [59, 60].
Structure activity relationship
The SAR of synthesized compounds (1–15) was established to examine the effect of substituents on biological activity and represented in Fig. 2. Compound 1 having a nitro group shows less activity, but after complexation with tin derivatives, there is significant enhancement in its biological activity. Dimethyltin (4) and diethyltin (5) complexes show moderate activity, while dibutyltin complex (6) showed good activity. Diphenyltin complex (7) was found to be the most active compound in the entire series. Compound 2 having a methoxy group shows very less activity, but its complexation with tin derivatives enhanced their biological activity. Diphenyl complex (11) exhibits higher activity than dibutyl complex (10), diethyl (9) and dimethyl (8) complexes exhibit moderate activity. Compound (3) having a methyl group shows very less activity but its diphenyl complex (15) exhibits good activity. Dibutyl complex (14) shows more potency than diethyl (13) and dimethyl (12) complexes. After evaluating the biological potential, it was found that metal complexes (4–15) show higher activity than parent ligands (1–3). The higher activity of the phenyl complexes as compared to other compounds was due to the delocalisation of the Π electron cloud above the ring. The enhanced activity of complexes may be a result of an increase in their lipophilicity.

Structure activity relationship (SAR) diagram for the synthesized compounds
Molecular docking studies
Organometallic complex 7 was found to be most active against all bacterial and fungal strains. So, it was chosen for computational study for assessing the probable binding conformation in the plausible enzyme responsible for its activity. Complex 7 was docked into the binding site of E. coli 3-oxoacyl-[acyl-carrier-protein] synthase 2 (FabF) and sterol 14-alpha demethylase of C. albicans. The binding site chosen was same as that of bound ligands.
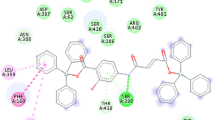
Figure 3 shows that complex 7 interacted with the active site residues of FabF through hydrophobic interactions mainly. One oxygen atom bonded with tin created carbon hydrogen bond with SER271 while three carbon atoms made carbon hydrogen bonds with ARG206, THR307 and GLY310. One of the phenyl ring connected with tin was involved in T-shaped pi-pi stacked interactions with PHE400.

Binding interactions of a complex 7 and b co-crystallized ligand in the active site of E. coli 3-oxoacyl-[acyl-carrier-protein] synthase 2 (FabF)
In the active site of enzyme sterol 14-alpha demethylase of C. albicans, this complex anchored itself through hydrophobic interactions. It interacted with HEME601 via nitrophenyl and phenyl rings by making pi-pi stacked interactions. TYR118 was stacked against the molecule via pi-pi interactions. There were two T-shaped pi-pi stacked interactions; one between TYR132 nitrophenyl ring electrons and other between PHE228 and phenyl ring attached with tin metal. These interactions along with other contacts between the active site residues and compound 7 are shown in Fig. 4.

Binding interactions of a complex 7 and b co-crystallized ligand in the active site of C. albicans sterol 14-alpha demethylase
The docked conformation of compound 7 along with cocrystallized ligand is shown in Fig. 5.

Cartoon diagrams of enzyme a E. coli 3-oxoacyl-[acyl-carrier-protein] synthase 2 and b C. albicans sterol 14-alpha demethylase along with respective co-crystallized ligand (magenta) and docked metal complex 7 (cyan)
Conclusion
In summary, herein we report the synthesis of new diorganotin(IV) complexes (4-15) of Schiff base ligands (1-3). The isolated compounds were very well characterized with the help of spectral and physico-analytical tools. The Schiff base ligands are bound to tin(IV) metal through nitrogen atom of azomethine and oxygen atoms of phenolic and enolic groups to form pentacoordinated complexes. The antimicrobial activity results displayed that metal complexes have enhanced potential as compared to ligands. Among the synthesized compounds, phenyl substituted compound (7) exhibits better activity against all the tested microbes. Compound Ph2SnL1 (7) demonstrated good binding modes in the active sites of E. coli 3-oxoacyl-[acyl-carrier-protein] synthase 2 (FabF) and C. Albicans sterol 14-alpha demethylase. Considering the in vitro and molecular docking studies, the present paper is a noteworthy scientific study for diorganotin(IV) complexes of hydrazone ligands and especially the Ph2SnL1 (7) complex provides a new vision for the development of antimicrobial medications.
Data availability
All data generated or analyzed in this study are included in this article.
References
A. Asadi, S. Razavi, M. Talebi, M. Gholami, Infection 47, 13 (2019)
L. Cantas, S.Q. Shah, L.M. Cavaco, C.M. Manaia, F. Walsh, M. Popowska, H. Garelick, H. Bürgmann, H. Sørum, Front. Microbiol. 4, 96 (2013)
A. Cusini, S.K. Rampini, V. Bansal, B. Ledergerber, S.P. Kuster, C. Ruef, R. Weber, PLoS ONE 5, e14011 (2010)
S. Abdel Halim, M. Shebl, J. Coord. Chem. 74(17–20), 2984 (2022)
P. Agarwal, S. Asija, Y. Deswal, N. Kumar, J. Indian Chem. Soc. 99, 100556 (2022)
M. Shebl, J. Coord. Chem. 69, 199 (2016)
O.M. Adly, M. Shebl, E.M. Abdelrhman, B.A. El-Shetary, J. Mol. Struct. 1219, 128607 (2020)
S. Saroya, S. Asija, N. Kumar, Y. Deswal, J. Indian Chem. Soc. 99, 100379 (2022)
S. Hajra, R. Ghosh, S. Chakrabarti, A. Ghosh, S. Dutta, T.K. Dey, R. Malhotra, S. Asijaa, S. Roy, S. Dutta, S. Basu, Adv. Synth. Catal. 354, 2433 (2012)
S. Nain, S. Asija, M. Malik, Y. Deswal, N. Kumar, Indian. J. Heterocycl. Chem. 32, 235 (2022)
J.V. Folgado, W. Henke, R. Allmann, H. Stratermeier, D. Beltran-Porter, T. Rajo, D. Reinen, Inorg. Chem. 29, 2035 (1990)
F. Samy, M. Shebl, Appl. Organometal. Chem. 34, e5502 (2020)
M. Shebl, J. Coord. Chem. 62, 3217 (2009)
M. Shebl, S.M. Khalil, M.A. Kishk, D.M. El-Mekkawi, M. Saif, Appl. Organomet. Chem. 33(10), e5147 (2019)
F. Samy, M. Shebl, Appl. Organomet. Chem. 36, e6650 (2022)
M. Shebl, A.A. Saleh, S.M. Khalil, M. Dawy, A.A. Ali, Inorg. Nano-Met. Chem. 51, 195 (2021)
J. Fergusson, Prog. Inorg. Chem. 12, 159 (1970)
J.R. Wasson, Spectrosc. Lett. 9, 95 (1976)
P.A. Storozhenko, A.V. Veselov, A.A. Grachev, N.I. Kirilina, V.I. Shiryaev, Catal. Ind. 12, 292 (2020)
T.N. da Silva, D.B. Batista, B.F. Braz, A.S. Luna, R.E. Santelli, M.A. dos Santos Fernandez, J.S. de Gois, Talanta 250, 123718 (2022)
P. Khatkar, S. Asija, Phosphorus Sulfur Silicon Relat. Elem. 192, 446 (2017)
S. Asijaa, N. Malhotra, R. Malhotra, Phosphorus Sulfur Silicon Relat. Elem. 187, 1510 (2012)
S. Shujha, A. Shah, N. Muhammad, S. Ali, R. Qureshi, N. Khalid, A. Meetsma, Eur. J. Med. Chem. 45, 2902 (2010)
S.S. Jawoor, S.A. Patil, S.S. Toragalmath, J. Coord. Chem. 71, 271 (2018)
M. Nath, S. Pokharia, G. Eng, X. Song, A. Kumar, J. Organomet. Chem. 669, 109 (2003)
V.K. Choudhary, A.K. Bhatt, D. Dash, N. Sharma, Appl. Organomet. Chem. 34, e5360 (2020)
J. Susperregui, A. Petsom, M. Bayle, G. Lain, C. Giroud, T. Baltz, G. Déléris, Europ. J. Med. Chem. 32, 123 (1997)
M. Nath, R. Yadav, G. Eng, P. Musingarimi, Appl. Organomet. Chem. 13, 29 (1999)
T. Sedaghat, M. Yousefi, G. Bruno, H.A. Rudbari, H. Motamedi, V. Nobakht, Polyhedron 79, 88 (2014)
N. Kumar, S. Asija, Y. Deswal, S. Saroya, A. Kumar, J. Devi, Phosphorus Sulfur Silicon Relat. Elem. 197, 952 (2022)
A.I. Vogel, Text Book of Quantitative Chemical Analysis (Addision Wesley Longman. Reading, New York, (1999)
Y. Deswal, S. Asija, A. Dubey, L. Deswal, D. Kumar, D.K. Jindal, J. Devi, J. Mol. Struct. 1253, 132266 (2022)
L. Deswal, V. Verma, D. Kumar, A. Kumar, M. Bhatia, Y. Deswal, A. Kumar, Future Med. Chem. 13, 975 (2021)
S. Nain, S. Asija, Y. Deswal, N. Kumar, P. Barwa, Indian J. Heterocycl. Chem. 32, 201 (2022)
L. Deswal, V. Verma, D. Kumar, Y. Deswal, A. Kumar, R. Kumar, M. Parshad, M. Bhatia, Chem. Pap. (2022). https://doi.org/10.1007/s11696-022-02436-1
M. Sketch, 20.3.0, ChemAxon (2020)
J.M. Yang, C.C. Chen, Prot. Struct. Funct. Bioinform. 55, 288 (2004)
D. Systèmes, J. Chem. Phys. 10, 21 (2016)
E.F. Pettersen, T.D. Goddard, C.C. Huang, E.C. Meng, G.S. Couch, T.I. Croll, J.H. Morris, T.E. Ferrin, Pettersen Prot. Sci. 30, 70 (2021)
T. Sedaghat, L. Tahmasbi, H. Motamedi, R. Reyes-Martinez, D. Morales-Morales, J. Coord. Chem. 66, 712 (2013)
M.A. Affan, S.W. Foo, I. Jusoh, S. Hanapi, E.R. Tiekink, Inorganica Chim. Acta. 362, 5031 (2009)
M. Hong, H. Geng, M. Niu, F. Wang, D. Li, J. Liu, H. Yin, Eur. J. Med. Chem. 86, 550 (2014)
H.D. Yin, M. Hong, G. Li, J. Organomet. Chem. 690, 3714 (2005)
F. Wang, H. Yin, J. Cui, Y. Zhang, H. Geng, M. Hong, J. Organomet. Chem. 759, 83 (2014)
Z.J. Zhang, H.T. Zeng, Y. Liu, D.Z. Kuang, F.X. Zhang, Y.X. Tan, W.J. Jiang, Inorg. Nano-Met. Chem. 48, 486 (2018)
H.D. Yin, J.C. Cui, Y.L. Qiao, Polyhedron 27, 2157 (2008)
L. Chen, L. Wang, W. An, R. Wang, L. Tian, Inorg. Nano-Met. Chem. 50, 872 (2020)
M. Sirajuddin, S. Ali, V. McKee, N. Akhtar, S. Andleeb, A. Wadood, J. Photochem. Photobiol. B Biol. 197, 111516 (2019)
J. Ordóñez-Hernández, R. Arcos-Ramos, H. García-Ortega, E. Munguía-Viveros, M. Romero-Ávila, M. Flores-Alamo, N. Farfán, J. Mol. Struct. 1180, 462 (2019)
S. Shujah, N. Muhammad, A. Shah, S. Ali, N. Khalid, A. Meetsma, J. Organomet. Chem. 741, 59 (2013)
S. Basu, C. Masharing, B. Das, Heteroat. Chem. 23, 457 (2012)
S. Sharma, S. Gupta, A.K. Narula, Indian J. Chem. 33A, 1119 (1994)
Y. Deswal, S. Asija, D. Kumar, D.K. Jindal, G. Chandan, V. Panwar, S. Saroya, N. Kumar, Res. Chem. Intermed. 48, 703 (2022)
S. Saroya, S. Asija, Y. Deswal, N. Kumar, A. Kumar, Res. Chem. Intermed. 48, 2949 (2022)
S. Saroya, S. Asija, Y. Deswal, N. Kumar, D. Kumar, D.K. Jindal, P. Puri, S. Kumar, Res. Chem. Intermed. (2022). https://doi.org/10.1007/s11164-022-04826-2
T.S. Basu Baul, Appl. Organomet. Chem. 22, 195 (2008)
J.O. Adeyemi, D.C. Onwudiwe, Pol. J. Environ. Stud. 29, 1 (2020)
M.A. Diab, G.G. Mohamed, W.H. Mahmoud, A.Z. El-Sonbati, S.M. Morgan, S.Y. Abbas, Appl. Organomet. Chem. 33, e4945 (2019)
P. Debnath, K.S. Singh, S. Sharma, P. Debnath, S.S. Singh, L. Sieroń, W. Maniukiewicz, J. Mol. Struct. 1223, 128971 (2021)
B. Nisha, M. Nidhi, A. Sonika, J. Chem. Pharm. Res. 6, 194 (2014)
Funding
The work was financially supported by Council of Scientific and Industrial Research (CSIR), New Delhi in terms of JRF-SRF fellowship with reference no. 09/752(0094)/2019-EMR-I.
Author information
Authors and Affiliations
Contributions
NK contributed to writing—original draft, conceptualization, methodology, validation, formal analysis, data curation, investigation, and funding acquisition. SA contributed to supervision, validation, data curation, and formal analysis. YD contributed to writing—review & editing, software, data curation, and formal analysis. SS contributed to formal analysis. AK provided the software.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
The authors can assure to comply with Springer’s ethical standards. The manuscript which is being submitted to Research on Chemical Intermediates has not been published previously in any form. Authors also assure that the manuscript is not under consideration for publication elsewhere and that its publication is approved by all authors and tacitly or explicitly by the responsible authorities where the work was carried out. The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. Each author knew that the submission also implies that, if accepted, it will not be published elsewhere in the same form, in English or in any other language, without the written consent of the copyright holder.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kumar, N., Asija, S., Deswal, Y. et al. Development of new tridentate ligands bearing hydrazone motif and their diorganotin(IV) complexes: Synthesis, spectral, antimicrobial and molecular docking studies. Res Chem Intermed 48, 5133–5154 (2022). https://doi.org/10.1007/s11164-022-04860-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-022-04860-0