
Abstract
Mesoporous silica materials of MCM-48 and MCM-41 types were synthetized by surfactant directed methods and modified with copper by template ion-exchange (TIE) protocol and its extended version including post-treatment of modified silicas with ammonia solution. Obtained samples were characterized with respect to their textural parameters (low-temperature N2 sorption), porous structure, chemical composition, surface acidity (NH3-TPD), form and aggregation of deposited copper species (UV–vis-DRS) as well as their reducibility (H2-TPR). It was shown that application of TIE method resulted in deposition of copper into MCM-48 in more dispersed forms comparing to MCM-41. Both, in the case of MCM-41 and MCM-48, treatment of the samples with ammonia directly after copper introduction resulted in deposition mainly monomeric copper cations. The catalysts containing such dispersed copper species were more active in the low-temperature NH3-SCR process. The NO conversion above 90% with selectivity to N2 above 97% was obtained in the temperature range of 225–325 °C. CuO aggregates, deposited on mesoporous silicas, were found to be active in the side process of direct ammonia oxidation at higher temperatures.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Mesoporous silicas, synthetised first time in 1992 by researchers of Mobil Oli company [1], due to their high surface area and uniform porous structures, were recognised as very promising materials with a great potential for various applications, also including catalysts. The synthesis of such materials is based on surfactant directed method [2], including formation of micellar structures by self-organisation of surfactants in solution and condensation of silica source around such organic templates. In the final step, organic surfactants must be removed from the pore system of mesoporous silica—most often by calcination [3], less often by extraction with suitable solvents [4]. Mesoporous silica materials, in contrast to zeolites, are characterized by significantly larger pores, what is important for ensuring effective internal diffusion of reactants in catalytic processes. On the other hand, mesoporous silica materials are made of amorphous silica and therefore are less thermally, hydrothermally, and mechanically stable comparing to zeolites. Moreover, pure silica mesoporous materials do not exhibit ion-exchange properties and therefore the spectrum of methods used for deposition of catalytically active metals is limited. Incorporation of guest elements, such as aluminium, into silica walls may result in generation of weak ion-exchange properties, however in contrast to zeolites, the location and density of such ion-exchange sites is not well defined and uniform [5]. The main approaches to solve the problem of limited stability of mesoporous silica materials have been based on synthesis of materials with thicker walls, e.g., SBA-15 [6], and controlled crystallisation of amorphous silica walls [7]. In this latter approach, crystallisation needs the presence of alkali elements and high temperature treatment, which unfortunately cause dramatic drop in surface area and porosity of these materials [7, 8]. Various methods have been used for deposition of catalytically active metals into mesoporous silica materials. One of them is incorporation of metal species into silica walls by co-condensation of silica source and precursor of metal during mesoporous materials synthesis [9]. In this case part of metal species is occluded inside silica walls and is not available for catalytic process. Moreover, synthesis of the samples with higher metal content may result in the formation of aggregated metal oxide species beside dispersed metal cations. On the other hand, this method is promising for the synthesis of catalysts operating with liquid reactants, e.g., slurry reactors [10], due to effective stabilisation of metal species in silica matrix and limited risk of their leaching into solution. Impregnation methods are relatively easy and cheap but controlling the form and aggregation of deposited metal species, important for high selectivity of the catalytic reaction, is very difficult or even impossible [11]. Moreover, in the case of using concentrated solution of metal precursors, partial plugging of pore system may occur. Methods based on grafting of metalorganic compounds followed by their thermal conversion into metal species, e.g., molecular designed dispersion (MDD) method [12], are relatively expensive and result in deposition of only limited amount of highly dispersed metal species [13]. The MDD method is based on the selective reaction of acetylacetonate complexes of various metals with silanol groups of silica support by ligand exchange mechanism [12, 13]. Promising method for metal species deposition on mesoporous silica materials in highly dispersed and uniform forms is template ion-exchange (TIE) method [4, 14]. This method is based on exchange of cationic surfactants, present in freshly prepared mesoporous silica materials, for metal cations [14]. It was shown that introduction of various transition metals, such as copper, iron, or manganese, into MCM-41 using TIE method results in deposition of highly dispersed metal species, mainly monomeric cations [4]. In the case of MCM-41 doped with larger amounts of copper the formation of larger CuO aggregates, located outside of mesoporous grains, was also observed [15]. This problem can be solved by treatment of the Cu-MCM-41 samples, directly after TIE, in ammonia solution [15]. Such extended TIE procedure resulted in the MCM-41 sample containing above 10 wt% of copper deposited nearly exclusively in the form of monomeric cations [4], which was found to be a very promising catalyst of the low-temperature NH3-SCR process. Up-till-now deposition of metals by TIE method and its extended version was tested only for MCM-41 silica materials with hexagonal porous structure. The presented studies verify application of the standard TIE method and extended TIE method with ammonia post-treatment for deposition of copper into MCM-48 with regular porous structure as well as application of such samples in the role of catalysts for the low-temperature NH3-SCR process.
Experimental
Catalysts preparation
MCM-41 and MCM-48 mesoporous silica materials were synthesized and after modification with copper by template ion-exchange method or its extended version used as catalysts of the NH3-SCR process.
Synthesis of MCM-41 was carried out according to the procedure presented in our previous paper [16]. A mixture of 45.3 cm3 hexadecyltrimethylammonium chloride (HDTMACl, 25% solution in water, Sigma-Aldrich), 44.0 cm3 of ammonia (25% solution of NH3 in water, Chempur) and 525 cm3 of distilled water was intensively stirred at room temperature for 30 min. Afterwards, 48.6 cm3 of tetraethyl orthosilicate (TEOS, 98%, Sigma Aldrich) was added dropwise (2 drops/s). The obtained mixture was agitated for 1 h. Subsequently, the solid product was filtered, wash with distilled water, and dried at 60 °C for 48 h.
MCM-48 silica material was prepared as follows [17]: 32.0 cm3 of hexadecyltrimethylammonium chloride (HDTMACl, 25% solution in water, Sigma-Aldrich), 60.0 cm3 of ammonia (25% solution of NH3 in water, Chempur), 134 cm3 of distillated water and 210 cm3 of methanol (Sigma-Aldrich) were mixed. The obtained mixture was vigorously stirring at room temperature for 15 min. Then, 18.0 cm3 of tetraethyl orthosilicate (TEOS, 98%, Sigma Aldrich) was slowly introduced (3 drops/s) and obtained mixture was stirred for 2 h. The solid product was separated by filtration washed with distillated water and dried at 60 °C for 48 h.
The first series of MCM-41 and MCM-48 samples was modified with copper using template ion-exchange (TIE) method. First, a series of CuCl2 (POCh) solutions in pure methanol was prepared. Then, the non-calcined silica sample was introduced into a CuCl2 methanol solution. The obtained slurry (50 cm3 liquid per 1 g of silica material) was stirred vigorously under reflux at 70 °C for 3 h. Afterwards, the modified material was filtered, washed with methanol, dried at 60 °C overnight and calcined at 550 °C for 8 h. The concentration and volumes of reagents used in the TIE modification process are presented in Table 1.
The second series of catalysts was prepared by the extended TIE method including treatment of the samples, directly after TIE, with ammonia (Chempur). In the first step, as-synthesized MCM-41 and MCM-48 samples were mixed with CuCl2 (POCh) methanol solutions and then vigorous stirring for 3 h. Subsequently, the samples were separated by filtration and washed with methanol. In the next step, the obtained materials were treated with aqueous solutions of ammonia (100 cm3) with intensive stirring for 1 h. The modified samples were separated by filtration, washed with distilled water, dried at 60 °C for 24 h and calcined at 550 °C for 8 h. The samples codes as well as concentrations of CuCl2 and NH3 solutions used are shown in Table 1.
Catalysts characterization
The X-ray diffraction patterns of the samples were obtained with a Bruker D2 Phaser diffractometer (Cu Kα radiation, λ = 1.54056 Å). Diffraction patterns were collected in the 2 theta angle ranges of 1–8 and 28–42 with a step of 0.02°. The count time for 2 theta values in the range 1–8 is 5 s per step, while in the range 28–42 is 1 s per step.
The textural parameters of MCM-41 and MCM-48 silicas were specified by N2 sorption at − 196 °C using a 3Flex v.1.00 (Micromeritics) automated gas adsorption system. Before the measurements, the samples were outgassed under vacuum at 350 °C for 24 h. The specific surface area was determined using the BET model, while the BJH model was used to calculate the pore size. The pore volume was estimated by means of the total amount of adsorbed N2 at relative p/p0 pressure of 0.98.
The form and aggregation of copper species deposited on surface of mesoporous silicas were determined by UV–Vis-DR spectroscopy. Spectra were recorded using Lambda 650 S (Perkin Elmer) spectrophotometer in the range of 190–900 nm with a resolution of 2 nm.
Inductively coupled plasma optical emission spectroscopy (ICP-OES) method was used for determination of the elemental composition of the samples. Measurements were made using iCAP 7000 instrument. Prior to the analysis the samples were dissolved in a solution containing 2 cm3 HCl (30%, Honeywell, Charlotte, NC, USA), 6 cm3 HNO3 (67–69%, Honeywell, Charlotte, NC, USA) and 2 cm3 HF (47–51%, Honeywell, Charlotte, NC, USA) at 190 °C using a microwave digestion system Ethos Easy (Milestone).
The content of surfactant in parent silica materials was determined by thermogravimetric analysis, using Mettler Toledo TGA/DSC 3 + thermogravimeter. The samples were heated in air flow of 80 cm3/min with temperature increase of 20 °C/min in temperature range of 30–1100 °C.
The reducing properties of catalysts were tested by temperature-programmed reduction method with hydrogen as a reductant (H2-TPR). The measurements were carried out in a flow microreactor with fixed bed equipped with thermal conductivity detector (TCD, Valco). Mass flow controllers (Brooks Instrument) were used to regulate and control the flow rate of gases supplied into the microreactor. Prior to the reduction process, each sample (25 mg) was outgassed in a flow of pure argon at 550 °C for 30 min. Subsequently, temperature was decreased to 50 °C and the H2-TPR measurements were carried out with the linear temperature increase of 10 °C/min from 50 to 950 °C in a flow of 5.0 V% H2 diluted in argon.
For the analysis of surface acidity of the catalysts, temperature-programmed desorption of ammonia (NH3-TPD) was applied. The measurements were carried out in a flow quartz microreactor system connected directly with quadrupole mass spectrometer (PREVAC-200). Prior to the NH3-TPD runs, the catalyst samples (100 mg) were outgassed in a flow of pure helium at 550 °C for 30 min. After cooling down to 70 °C, the sample was saturated in a flow of gas mixture containing 1.0 V% of NH3 diluted in helium for 2.5 h. In the next step, the catalyst was purged in a helium flow, to remove physiosorbed ammonia molecules, until a stable baseline level was reached. Ammonia desorption was performed with a linear heating rate of 10 °C/min in a flow of pure helium (20 cm3/min).
Catalytic tests
The catalytic activity of the obtained silica-based materials modified with copper was investigated in the selective catalytic reduction of NO with ammonia (NH3-SCR, DeNOx) process. Catalytic experiments were carried out in a quartz microreactor with a fixed bed in a temperature range from 100 to 400 °C under atmospheric pressure. The continuous measurement of reactant concentrations was performed using a quadrupole mass spectrometer (PREVAC-200) directly connected to the microreactor outlet. Prior to catalytic runs, the samples (100 mg) with particle size in the range of 0.250–0.315 mm were placed in the microreactor outgassed in a helium stream at 550 °C for 30 min. The NH3-SCR process involved the utilization of a gas mixture containing 0.25 V% of NO, 0.25 V% of NH3 and 2.5 V% of O2 diluted with pure helium. Additionally, the modified silica materials were tested in NH3-SCR process in a wet atmosphere with a gas mixture containing 0.25 V% of NO, 0.25 V% of NH3, 2.5 V% of O2, 5 V% H2O and 92 V% He.
Furthermore, the modified MCM-41 and MCM-48 samples were tested in the role of catalysts for the selective catalytic oxidation of ammonia to NO and H2O (NH3-SCO). The catalytic tests were performed in a fixed-bed quartz microreactor at temperatures ranging from 225 to 400 °C under atmospheric pressure. Similar to the previous catalytic tests, the reactant concentrations were constantly monitored by a quadrupole mass spectrometer (PREVAC) connected directly to the reactor outlet. 100 mg of the sample with particle sizes in the range from 0.250 to 0.315 mm was subjected to outgassing in a stream of pure helium at 550 °C for 30 min. In the catalytic tests a gas mixture consisting of 0.5 V% of NH3 and 2.5 V% of O2 diluted in pure helium was used.
All NH3-SCR (dry and wet reaction mixtures) and NH3-SCO tests were conducted at a total flow rate of 40 cm3/min. The VHSV (volumetric hourly space velocity), applied in all catalytic tests, was about 24 000 cm3 h−1 g−1.
Results and discussion
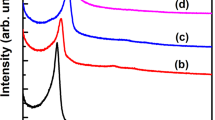
Mesoporous silicas, including materials of MCM-41 and MCM-48 types, are characterized by amorphous silica walls and the reflections, presented in Fig. 1, indicate the ordered porous structure of this type of materials. In diffractogram recorded for mesoporous silica of MCM-41 type, (100), (110), and (200) diffraction peaks characteristic of the porous hexagonal structure, typical of this group of silica materials, are observed (Fig. 1A) [18]. Deposition of copper by template ion-exchange (TIE) method, resulting in the MCM-41-Cu sample, shifted diffraction peaks into higher values of 2 theta angle (Fig. 1A). This effect is possibly related to decrease in the pore size due to deposition of copper species on the internal walls of porous silica. On the other hand, deposition of copper by TIE method followed by ammonia treatment, resulting in MCM-41-Cu-A, significantly decreased intensity of the diffraction peaks and their shift to lower values of 2 theta angles comparing to their positions in diffractogram recorded for MCM-41-Cu (Fig. 1A). These effects are possibly related to partial hydrolysis of siloxane bridges to silanol groups (≡Si–O–Si≡ + H2O → 2 ≡Si–OH) catalysed by ammonia and therefore partial destruction of mesoporous structure of MCM-41. Similar effects were reported in our previous studies for the MCM-41 silica with spherical [15] and cylindrical morphology [4]. In the case of MCM-48, (211), (220) and (420) diffraction peaks, characteristic of the regular porous structure, can be identified (Fig. 1B) [19]. Similarly to MCM-41, deposition of copper by TIE method, resulting in MCM-48-Cu, shifted diffraction peaks into higher 2 theta values. This effect, indicating decrease in pore size, is possibly related to deposition of copper species inside pores. On the other side, post-treatment of such sample with ammonia, resulting in the MCM-48-Cu-A sample, shifted reflections into lower 2 theta values and significantly reduced their intensity (Fig. 1B). Similarly to MCM-41-Cu-A, these effects are possibly assigned to partial hydrolysis of siloxane bridges in ammonia presence and therefore partial destruction of the MCM-48 mesoporous structure.

X-ray diffractograms of MCM-41 (A) and MCM-48 (B) and their modifications with copper (Cu Kα radiation, λ = 1.54056 Å, 2 theta angle ranges of 1–8 and 28–42 with a step of 0.02°, the count time for 2 theta values in the range 1–8 is 5 s per step and in the range 28–42 is 1 s per step)
The 2 theta range characteristic of copper oxide is presented in inserts of Fig. 1A and B. Diffraction peaks indicating the presence of CuO crystallites were found in diffractograms of MCM-41-Cu and MCM-48-Cu, obtained by TIE method. However, it should be noted that the intensity of these reflections is significantly lower for MCM-48-Cu comparing to MCM-41-Cu, containing 4.9 and 4.5 wt% of copper, respectively (Table 2). Thus, it could be postulated that deposition of aggregated copper species by TIE method is significantly limited in the case of MCM-48 comparing to MCM-41. It could be assigned to differences in porous structures and more effective internal diffusion of copper species in straight channels of MCM-41 comparing to twisted channels of MCM-48. Post-treatment of these samples with ammonia resulted in disappearance of the reflections characteristic of copper oxide crystallites (Fig. 1A and B). This effect, indicating deposition of copper in the form of highly dispersed species, has been already reported for deposition of copper by TIE method followed by ammonia treatment for the MCM-41 silicas [4, 15], but up till now has not be verified for MCM-48. Thus, the obtained results prove our hypothesis related to post-treatment of MCM-41 samples with ammonia after TIE protocol and extend this hypothesis for MCM-48 materials.
Nitrogen adsorption–desorption isotherms and pore size distribution (PSD) determined for the studied samples are presented in Figs. 2 and 3, while their textural parameters are compared in Table 2. The isotherms of MCM-41 and MCM-48 samples as well as their modifications with copper are classified as type IV according to the IUPAC classification [20, 21]. The characteristic feature of the isotherms recorded for both series of the mesoporous materials is a steep increase in nitrogen uptake at relative pressure of 0.15–0.30 assigned to the capillary condensation of nitrogen inside mesopores. Deposition of copper by TIE method (MCM-41-Cu and MCM-48-Cu) resulted in a small decrease in nitrogen uptake assigned to decreased mesopore volume, possibly due to deposition of copper species inside pores. More significant decrease in nitrogen uptake, especially in the region characteristic for capillary condensation inside mesopores, was observed for the silica samples modified with copper by TIE method with ammonia post-treatment—MCM-41-Cu-A and MCM-48-Cu-A. This effect is related to partial destruction of the mesoporous structures of MCM-41 and MCM-48 by ammonia. Moreover, an increase in nitrogen sorption, observed at the relative pressure above 0.45, is possibly related to the presence of larger pores located between sticked silica spheres. Such sticking effect is a result of hydrolysis of siloxane bridges to silanol groups and condensation of ≡Si–OH species located on the outer surface of adjacent silica grains. The hysteresis loops observed in adsorption–desorption isotherms belong to H3 category according to the IUPAC classification and is characteristic for the pore network consisting of large pores only partially filled with condensate [20, 21]. PSD profiles, presented in Fig. 3, show high homogeneity of pores diameters in the studied samples. The maxima of PSD are at about 3.35 and 3.15 nm for MCM-41 and MCM-48, respectively. Introduction of copper species into MCM-41 and MCM-48 by TIE method slightly reduced the intensity of the PSD maxima, possibly due to deposition of metal species inside pores. This effect was more significant for MCM-48-Cu and additionally, the shift of PSD maximum into about 2.9 nm was observed in this case (Fig. 3B). Much more significant decrease in intensity of PSD maximum was observed for the samples post-treated with ammonium, MCM-41-Cu-A and MCM-48-Cu-A, indicating partial degradation of the porous structure of these samples. Moreover, broad maxima located at about 13 and 18 nm for MCM-41-Cu-A and MCM-48-Cu-A, respectively, were found in the PSD profiles (Fig. 3A and B). These additional maxima are caused by sticking of silica grains with the formation of secondary macroporous structures in inter-grain spaces. Possibly, silanol groups formed by ammonia catalysed hydrolysis of siloxanes (≡Si–O–Si≡ + H2O → 2 ≡Si–OH) re-condensate with the formation inter-grain links.

Nitrogen adsorption–desorption isotherms of MCM-41 (A) and MCM-48 (B) and their modifications with copper (textural parameters were specified by N2 sorption at − 196 °C, the specific surface area was determined by the BET model)

Pore size distribution (PSD) profiles of MCM-41 (A) and MCM-48 (B) and their modifications with copper (the BJH model was used to calculate the pore size)
Specific surface areas, determined by BET method, are 1200 and 1363 m2/g for MCM-41 and MCM-48, respectively (Table 2). Deposition of copper by TIE method decreased specific surface area by about 12% in the case of MCM-41-Cu as well as MCM-48-Cu. Also decrease of pore volume was observed after copper deposition into these silica materials. More significant decrease in specific surface area, at around 45–50%, occurred for the MCM-41-Cu-A and MCM-48-Cu-A samples obtained by TIE method with ammonia post-treatment (Table 2). Thus, the reduction of specific surface area observed as a result of copper doping depends on various factors. First, specific surface area of the silica samples decreased due to pore blocking by deposited copper species. This effect is more distinct in the case of CuO aggregates deposition, which may more effectively clog the channels of mesoporous silicas. Secondly, a very significant decrease of specific surface area is observed for the samples post-treated with ammonia (MCM-41-Cu-A and MCM-41-Cu-A, Table 2). This effect is related to partial destruction of mesoporous silica porous structure by basic ammonia solution. On the other side, pore volume determined for these samples significantly increased under conditions of ammonia post-treatment. As it was already mentioned, these effects could be explained by partial destruction of mesoporous structure by ammonia resulting in decrease of specific surface area as well as sticking of silica grains with the formation of the secondary macroporous structures in inter-grain spaces, resulting in increased pore volume.
The forms and aggregation of copper species deposited on MCM-41 and MCM-48 were verified by UV–Vis-DR spectroscopy (Fig. 4). In spectra recorded for all the samples, the band at about 220–230 nm, indicating the presence of monomeric Cu2+ ions interacting with oxygens of silica (O2− → Cu2+) [22, 23], dominate. The presence of such monomeric cations is also proved by the broad band located above 550 nm and related to d-d transition in Cu2+ ions in pseudo-octahedral coordination, such as Cu(H2O)62+ [22, 23]. In the case of the spectrum recorded for MCM-41-Cu (Fig. 4A) and, to a lesser extent, for the MCM-48-Cu sample (Fig. 4B), the increased level of absorbance above 300 nm indicates the presence of aggregated copper oxide species of different size, such as oligomeric and small crystallites [23, 24]. It should be noted that CuO crystallites were detected in the MCM-41-Cu and MCM-48-Cu samples by XRD method (Fig. 1A and B).

UV–Vis DR spectra of MCM-41 (A) and MCM-48 (B) modified with copper (measurement range of 190–900 nm with a resolution of 2 nm)
Surface acidity of the studied samples was analysed by temperature-programmed desorption of ammonia (NH3-TPD). No acidity was found for the silica samples non-modified with copper, MCM-41 and MCM-48 (results not shown). Thus, ammonia chemisorption was possible only on the copper species introduced to the silica samples. Interaction of copper with ammonia molecule is due to the formation of donor–acceptor bound by transfer of free electron pair of ammonia into unoccupied d-orbitals of copper (NH3 → Cu). Thus, the amount of chemisorbed ammonia indicates the surface available copper cations, that can participate in catalytic surface reaction. As it can be seen in Fig. 5, ammonia desorption profiles are similar for all the studied samples and consist of broad maximum, possibly superposition of at least two peaks, at temperature below 450 °C. The intensity of these maxima depends on the amount of surface exposed copper cations able to chemisorb ammonia molecules. Surface concentration (CA) and surface density (DA) of chemisorbed ammonia determined from NH3-TPD measurements are compared in Table 1. The lowest surface concentration of chemisorbed ammonia was determined for MCM-41-Cu, while significantly larger amount of ammonia was chemisorbed on the surface of MCM-48-Cu. Considering similar copper loading in both these samples (Table 2) it shows that copper was introduced into MCM-48 in the form of more dispersed species comparing to MCM-41. It is proved by the ratio of the amount of chemisorbed ammonia (determined from NH3-TPD measurements) and copper content in the samples (determined by chemical analyses—ICP-OES), CA/[Cu], presented in Table 2. The CA/[Cu] value determined for MCM-48-A is significantly higher than for MCM-41-A, indicating higher contribution of surface exposed copper cations in the MCM-48-A sample. Post-treatment of the samples with ammonia increased the CA/[Cu] ratio, proving beneficial role of this procedure for deposition of copper in highly dispersed forms. These results are in-line with the results of XRD (Fig. 1) and UV–Vis-DRS (Fig. 4) studies.

Ammonia desorption profiles of MCM-41 (A) and MCM-48 (B) modified with copper (100 mg of sample, the sample was saturated at 70 °C in a flow of gas mixture containing 1.0 V% of NH3, 99 V% He for 2.5 h; after ammonia physiosorbed molecules removal, NH3 desorption was performed with with linear heating rate of 10 °C/min in a flow of pure He)
Temperature-programmed reduction with hydrogen (H2-TPR) is very useful experimental method for distinguishing between aggregated and monomeric copper species. Cu2+ cations in copper oxide species are reduced in one step directly to Cu0 (Cu2+ → Cu0) at relatively low temperatures (typically below 350 °C) [25, 26]. In similar temperature range monomeric Cu2+ cations are reduced to Cu+ (Cu2+ → Cu+), while monomeric Cu+ cations are reduced to Cu0 (Cu+ → Cu0) at higher temperatures (typically above 400 °C) [25, 26]. Reduction profiles, presented in Fig. 6, show the presence of low-temperature maximum centred at about 245–275 °C related to reduction of Cu2+ to Cu0 in aggregated species as well as monomeric Cu2+ cations to Cu+ and high temperature maximum assigned to the second step of monomeric copper cations reduction from Cu+ to Cu0. Integration of the area under reduction peaks and simple recalculation result in estimation of contribution of copper cations in the form of monomeric and aggregated species (Table 3). In the calculation it was assumed that copper in all species present in the samples prior to the H2-TPR runs exist as Cu2+ cations. Deposition of copper into MCM-48 by TIE method resulted in a significantly higher dispersion of this metal comparing to its deposition into MCM-41. Moreover, post-treatment of the samples with ammonia improved copper dispersion in both MCM-41 and MCM-48 silica materials (Table 3). In the case of the MCM-41-Cu sample, with the dominating contribution of aggregated copper species (Table 3), low-temperature reduction maximum is located at temperature higher by about 25–30 °C comparing to other samples containing copper mainly in the form of monomeric cations (Fig. 6). Thus, it could be supposed that reduction for copper cations in aggregated species (Cu2+ → Cu0) occurs at higher temperatures than reduction of monomeric copper cations (Cu2+ → Cu+).

H2-TPR profiles of MCM-41 and MCM-48 modified with copper (25 mg of sample, linear temperature increase of 10 °C/min, 5.0 V% H2 diluted in Ar)
Results of the NH3-SCR catalytic tests in dry and wet atmosphere are shown in Fig. 7A and B, respectively. The NO conversion with ammonia in the absence of water vapour starts at about 100 °C, increases to about 225 °C reaching the level of 85–95% and decreases above 325 °C due to the side process of direct ammonia oxidation by oxygen present in the reaction mixture (Fig. 7A). The catalysts containing mainly highly dispersed forms of copper, especially MCM-48-Cu-A, presented higher catalytic activity both in the low- and high-temperature range comparing to MCM-41-Cu containing mainly aggregated copper oxide species. The NO conversion above 90% with selectivity to N2 above 97% was obtained in the temperature window from 225 to 325 °C for the MCM-48-Cu-A, MCM-41-Cu-A and MCM-48-Cu-A catalysts containing copper mainly in the form of monomeric copper cations. Thus, the obtained results support hypothesis about high catalytic activity of monomeric copper cations in the low-temperature NH3-SCR process [27]. To verify activity of the catalysts in the side reaction of direct ammonia oxidation, limiting efficiency of the NH3-SCR process at higher temperatures, additional tests of ammonia oxidation were done (Fig. 8). As can be seen, the MCM-41-Cu, containing copper mainly in the form of aggregated copper oxide species, is significantly more active in direct ammonia oxidation comparing to other catalysts containing copper mainly in the form of monomeric copper cations. Thus, it is supposed that aggregated copper oxide species are more active in ammonia oxidation comparing to highly dispersed copper species. Comparison of the NH3-SCR tests conducted in the absence (Fig. 7A) and presence (Fig. 7B) of water vapour shows that efficiency of the reaction in wet reaction mixture is limited mainly in the lower temperatures, possibly due to competitive adsorption of ammonia and water molecules for these same sites. Similarly, the lower activity in the side reaction of direct ammonia oxidation, observed in the wet reaction mixture (Fig. 7B), is a result of such competition.

Results of NH3-SCR tests in dry (A) and wet (B) atmosphere (100 mg of sample, composition of dry atmosphere: 0.25 V% of NO, 0.25 V% of NH3 and 2.5 V% of O2, 97 V% He, composition of wet atmosphere: 0.25 V% of NO, 0.25 V% of NH3, 2.5 V% of O2, 5 V% H2O and 92 V% He)

Results of NH3-SCO tests (100 mg of sample, 0.5 V% of NH3, 2.5 V% of O2 and 97 V% He)
Conclusions
The applicability of template ion-exchange (TIE) method for deposition metal species into MCM-48 was verified for the first time. It was shown that deposition of copper into MCM-48 by TIE method resulted in introduction of this metal in highly dispersed forms, mainly monomeric copper cations. In contrast, applying this same method for deposition of copper into MCM-41 resulted mainly in CuO aggregates. Post-treatment of the samples with ammonia solution, directly after TIE, resulted in an increased contribution of monomeric copper cations both in MCM-41 and MCM-48. This effect was more significant for MCM-41 (increase of monomeric cations contribution from 40 to 75% after ammonia posttreatment). Unfortunately, treatment of the silica samples with ammonia resulted in partial destroying of their porous structure. Mesoporous silicas containing highly dispersed forms of copper (samples modified with copper by TIE method including post-treatment with ammonia solution) were found to be significantly more catalytically active in the low-temperature NH3-SCR process comparing to the catalysts containing CuO aggregates. The NO conversion above 90% with selectivity to N2 above 97% was obtained in the temperature window from 225 to 325 °C for the catalysts containing copper mainly in the form of monomeric copper cations. On the other hand, copper oxide aggregates presented activity in the side process of direct ammonia oxidation and therefore decrease of the NH3-SCR efficiency at higher temperatures was significantly limited for the catalysts containing such aggregated species. Introduction of water vapour into the reaction mixture resulted in a shift of the NO conversion profiles into higher temperatures by about 30–50 °C. Possibly, this effect is related to the competition of ammonia and water molecules for adsorption on these same sites in the low-temperature range. Presence of water also decreased the efficiency of the side process of direct ammonia oxidation at higher temperatures and therefore improve the NO reduction in this temperature range. The competition of water and ammonia for the same adsorption sites could be also explanation of this effect.
Data availability
On request of those interested.
References
Kresge CT, Leonowicz ME, Roth WJ, Vartuli JC (1992) Beck JS Nature 252:710–712
Kresge CT, Vartuli JC, Roth WJ, Leonowicz ME (2004) Stud Surf Sci Catal 148:53–72
Chen H, Wang Y (2002) Ceram Inter 28:541–547
Kowalczyk A, Święs A, Gil B, Rutkowska M, Piwowarska Z, Borcuch A, Michalik M, Chmielarz L (2018) Appl Catal B Environ 237:927–937
Chmielarz L, Rutkowska M, Kowalczyk A (2018) Adv Inorg Chem 72:323–383
Macina D, Opioła A, Rutkowska M, Basąg S, Piwowarska Z, Michalik M, Chmielarz L (2017) Mater Chem Phys 187:60–71
Hu G, Li W, Xu J, He G, Ge Y, Pan Y, Wang J, Yao B (2016) Mater Lett 170:179–182
Park SS, Chu SW, Shi L, Yuan S, Ha CS (2021) Materials 14:5270
Szczepanik N, Kowalczyk A, Piwowarska Z, Chmielarz L (2022) Catalysts 12:1324
Jibril ZI, Ramli A, Jumbri K (2018) J Jpn Inst Energy 97:200–204
Munnik P, de Jongh PE, de Jong KP (2015) Chem Rev 115:6687–6718
Chmielarz L, Kuśtrowski P, Dziembaj R, Cool P, Vansant EF (2010) Micropor Mesopor Mat 127:133–141
Chmielarz L, Kuśtrowski P, Drozdek M, Rutkowska M, Dziembaj R, Michalik M, Cool P, Vansant EF (2011) J Porous Mat 18(4):483–491
Kowalczyk A, Piwowarska Z, Macina D, Kuśtrowski P, Rokicińska A, Michalik M, Chmielarz L (2016) Chem Eng J 295:167–180
Jankowska A, Chłopek A, Kowalczyk A, Rutkowska M, Mozgawa W, Michalik M, Liu S, Chmielarz L (2021) Micropor Mesopor Mater 315:110920
Kowalczyk A, Borcuch A, Michalik M, Rutkowska M, Gil B, Sojka Z, Indyka P, Chmielarz L (2017) Micropor Mesopor Mater 240:9–21
Machowski K, Natkański P, Białas A, Kuśtrowski P (2016) J Therm Anal Calorim 126:1313–1322
Wu G, Jiang S, Li L, Zhang F, Yang Y, Guan N, Mihaylov M, Knözinger H (2010) Micropor Mesopor Mater 135:2–8
Solovyov LA, Belousov OV, Dinnebier RE, Shmakov AN, Kirik SD (2005) J Phys Chem B 109:3233–3237
Leofanti G, Padovan M, Tozzola G, Venturelli B (1998) Catal Today 41:207–219
Thommes M, Kaneko K, Neimark AV, Olivier JP, Rodriguez-Reinoso F, Rouquerol J, Sing KSW (2015). Pure Appl Chem. https://doi.org/10.1515/pac-2014-1117
Martins L, Peguin RPS, Wallau M, Urquieta EA (2004) Stud Surf Sci Catal 154:2475–2483
Rutkowska M, Piwowarska Z, Micek E, Chmielarz L (2015) Micropor Mesopor Mater 209:54–65
Kumar MS, Schwidder M, Grunet W, Bruckner A (2004) J Catal 227:384–397
Huo Ch, Ouyang J, Yang H (2014) Sci Rep 4:3682
Jankowska A, Chłopek A, Kowalczyk A, Rutkowska M, Michalik M, Liu S, Chmielarz L (2020) Molecules 25:5651
Jankowska A, Bobowska A, Gil B, Chmielarz L (2023). ChemNanoMat. https://doi.org/10.1002/cnma.202300167
Acknowledgements
The studies were carried out in the frame of project 2018/31/B/ST5/00143 from the National Science Centre (Poland).
Funding
Funding was supported by National Science Centre (Poland), 2018/31/B/ST5/00143.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pietraszek, A., Kowalczyk, A., Piwowarska, Z. et al. Mesoporous silicas of MCM-48 and MCM-41 types doped with copper by modified template ion-exchange method as effective catalysts for low-temperature reduction of nitrogen oxide with ammonia. Reac Kinet Mech Cat 136, 2259–2276 (2023). https://doi.org/10.1007/s11144-023-02461-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-023-02461-5