
Abstract
Quinones can accept two electrons and two protons, and are involved in electron transfer and proton transfer reactions in photosynthetic reaction centers. To date, the pK a of these quinones in aqueous solution have not been reported. We calculated the pK a of the initial protonation (Q·− to QH·) and the second protonation (QH− to QH2) of 1,4-quinones using a quantum chemical approach. The calculated energy differences of the protonation reactions Q·− to QH· and QH− to QH2 in the aqueous phase for nine 1,4-quinones were highly correlated with the experimentally measured pK a(Q·−/QH·) and pK a(QH−/QH2), respectively. In the present study, we report the pK a(Q·−/QH·) and pK a(QH−/QH2) of ubiquinone, menaquinone, phylloquinone, plastoquinone, and rhodoquinone in aqueous solution.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Quinones can accept two electrons and two protons via the initial protonation of semiquinone, Q·− to QH·, and second protonation of hydroquinone, QH− to QH2. Ubiquinone serves as an electron acceptor at the QA and QB binding sites in reaction centers of purple bacteria (PbRC) from Rhodobacter sphaeroides (Fig. 1). Similarly, menaquinone (vitamin K2) is the acceptor at the QA site in PbRC from Blastochloris viridis, while phylloquinone (vitamin K1) is the active center at the A1A and A1B sites in photosystem I (PSI). In reaction centers of green non-sulfur bacteria from Chloroflexus aurantiacus, menaquinones are also located at both the QA and QB sites (Hale et al. 1983). It should be noted that phylloquinone and menaquinone have the same head-group structure (Fig. 2). Plastoquinone serves as an electron acceptor at the QA and QB sites in photosystem II (PSII) (Fig. 1) (Robinson and Crofts 1984; Rutherford et al. 1984; Okamura et al. 2000; Brettel and Leibl 2001; Wraight 2004). Rhodoquinone is a required cofactor for anaerobic respiration in Rhodospirillum rubrum (Okayama et al. 1968). Because rhodoquinone is assumed to have a higher pK a(Q·−/QH·) than ubiquinone, rhodoquinone-substituted PbRC has been used to investigate the mechanism of proton transfer to QB (e.g., Graige et al. 1999; Maroti et al. 2015).

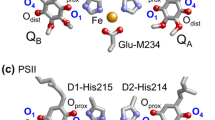
Quinones in photosynthetic reaction centers: a menaquinone as QA and ubiquinone as QB in bacterial photosynthetic reaction centers from Blastochloris viridis (Blc. viridis, PDB ID: 2I5N) (Li et al. 2006); b ubiquinone as QA and QB in bacterial photosynthetic reaction centers from Rhodobacter sphaeroides (Rb. sphaeroides, PDB ID: 3I4D), where the 2-methoxy group of each ubiquinone is red circled; c plastoquinone as QA and QB in PSII (PDB ID: 3ARC) (Umena et al. 2011); and d phylloquinone as A1A and A1B in PSI (PDB ID:1JB0) (Jordan et al. 2001). Red and blue balls indicate O and N atoms, respectively. In PbRC and PSII, Oprox and Odist stand for O atoms of the quinones at the proximal and distal positions with respect to the non-heme Fe complex, respectively. Note that except for QB in PSII, Oprox is O1 and Odist is O4 in PbRC and PSII

Molecular structures of a ubiquinone (n = 10), b menaquinone/phylloquinone (n = 3–9), c plastoquinone (n = 6–9), and d rhodoquinone (n = 10), where n is the number of isoprene units
In PbRC and PSII, QA/QA ·− acts as a one-electron redox couple and donates an electron to the second quinone QB, without undergoing protonation itself. In contrast, QB reduction involves two consecutive one-electron reduction reactions with a series of associated proton uptake reactions (reviewed in Diner and Rappaport 2002; Renger and Renger 2008; Holzwarth 2008; Cardona et al. 2012; Muh et al. 2012; Petrouleas and Crofts 2005). Both QA and QB are located near the non-heme Fe2+ and the ligands to the Fe2+ (i.e., His-L190 and His-M217 (or M219) in PbRC and D1-His215 and D2-His214 in PSII) donate an H-bond to the carbonyl O atoms of quinones that are nearer to the Fe complex (Oprox) (Fig. 1a–c). The carbonyl O atoms of quinones at the distal position (Odist) also form H-bonds with the proteins. On the other hand, in PSI, the non-heme Fe2+ is absent, but the Fe4S4 cluster FX is located near the two A1 binding sites (Fig. 1d). For many 1,4-quinones, experimentally measured pK a(Q·−/QH·) in aqueous solution were summarized by Swallow (1982) and experimentally measured pK a(QH−/QH2) in aqueous solution were reported by Bishop and Tong (1965).
As far as we are aware, experimentally measured pK a(Q·−/QH·) and pK a(QH−/QH2) for ubiquinone, menaquinone, plastoquinone, and rhodoquinone in aqueous solution have not been reported, because of their insoluble hydrophobic isoprene side-chains (Fig. 2). The pK a(Q·−/QH·) for ubiquinone was roughly estimated to be 4.9 in aqueous solution (Swallow 1982) or measured to be 6.5 in methanol (Land and Swallow 1970; Swallow 1982). In theoretical studies by Cape et al. (2006), pK a(Q·−/QH·) for ubiquinone, plastoquinone, and rhodoquinone were calculated to be 5.35, 4.86, and 5.09, respectively (Cape et al. 2006), although pK a(Q·−/QH·) of rhodoquinone is assumed to be higher than pK a(Q·−/QH·) of ubiquinone (Graige et al. 1999). The pK a(QH−/QH2) for ubiquinone were measured to be 13.3 in 80% ethanol (Morrison et al. 1982). On the other hand, to calculate pK a(QH−/QH2) of QB in PbRC from R. sphaeroides, Zhu and Gunner used a pK a(QH−/QH2) of 10.7 for ubiquinone in aqueous solution in their theoretical studies (Zhu and Gunner 2005). Reliable pK a(Q·−/QH·) and pK a(QH−/QH2) values for ubiquinone, menaquinone, and plastoquinone in aqueous solution are required for understanding the mechanisms of reactions involving quinones in PbRC, PSI, and PSII. Here, we report the pK a(Q·−/QH·) and pK a(QH−/QH2) for ubiquinone, menaquinone, phylloquinone, and plastoquinone in aqueous solution, obtained by adopting a quantum chemical approach.
Computational procedures
In the deprotonation reaction of the protonated state (AH) to deprotonated state (A−) in aqueous solution, pK a is defined as
where ΔG aq is the free energy difference between (AH) and (A− + H+) (i.e., ΔG aq = G aq(A−) − G aq(AH) + G aq(H+)), R is the gas constant, and T is the temperature. ΔG aq can also be approximated as
where k is the scaling factor, ΔE QM/PCM is the energy difference between AH and A− in aqueous phase (i.e., ΔE QM/PCM = E QM/PCM(A−) − E QM/PCM(AH)), which can be calculated using a quantum chemical (QM) approach with the polarizable continuum model (PCM) method, and C is the constant [“simple pK a estimation with energy of the optimized geometry scheme” (Matsui et al. 2012)]. If pK a of molecules are obtained at the same temperature, Eq. 1 can be written into Eq. 3 using Eq. 2,
where k′ is the scaling factor and C′ is constant. To determine k′ and C′, we calculated ΔE QM/PCM for nine 1,4-quinones whose experimentally measured pK a(Q·−/QH·) (Swallow 1982) and pK a(QH−/QH2) (Bishop and Tong 1965) are reported. We employed the unrestricted DFT method with the B3LYP functional and 6-31g++** basis sets, using the Gaussian (Frisch et al. 2004) program code with the PCM method, i.e., water molecules were considered implicitly. This treatment is more appropriate to describe H-bonds between quinones and bulk water molecules, in which the H-bond patterns are not unique. We evaluated all possible conformations of each protonated quinone (i.e., QH· and QH2) regarding the –OH group orientation, and we took the energetically lowest conformation as the relevant structure. In the resulting structures, the –OH group was essentially in the plane of the quinone ring (SI Dataset 1 for atomic coordinates). Because the isoprene units are not composed of conjugated double bonds, the isoprene side-chain length n (Fig. 2) was set to 1 for the calculations of ubiquinone, menaquinone, phylloquinone, and plastoquinone. This could also reduce the number of the possible conformations. In fact, the length of the ubiquinone does not practically affect the energetics of ubiquinones, as demonstrated by the experimentally measured redox potential values of ubiquinone-1 and -10 in dimethylformamide being −611 and −602 mV versus saturated calomel reference electrode, respectively, essentially no difference (Prince et al. 1983).
Results and discussion
Correlation of calculated energies with pK a for 1,4-quinones
The calculated ΔE QM/PCM for deprotonation of QH· to Q·− for nine 1,4-quinones were highly associated with the experimentally measured pK a(Q·−/QH·) ranging from 4.0 to 5.1 (summarized in Swallow 1982), which was best fitted in the following equations (Fig. 3):

Correlation between experimentally measured pK a(Q·−/QH·) and calculated ΔE QM/PCM (coefficient of determination R 2 = 0.99). ΔE QM/PCM can be calculated using a quantum chemical approach with the PCM method. The solid line was drawn according to Eq. 4
The term of −268.48 kcal/mol corresponds to the proton solvation energy, which typically ranges from −252.6 to −271.7 kcal/mol [see Schmidt am Busch and Knapp (2004) and references therein]. Using Eq. 4, the calculated pK a(Q·−/QH·) for nine 1,4-quinones are listed in Table 1, which confirms that Eq. 4 can reproduce the experimentally measured pK a(Q·−/QH·). It should be noted that the accuracy of the experimentally measured pK a values is generally considered to be within 0.2 units (Swallow 1982).
Symmetrically shaped quinones (e.g., benzoquinone and 2,3-dimethylbenzoquinone) had a single pK a value, whereas asymmetrically shaped quinones (e.g., methylbenzoquinone and 2,6-dimethylbenzoquinone) had two distinguishable pK a values (Table 1). The difference in pK a values is caused by the difference in the chemical environment of the two O sites (O1 and O4). For example, the calculated pK a(Q·−/QH·) of the O4 site in 2,6-dimethylbenzoquinone was larger than the calculated pK a(Q·−/QH·) of the O1 site by 0.46 (Table 1), because the protonation at O1 (i.e., the formation of –OH at O1) increases the steric hindrance with the two methyl groups, which results in a decrease in pK a at O1. Since protonation of Q·− to QH· occurs predominantly at one of the two O sites with a higher pK a(Q·−/QH·), the higher pK a(Q·−/QH·) can be considered to be relevant for asymmetrically shaped quinones and are used to obtain Eq. 4.
The calculated ΔE QM/PCM for deprotonation of QH2 to QH− for nine 1,4-quinones were also highly correlated with the experimentally measured pK a(QH−/QH2) ranging from 9.85 to 11.25 (Bishop and Tong 1965), which was best fitted in the following equations (Fig. 4):

Correlation between experimentally measured pK a(QH−/QH2) and calculated ΔE QM/PCM (coefficient of determination R 2 = 0.96). ΔE QM/PCM can be calculated using a quantum chemical approach with the PCM method. The solid line is drawn according to Eq. 5
The pK a(QH−/QH2) calculated for nine quinones using Eq. 5 are listed in Table 2, which confirms that Eq. 5 can reproduce the experimentally measured pK a(QH−/QH2) when ΔE QM/PCM can be calculated appropriately.
pK a(Q·−/QH·) and pK a(QH−/QH2) for ubiquinone, menaquinone, phylloquinone, plastoquinone, and rhodoquinone in aqueous solution
Using Eqs. 4 and 5, we also calculated pK a(Q·−/QH·) and pK a(QH−/QH2) for ubiquinone, menaquinone, phylloquinone, and plastoquinone in aqueous solution, respectively. The calculated pK a(Q·−/QH·) were 5.31 for ubiquinone, 4.92 for menaquinone/phylloquinone, and 5.11 for plastoquinone in aqueous solution (Table 1). pK a(Q·−/QH·) did not significantly differ between O1 and O4 in plastoquinone and ubiquinone (Table 1). This result suggests that which of the two O sites, O1 and O4, serves as the initial (i.e., Q·− to QH·) and second (i.e., QH− to QH2) protonation sites of QB in PbRC and PSII, is predominantly determined by the protein environments.
The calculated pK a(Q·−/QH·) for ubiquinone was closer to the value of 4.9 roughly estimated for aqueous solution (Swallow 1982) than the value of 6.5 measured in methanol (Land and Swallow 1970; Swallow 1982). Because methanol (dielectric constant = 33) is less polar than water (dielectric constant = 80), the negatively charged Q·− state is less stable in methanol than in water; this could explain the high pK a(Q·−/QH·) in methanol with respect to water.
In theoretical studies by Cape et al., the calculated pK a(Q·−/QH·) of 5.09 for rhodoquinone was lower than their calculated pK a(Q·−/QH·) of 5.35 for ubiquinone (Cape et al. 2006). In contrast, in the present study, the calculated pK a(Q·−/QH·) of rhodoquinone was significantly high, 5.78 (Table 1). It should be noted that pK a(Q·−/QH·) of rhodoquinone is assumed to be higher than pK a(Q·−/QH·) of ubiquinone (e.g., Graige et al. 1999), which is consistent with the present result.
The calculated pK a(QH−/QH2) of 10.86 for ubiquinone (Table 2) is very close to pK a(QH−/QH2) of 10.8 measured for trimethylbenzoquinone (Bishop and Tong 1965). Zhu and Gunner considered pK a(QH−/QH2) for trimethylbenzoquinone being more relevant to the pK a(QH−/QH2) for ubiquinone in aqueous solution (Zhu and Gunner 2005) than pK a(QH−/QH2) = 13.3 in 80% ethanol (Morrison et al. 1982). The present study supports their conclusion. The calculated pK a(QH−/QH2) of 10.86 for ubiquinone is also close to the calculated pK a(QH−/QH2) of 10.74 for plastoquinone (Table 2), which might be associated with the similarity in the QB protonation events for PbRC and PSII (Robinson and Crofts 1984; Rutherford et al. 1984; Okamura et al. 2000; Wraight 2004; Ishikita and Knapp 2005).
Influence of the 2-methoxy group orientation in pK a(Q·−/QH·) for ubiquinone
It was proposed that difference in the 2-methoxy orientation of ubiquinone (Fig. 1b) was responsible for (i) the E m difference of more than 160 mV between QA and QB in PbRC (Taguchi et al. 2013) or (ii) the quantum chemically obtained electron affinity difference of more than 170 meV between QA and QB in PbRC (de Almeida et al. 2014). On the other hand, FTIR studies suggested that the QA binding to PbRC (i.e., interaction between QA and the PbRC protein environment) was affected by the H-bond interaction with the protein environment but not by the methoxy orientation (Remy et al. 2003). In the present study, using the crystal structure (PDB: 3I4D; Fig. 1b) whose different 2-methoxy orientations between QA and QB were highlighted in ref. (de Almeida et al. 2014), pK a(Q·−/QH·) were calculated. The calculated pK a(Q·−/QH·) were the same, 5.30 at the O4 site in the QA conformation and 5.31 at the O4 site in the QB conformation in water (Table S1), irrespective of the different 2-methoxy orientations in the quantum chemically optimized quinone structures (SI Dataset 2 for atomic coordinates). In the protonated QH· state, the energetically lowest conformations showed that the –OH group was oriented toward the 2-methoxy O atom (–O–H⋯Omethoxy angle = 116° and –O⋯Omethoxy distance = 2.1 Å in both QA and QB conformations). When the –OH group was oriented away from the 2-methoxy O atom, the calculated pK a(Q·−/QH·) were lowered by ~0.6 to 1 pK a unit from the pK a(Q·−/QH·) value of the energetically lowest conformation (Table S1). In the quantum chemically optimized QA and QB conformations, the 2-methoxy group was out of the quinone ring, as identified in the crystal structure (PDB: 3I4D). These results suggest that pK a(Q·−/QH·) is more affected by the difference in the –OH orientation than the difference in the 2-methoxy orientation. The influence of the 2-methoxy orientation might be more pronounced when calculated in vacuum (i.e., in the absence of the protein environment), where H-bond partners of ubiquinone (e.g., bulk water molecules and protein environments) are absent. In vacuum, alteration in the molecular configuration is the only way to alter the stability of the Q·− and QH· states. It should be noted that the influence of –OH orientation can be ignored when considering E m(Q/Q·−), because –OH is absent in both the Q and Q·− states.
Evolutionary transition of quinones in photosynthetic reaction centers
Among all of the 1,4-quinones investigated here, plastoquinone and ubiquinone employed at QB in PbRC and PSII have the second and third largest pK a(Q·−/QH·) (Table 1). This might be a reason why nature uses these quinones exclusively as the terminal electron acceptor QB in PbRC and PSII. The large pK a(Q·−/QH·) would result in a larger population of QH·. This will then enable it to accept the second electron, leading to the fully protonated QBH2 state. Thus, the large pK a(Q·−/QH·) is advantageous in fixing the two transferred electrons using protonation in the form of QBH2. Remarkably, QA in PbRC from B. viridis and A1A and A1B in PSI, which serve as electron donors (to QB and the Fe4S4 cluster FX, respectively, Fig. 1) without altering the protonation states, are menaquinone/phylloquinone with lower pK a(Q·−/QH·) = 4.9 (Table 1).
Menaquinone is assumed to represent the ancestral type of quinones in bioenergetics systems, whereas ubiquinone and plastoquinone are assumed to represent “more recent” quinones (Schoepp-Cothenet et al. 2009). Intriguingly, E m(Q/Q·−) of the “more recent” quinones are higher [by ~100 mV (Prince et al. 1983)] than E m(Q/Q·−) of the “ancestral” quinone. Reduced menaquinone becomes rapidly oxidized in the presence of O2, whereas ubiquinone and plastoquinone are less sensitive toward oxygen, minimizing loss of the unproductive reduced power (Schoepp-Cothenet et al. 2009); it seems plausible that the difference in E m(Q/Q·−) is associated with rising levels of dioxygen 2.5 billion years ago. In most PbRC, the tightly bound cofactor at the QA site can be either menaquinone or ubiquinone, whereas the cofactor at the terminal electron acceptor QB site is ubiquinone [except for, e.g., PbRC from Halorhodospira halophile, which has menaquinone at both the QA and QB site (Schoepp-Cothenet et al. 2009)]. The higher E m(Q/Q·−) of ubiquinone at the QB site would contribute to the driving force of the electron transfer from QA to QB when menaquinone is the cofactor at the QA site (e.g., B. viridis). The present results show that the “more recent” quinones, ubiquinone and plastoquinone, have larger pK a(Q·−/QH·) and pK a(QH−/QH2) values than the “ancestral” quinone, menaquinone/phylloquinone (Tables 1, 2), which is also consistent with a role of QB, fixation of two electrons and two protons in the form of QBH2.
In photosynthetic reaction centers of the green non-sulfur bacteria (e.g., Chloroflexus aurantiacus), which is assumed to have evolved first as the type-II reaction center (Gupta 2003), menaquinone occupies at both the QA and QB sites (Hale et al. 1983). One of the simplest ways to generate the driving force for electron transfer between the identical menaquinone cofactors would be to alter the solvent accessibility between the QA and QB sites. As the quinone-binding site is more exposed to the bulk water region, it can stabilize the Q·− state with respect to Q state, which results in an increase in E m(Q), i.e., the case for E m(QB), even if the amino acid sequences near the QA and QB binding sites were similar. In addition, the exposure of the quinone-binding site makes (P)bRC easier to hire external quinones. The effort to make the solvent accessibility of the quinone-binding sites different between the two quinone-binding sites in the homodimeric type-I reaction center would lead to formation of the heterodimeric type-II reaction center with E m(QB) > E m(QA) and might also explain why the cofactor can be ubiquinone at the QB site in most PbRC.
Conclusions
Experimentally measured pK a(Q·−/QH·) (Swallow 1982) and pK a(QH−/QH2) (Bishop and Tong 1965) values of nine 1,4-quinones in aqueous solution were highly correlated with the quantum chemically calculated energy differences (ΔE QM/PCM) between the protonated and deprotonated states (Figs. 3, 4). They can be best fitted to Eqs. 4 and 5, respectively. The calculated pK a(Q·−/QH·) values were 5.31 for ubiquinone, 4.92 for menaquinone/phylloquinone, 5.11 for plastoquinone, and 5.78 for rhodoquinone in aqueous solution (Table 1). pK a(Q·−/QH·) for plastoquinone and ubiquinone in aqueous solution are the largest among all of the 1,4-quinones but rhodoquinone listed in Table 1, partially explaining why nature employs these two 1,4-quinones specifically as the terminal electron acceptors QB in PbRC and PSII.
In PbRC and PSII, the initial protonation of Q·− to QH· predominantly occurs at the distal carbonyl site Odist of QB (Fig. 1) (Okamura et al. 2000; Wraight 2004; Saito et al. 2013). The pK a(Q·−/QH·) at the O1 and O4 sites did not differ significantly for each quinone (Table 1), suggesting that the protein environments predominantly determine the initial protonation O site of QB.
The pK a(QH−/QH2) values were calculated to be 10.86 for ubiquinone, 9.16 for menaquinone/phylloquinone, 10.74 for plastoquinone, and 9.81 for rhodoquinone in aqueous solution using Eq. 5 (Table 2). The calculated pK a(QH−/QH2) for ubiquinone was closer to the experimentally measured value of 10.8 for trimethylbenzoquinone (Bishop and Tong 1965) than the experimentally measured pK a(QH−/QH2) of 13.3 for ubiquinone in 80% ethanol (Morrison et al. 1982), as already pointed out (Zhu and Gunner 2005).
The pK a(Q·−/QH·) and pK a(QH−/QH2) for ubiquinone, menaquinone/phylloquinone, and plastoquinone in aqueous solution, which were determined in the present study, will help understand the mechanisms of quinone-mediated reactions in photosynthetic reaction centers, such as formation of QBH2 in PbRC and PSII (Robinson and Crofts 1984; Rutherford et al. 1984; Okamura et al. 2000; Wraight 2004; Saito et al. 2013), formation of QAH2 in PSII (van Mieghem et al. 1995; Noguchi 2002), and evolutionary relationship between the type-I and type-II photosynthetic reaction centers (Gupta 2003; Rutherford and Faller 2003; Schoepp-Cothenet et al. 2009).
References
Adams GE, Michael BD (1967) Pulse radiolysis of benzoquinone and hydroquinone. Semiquinone formation by water elimination from trihydroxy-cyclohexadienyl radicals. Trans Faraday Soc 63(0):1171–1180. doi:10.1039/tf9676301171
Bishop CA, Tong LKJ (1965) Equilibria of substituted semiquinones at high pH. J Am Chem Soc 87:501–505
Brettel K, Leibl W (2001) Electron transfer in photosystem I. Biochim Biophys Acta 1507:100–114
Cape JL, Bowman MK, Kramer DM (2006) Computation of the redox and protonation properties of quinones: towards the prediction of redox cycling natural products. Phytochemistry 67(16):1781–1788. doi:10.1016/j.phytochem.2006.06.015
Cardona T, Sedoud A, Cox N, Rutherford AW (2012) Charge separation in photosystem II: a comparative and evolutionary overview. Biochim Biophys Acta 1817(1):26–43
de Almeida WB, Taguchi AT, Dikanov SA, Wraight CA, O’Malley PJ (2014) The 2-methoxy group orientation regulates the redox potential difference between the primary (QA) and secondary (QB) quinones of Type II bacterial photosynthetic reaction centers. J Phys Chem Lett 5(15):2506–2509. doi:10.1021/jz500967d
Diner BA, Rappaport F (2002) Structure dynamics, and energetics of the primary photochemistry of photosystem II of oxygenic photosynthesis. Annu Rev Plant Biol 53:551–580
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr., Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004). Gaussian 03. Gaussian, Inc., Wallingford
Graige MS, Paddock ML, Feher G, Okamura MY (1999) Observation of the protonated semiquinone intermediate in isolated reaction centers from Rhodobacter sphaeroides: implications for the mechanism of electron and proton transfer in proteins. Biochemistry 38(35):11465–11473. doi:10.1021/bi990708u
Gupta RS (2003) Evolutionary relationships among photosynthetic bacteria. Photosynth Res 76(1–3):173–183. doi:10.1023/A:1024999314839
Hale MB, Blankenship RE, Fuller RC (1983) Menaquinone is the sole quinone in the facultatively aerobic green photosynthetic bacterium Chloroflexus aurantiacus. Biochimica et Biophysica Acta 723(3):376–382. doi:10.1016/0005-2728(83)90044-0
Holzwarth AR (2008) Ultrafast primary reactions in the photosystems of oxygen-evolving organisms. In: Braun M, Gilch P, Zinth W (eds) Ultrashort laser pulses in biology and medicine. Springer, Dordrecht, pp 141–164
Ishikita H, Knapp E-W (2005) Control of quinone redox potentials in photosystem II: electron transfer and photoprotection. J Am Chem Soc 127:14714–14720
Jordan P, Fromme P, Witt HT, Klukas O, Saenger W, Krauβ N (2001) Three-dimensional structure of cyanobacterial photosystem I at 2.5 Å resolution. Nature 411:909–917
Land EJ, Swallow AJ (1970) One-electron reactions in biochemical systems as studied by pulse radiolysis. 3. Ubiquinone. J Biol Chem 245(8):1890–1894
Li L, Mustafi D, Fu Q, Tereshko V, Chen DL, Tice JD, Ismagilov RF (2006) Nanoliter microfluidic hybrid method for simultaneous screening and optimization validated with crystallization of membrane proteins. Proc Natl Acad Sci USA 103(51):19243–19248. doi:10.1073/pnas.0607502103
Maroti A, Wraight CA, Maroti P (2015) Protonated rhodosemiquinone at the QB binding site of the M265IT mutant reaction center of photosynthetic bacterium Rhodobacter sphaeroides. Biochemistry 54(12):2095–2103. doi:10.1021/bi501553t
Matsui T, Baba T, Kamiya K, Shigeta Y (2012) An accurate density functional theory based estimation of pK a values of polar residues combined with experimental data: from amino acids to minimal proteins. Phys Chem Chem Phys 14(12):4181–4187
Morrison LE, Schelhorn JE, Cotton TE, Bering CL, Loach PA (1982) Electrochemical and spectral properties of ubiquinone and synthetic analogs: relevance to bacterial photosynthesis. In: Trumpower BL (ed) Function of quinones in energy conserving systems. Academic Press, New York, pp 35–58
Muh F, Glockner C, Hellmich J, Zouni A (2012) Light-induced quinone reduction in photosystem II. Biochim Biophys Acta 1817(1):44–65
Noguchi T (2002) Dual role of triplet localization on the accessory chlorophyll in the photosystem II reaction center: photoprotection and photodamage of the D1 protein. Plant Cell Physiol 43:1112–1116
Okamura MY, Paddock ML, Graige MS, Feher G (2000) Proton and electron transfer in bacterial reaction centers. Biochim Biophys Acta 1458:148–163
Okayama S, Yamamoto N, Nishikawa K, Horio T (1968) Roles of ubiquinone-10 and rhodoquinone in photosynthetic formation of adenosine triphosphate by chromatophores from Rhodospirillum rubrum. J Biol Chem 243(11):2995–2999
Patel KB, Willson RL (1973) Semiquinone free radicals and oxygen. Pulse radiolysis study of one electron transfer equilibria. J Chem Soc Faraday Trans 1 69(0):814–825. doi:10.1039/f19736900814
Petrouleas V, Crofts AR (2005) The quinone iron acceptor complex. In: Wydrzynski T, Satoh K (eds) Photosystem II: the water/plastoquinone oxido-reductase in photosynthesis. Springer, Dordrecht, pp 177–206
Prince RC, Dutton PL, Bruce JM (1983) Electrochemistry of ubiquinones: menaquinones and plastoquinones in aprotic solvents. FEBS Lett 160:273–276
Rao PS, Hayon E (1973) Ionization constants and spectral characteristics of some semiquinone radicals in aqueous solution. J Phys Chem 77:2274–2276
Remy A, Boers RB, Egorova-Zachernyuk T, Gast P, Lugtenburg J, Gerwert K (2003) Does different orientation of the methoxy groups of ubiquinone-10 in the reaction centre of Rhodobacter sphaeroides cause different binding at QA and QB? Eur J Biochem 270(17):3603–3609
Renger G, Renger T (2008) Photosystem II: the machinery of photosynthetic water splitting. Photosynth Res 98(1–3):53–80
Robinson HH, Crofts AR (1984) Kinetics of proton uptake and the oxidation-reduction reactions of the quinone acceptor complex of photosystem II from pea chloroplasts. In: Sybesma C (ed) Advances in photosynthesis research, vol 1. Martinus Nijhoff/Dr. W. Junk Publishers, The Hague, pp 477–480
Rutherford AW, Faller P (2003) Photosystem II: evolutionary perspectives. Phil Trans R Soc Lond B 358:245–253
Rutherford AW, Renger G, Koike H, Inoue Y (1984) Thermoluminescence as a probe of photosystem II. The redox and protonation states of the secondary acceptor quinone and the O2-evolving enzyme. Biochim Biophys Acta 767(3):548–556
Saito K, Rutherford AW, Ishikita H (2013) Mechanism of proton-coupled quinone reduction in Photosystem II. Proc Natl Acad Sci USA 110(3):954–959
Schmidt am Busch M, Knapp E-W (2004) Accurate pK a determination for a heterogeneous group of organic molecules. ChemPhysChem 5:1513–1522
Schoepp-Cothenet B, Lieutaud C, Baymann F, Vermeglio A, Friedrich T, Kramer DM, Nitschke W (2009) Menaquinone as pool quinone in a purple bacterium. Proc Natl Acad Sci USA 106(21):8549–8554. doi:10.1073/pnas.0813173106
Steenken S, O’Neill P (1977) Oxidative demethoxylation of methoxylated phenols and hydroxybenzoic acids by the hydroxyl radical. An in situ electron spin resonance, conductometric pulse radiolysis and product analysis study. J Phys Chem 81:505–508
Swallow AJ (1982) Physical chemistry of semiquinones. Trumpower BL (ed) Function of quinones in energy conserving systems. Academic Press, New York, pp 59–72
Taguchi AT, Mattis AJ, O’Malley PJ, Dikanov SA, Wraight CA (2013) Tuning cofactor redox potentials: the 2-methoxy dihedral angle generates a redox potential difference of >160 mV between the primary (QA) and secondary (QB) quinones of the bacterial photosynthetic reaction center. Biochemistry 52(41):7164–7166. doi:10.1021/bi4011896
Umena Y, Kawakami K, Shen J-R, Kamiya N (2011) Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 473:55–60
van Mieghem F, Brettel K, Hillman B, Kamlowski A, Rutherford AW, Schlodder E (1995) Charge recombination reactions in photosystem II. 1. Yields, recombination pathways, and kinetics of the primary pair. Biochemistry 34:4798–4813
Willson RL (1971) Semiquinone free radicals: determination of acid dissociation constants by pulse radiolysis. J Chem Soc D 20:1249–1250. doi:10.1039/c29710001249
Wraight CA (2004) Proton and electron transfer in the acceptor quinone complex of photosynthetic reaction centers from Rhodobacter sphaeroides. Front Biosci 9:309–337
Zhu Z, Gunner MR (2005) Energetics of quinone-dependent electron and proton transfers in Rhodobacter sphaeroides photosynthetic reaction centers. Biochemistry 44:82–96
Acknowledgements
This research was supported by JST CREST (JPMJCR1656 to H.I.), JSPS KAKENHI (JP26800224 to K.S., JP15H00864, JP16H06560, JP26105012, and JP26711008 to H.I., and JP22740276 to K.S. and H.I.), Materials Integration for engineering polymers of Cross-ministerial Strategic Innovation Promotion Program (SIP, H.I.), and Interdisciplinary Computational Science Program in CCS, University of Tsukuba. Theoretical calculations were partly performed using Research Center for Computational Science, Okazaki, Japan.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hasegawa, R., Saito, K., Takaoka, T. et al. pK a of ubiquinone, menaquinone, phylloquinone, plastoquinone, and rhodoquinone in aqueous solution. Photosynth Res 133, 297–304 (2017). https://doi.org/10.1007/s11120-017-0382-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11120-017-0382-y