
Abstract
Aims and background
The resurrection plant Myrothamnus flabellifolia tolerates complete desiccation and is a great model for studying how plants cope with extreme drought. Root-associated microbes play a major role in stress tolerance and are an attractive target for enhancing drought tolerance in staple crops. However, how these dynamics play out under the most extreme water limitation remains underexplored. This study aimed to identify bacterial and fungal communities that tolerate extreme drought stress in the bulk soil, rhizosphere, and endosphere of M. flabellifolia.
Methods
High-throughput amplicon sequencing was used to characterise the microbial communities associated with M. flabellifolia.
Results
The bacterial phyla that were most abundant across all compartments were Acidobacteriota, Actinobacteriota, Chloroflexota, Planctomycetota, and Pseudomonadota, while the most abundant fungal phyla were Ascomycota and Basidiomycota. Although the bulk soil hosted multiple beneficial root-associated microbes, the rhizosphere compartment showed the highest functional diversity of bacteria and fungi. In contrast, the endosphere exhibited a low abundance and diversity of microbes. These findings share consistent with the theory that M. flabellifolia recruits soil microbes from the bulk to the rhizosphere and finally to the endosphere. It is possible that these microbes could promote drought tolerance in associated plant tissues.
Conclusion
We find that compartments act as the major driver of microbial diversity, but the soil physicochemical factors also influence microbial composition. These results suggest that the root-associated microbiome of M. flabellifolia is highly structured and may aid in plant function.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Many plants are threatened by climate change and fluctuating rainfall patterns (Lizumi and Ramankutty 2016). Although plants have evolved mechanisms to cope with periods of drought, the extreme weather patterns brought on by global change are likely to push some species beyond the limits of their tolerance. However, some unique plants that evolved in habitats with seasonal aridity have developed the ability to tolerate complete desiccation of their vegetative tissues. These species, commonly called resurrection plants, may provide critical insight into how organisms survive the extreme drought that can be used to protect vulnerable species and mitigate drought induced losses (Bewley 1979; Gaff 1989; Hilhorst and Farrant 2018).
Myrothamnus flabellifolia is one of the most iconic resurrection plants and can tolerate desiccation for 9–12 months (Farrant and Kruger 2001). Most of the research on M. flabellifolia has focused on its leaf phytochemical properties, medicinal value, and the physiological and biochemical mechanisms of vegetative desiccation tolerance (Bentley et al. 2019; Erhabor et al. 2020; Marks et al. 2022; Moore et al. 2005, 2006, 2011; Sherwin et al. 1998). However, the belowground mechanisms associated with desiccation tolerance in M. flabellifolia remain largely unknown (Tebele et al. 2021). Processes happening in the soil and roots play a major role in sensing and responding to drought stress. The occurrence of root elongation, branching, and rhizogenesis are drought-adaptive traits in multiple models and crop species (Kato and Okami 2011). However, little is known about parallel processes in resurrection plants, and they may respond to water deficit in a very different way as a result of their unique adaptations to extreme environmental stresses.
Plant-microbe interactions are another critical aspect of underground drought response mechanisms. Plants can be considered holobionts because they are the cumulative association of multiple living species. In addition, plants can actively select microbial populations that are involved in various beneficial processes to inhabit their cells and surfaces, thereby supporting overall holobiont growth and health. For example, soybean primed with arbuscular mycorrhizal fungi (AMF) and Bradyrhizobium exhibited enhanced shoot growth, root biomass, and higher seed yield compared to non-inoculated plants under drought stress conditions (Sheteiwy et al. 2021b). The roots of nearly all plants support a vast community of microorganisms internally and on their external surfaces, which could aid during environmental stresses. In fact, under drought stress, root microbes not only protect themselves but also minimise drought induced damage to the plant host (Hartman and Tringe). The mechanisms by which microbes improve plant drought tolerance are complex but likely involve reactive oxygen species (ROS) scavenging systems. For example, the upregulation of ROS genes in Burkholderia phytofirmans associated with Solanum Tuberosum roots was observed under drought stress (Sheibani-Tezerji et al. 2015). Additionally, wheat primed with a Paenibacillus polymyxa mutant with an inactivated non-ribosomal peptide synthases greatly improved water use efficiency, water relative content, and increased the activity of antioxidant enzyme activities (Timmusk et al. 2015). The antioxidant enzymes such as superoxide dismutase, glutathione reductase, catalase, and monodehydroascorbate reductase play a significant role in scavenging ROS caused by drought and this defense mechanism enhances drought tolerance in plants. Resurrection plants have robust antioxidant systems, in which there is high activity of ascorbate, catalase, glutathione (GSH), peroxidase (POD), superoxide dismutase (SOD) and that scavenge ROS to combat drought effects (Kranner et al. 2002). How host and microbiome ROS scavenging systems interact to improve drought tolerance is not known, but differences in the ability to recruit and retain beneficial microorganisms can have major impacts on holobiont performance and survival.
Soil microbiomes have a magnitude of functions that are significant to plant health, including nutrient cycling, breakdown of soil organic matter, defense against plant pathogens, and enhancing abiotic stress tolerance (Hartman and Tringe 2019; Yadav et al. 2021). A consortium of microbes is more efficient in enhancing a crop’s drought tolerance than one inoculum. A recent study showed that co-inoculation of soybean with Bacillus amyloliquefaciens and AMF alleviated drought stress by increasing the production of sugars such as sucrose, fructose, and stachyose, as well as increased protein content compared to non-inoculated plants (Sheteiwy et al. 2021a). Undoubtedly, microbes regulate the physio-biochemical metabolism of the plant under water deficit conditions, which results in metabolic changes and increment of primary metabolites of primed compared to non-inoculated plants in drought stress.
A substantial amount of research has gone into studying these interactions in model and crop species, but the interaction between root and soil microbiomes in resurrection plants remains largely unknown. There is only one study that investigated rhizospheric bacteria of Ramonda serbica and Ramonda nathaliae resurrection plants using high-throughput sequencing (Đokić et al. 2010). Therefore, exploring the interaction of bacteria and fungi associated with the roots of the resurrection plant M. flabellifolia provides novel insight into how plant-microbe interactions function in the most extreme drought events. M. flabellifolia occurs in shallow rocky soil and high-temperature environments with limited rainfall in summer only. It is thus highly likely to harbour diverse microbial communities that possess desiccation tolerance traits.
The soil microbiome is typically divided into multiple compartments based on its proximity to the host plant from bulk soil to rhizosphere to endosphere (Fig. 1). The rhizosphere is the region surrounding plant roots and hosts a wide range of microorganisms, from algae, archaea, bacteria, fungi, to nematodes, protozoa and viruses (Kennedy and de Luna 2005), but the endosphere and bulk soil can also host a diversity of microbes. The most abundant clades in the rhizosphere are plant-growth-promoting rhizobacteria and mycorrhizal fungi that reside within the proximity of the root system and significantly influence plant physiological processes (Tomer et al. 2016). The microbial composition of all three compartments is influenced by multiple factors such as environmental conditions, type of habitat, contribution to plant health, and stress. In some cases, microbial colonisation of the roots leads to endocytosis and it can confer abiotic and biotic stress tolerance within the host plant (Fitzpatrick et al. 2018a; Leborgne-Castel et al. 2010).

Soil microbiome compartments are typically categorised into three zones based on their respective proximity to the plant roots. The rhizosphere is the region around the roots into which the root hairs extend. The rhizoplane is a narrow area on the root’s surface, and the endosphere is the area inside the roots. Plants actively recruit beneficial bacterial and fungal species into these compartments, with increasing specificity from rhizosphere to endosphere. The upper root shows the secretion of metabolites, and the bottom root shows the uptake of nutrients, water, and the process of microbes entering the root endosphere (endocytosis)
To better understand the role of the soil microbiome in extreme drought stress, we quantified the bacterial and fungal communities in the bulk soil, rhizosphere, and endosphere of M. flabellifolia. The aims of this study were to (1) identify the microbial taxa associated with M. flabellifolia under extreme drought stress, (2) quantify the differential abundance of microbial clades and explore the ecological function of the microbiome based on previous characterisations of the specific microbiota in each compartment.
Materials and methods
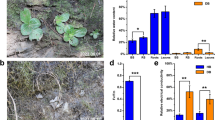
Five Myrothamnus flabellifolia plants were collected from Swebeswebe Nature Preserve in the Waterberg, Limpopo Province, South Africa in the autumn of 2021. Individual M. flabellifolia plants and surrounding native soil (~2 kg) were harvested from five discrete locations (within two metres of one another) while the plants were in the state of desiccation (Fig. S1A). These plants were transported to the University of Cape Town for experimental procedures. First, each plant’s root system was gently pulled out of the ground to avoid tissue damage. Loose soil attached to the roots was removed by vigorously shaking the plant, and it was classified as bulk soil. Additionally, three replicates of bulk soil were used for soil physicochemical and DNA analysis. Next, a sterilised scissor was used to cut ~5 cm roots with rhizosphere soil attached from the shoot-root joint. Five replicates of the rhizosphere were collected for soil physicochemical analysis by vigorously shaking to dislodge the rhizosphere from the roots. To prepare samples for microbial DNA extraction, five replicates of roots with rhizosphere soil attached were placed in a 50 mL conical centrifuge tube containing 15 mL of autoclaved phosphate-buffered saline (PBS). The rhizosphere soil, which was firmly attached to the root system, was removed according to Edwards et al. (2018). Briefly, tubes were vortexed at the maximum speed for 20 seconds to dislodge the rhizosphere soil from the roots (Fig. S1B). The roots were transferred using a sterile tweezer into a new 50 ml falcon tube with 25 ml PBS. The rhizosphere soil was briefly centrifuged, and the PBS buffer was discarded. Roots were washed with 20 ml of PBS before isolating the endosphere compartment and vortexed at maximum speed for 20–30 seconds. The PBS buffer was discarded, and another 20 ml of PBS was added and vortexed for 20–30 seconds. This step was repeated until there were no soil residues at the bottom of the tube. A fresh 20 ml PBS buffer was added to the root tissues, and they were cleaned by sonicating at 40 Hz for 30 seconds, and this step was repeated twice. Root tissues with no traces of soil were pulverised in liquid nitrogen using a sterile pestle and mortar. All samples were stored at -80 °C for DNA extraction and 4 °C soil physicochemical analysis, respectively.
Physicochemical soil analysis
Soil samples were prepared in microwave-assisted acid digestion (Mars 6 Microwave Digester) using concentrated boric, hydrofluoric, and nitic acid (Al-Harahsheh et al. 2009). Before the elemental analysis, the method was validated using certified reference materials (CRMs), and calibration curves for each analyte were constructed. The targeted elements of, calcium (Ca), copper (Cu), iron (Fe), potassium (K), magnesium, manganese (Mn), sodium (Na), phosphorus (P), and zinc (Zn) were analysed using Varian ES 730 inductively coupled plasma-optical emission spectroscopy (Agilent Technologies, Inc). The organic carbon (OC) and organic matter (OM) measurements/analysis of the soil were performed based on the established Walkley Black protocol (Walker-Black 1934). The soil pH, moisture content, exchangeable acidity, and aluminium ions were also determined according to (Robertson et al. 1999). We used a one-way analysis of variance (ANOVA) with Turkey’s pairwise comparison test to test for differences in physicochemical soil analysis between bulk and rhizosphere soil compartments.
DNA extraction, PCR amplification, library preparation, and sequencing
Total DNA was extracted from the bulk soil, rhizosphere, and endosphere samples using DNeasy PowerSoil (QIAGEN, 12888–50) kit following the manufacturer’s protocol. The purity and quantity of the extracted DNA were assessed using gel electrophoresis and a Nanodrop ND-2000 UV-Vis spectrophotometer (Thermo Fisher Scientific, USA). The V3-V4 hypervariable region of the 16S rRNA gene from each sample was amplified using the 341F (5’-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-3′) and the 505R (5-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-3′) primers. The mitochondrial peptide nucleic acid (mPNA, 5-GGCAAGTGTTCTTCGGA-3) and plastid peptide nucleic acid (pPNA, 5-GGCTCAACCCTGGACAG-3) clamps (PNA Bio, Newbury Park, CA, USA) were included to minimise the amplification of host plant DNA in the PCR reaction (Fitzpatrick et al. 2018b). Each 25 μL PCR reaction consisted of the following ingredients: 12.5 μL of 2x KAPA HiFi HotStart mix, 1.25 μL of 0.25 μM mPNA, 1.25 μL of 0.25 μM pPNA, 5 μL of 1 μM forward primer, 5 μL of 1 μM forward reverse, and 2.5 μL microbial genomic DNA. The reaction mixture was placed in the thermocycler for PCR program: denature at 95 °C for 3 min, followed by 25 cycles of 95 °C for 30 sec denaturation, 75 °C for 10 sec PNAs annealing, 55 °C for 30 sec primer annealing, 72 °C for one min elongation, and 72 °C for five min final extension before holding at 4 °C. The ITS1 and ITS2 regions were amplified using the NSA3 (5’-AAACTCTGTCGTGCTGGGGATA-3′) and NLC2 (5’-GAGCTGCATTCCCAAACAACTC-3′) primers. These Dikaryomycota-specific primers were developed to reduce the amplification of host plant DNA (Martin and Rygiewicz 2005). All reactions, including negative (sterile H2O) and positive (ZymoBIOMICS standard) controls, were performed in duplicates. The PCR amplicons of both bacterial and fungal species were purified using Agencourt AMPure XP beads (Beckman Coulter) to remove free primers and primer-dimers molecules. The PCR amplicons were analysed using an Agilent D1000 ScreenTape. Libraries were generated using the Illumina 16S rRNA and fungal metagenomic protocol. For library preparations, the 16S rRNA and ITS1 regions were amplified using a pool of nested primers modified to include the binding site for the sequencing primer and regions for index binding. Similar PCR conditions were used for the index PCR. The ITS1 amplicon varied in length relative to taxa. Library quantification, normalisation, and pooling were performed prior to Illumina sequencing. The 16S V3-V4 libraries were sequenced on an Illumina MiSeq sequencing instrument (Illumina, Inc) using a MiSeq reagent v2 kit (500 cycles) with 2 × 250 bp paired-end reads. The ITS1 libraries were sequenced using a MiSeq reagent nano v2 kit (500 cycles) with 2 × 250 bp paired-end reads.
Bioinformatics analysis of 16S rRNA and ITS data
The resulting fastq files were processed using R for mac OS X GUI version 4.1.2 (Urbanek 2022). Raw sequencing paired-end reads were imported to RStudio for pre-processing steps. The quality profile of the reads was inspected using the DADA2 version 1.22.0 (Callahan et al. 2016), and the raw sequence reads with poor quality average scores (< 30) were discarded. The bacterial and fungal reads were filtered and trimmed using DADA2 to eliminate primer and adaptor sequences. In addition, error rates for each consensus quality score were evaluated. Samples were processed independently after sharing information between samples using pool ‘pseudo’. The denoised forward and reverse reads longer than ten bp were merged into a multiple sequence alignment using DECIPHER package, and amplicon sequence variances (ASVs) were obtained. Chimeric sequences were identified and removed using DADA2. Taxonomic annotation was performed using the most updated and extensive SILVA and UNITE databases for bacteria and fungi, respectively (Nilsson et al. 2019; Quast et al. 2012).
The bacterial and fungal alpha-diversity were calculated based on the pairwise Wilcoxon test metric at the ASV level and was measured using phyloseq, picante, and stat R packages. The beta-diversity of bacterial and fungal communities was assessed by computing Bray-Curtis distance across different microbial taxa of three compartments. The ordination was performed using non-metric multidimensional scaling (NMDS). The differences in microbial composition between compartments were transformed with total sum scaling (Nilsson et al. 2019) from 16S rRNA and ITS data. A non-parametric paired samples Wilcoxon test was performed to evaluate the alpha-diversity and taxonomic differences among various compartments. The microbiome dissimilarity distance metrics were also analysed by the multivariate permutation analysis of variance (PERMANOVA), together with the nonparametric statistical Adonis test (vegan package) to establish the homogeneity of dispersion among three compartments. The PERMANOVA test was performed using 999 permutations. A Dirichlet-Multinomial distribution test was used for relative abundance using the HMP package and this test is analogous to a paired sample t-test. However, it evaluates whether taxa frequencies observed in all groups of metagenomic samples are equal. Differentially abundant ASVs were identified using the DESeq2 package and a pseudo count was added to bypass the errors in the logarithm transformation. Taxa on the heatmap were selected from the annotation table fitted to a negative binomial generalised linear model (nbFLM) using analysis of deviance (alpha = 0.05) at the genus level.
Results
Soil physicochemical analysis
There were significant physiochemical and elemental differences between bulk and rhizosphere soil (Table 1). The bulk soil had significantly lower moisture content than the rhizosphere soil (P < 0.01), and exchangeable hydrogen ions were significantly (P < 0.05) lower in the rhizosphere than the bulk soil. Thus the bulk soil was more acidic compared to the rhizosphere. In general, with the exception of Fe and Mn, the rhizosphere had higher concentrations of elements tested (Table 1).
Amplicon 16S rRNA and ITS sequencing data
A total of 21,871 bacterial ASVs and 1332 fungal ASVs were retained after filtering across all compartments. The Venn diagrams of bacteria and fungi were constructed by creating a presence/absence matrix summarising the total number of ASVs in all samples for each compartment. The ASVs that were present in the bulk soil, rhizosphere, and endosphere were compared. We identified unique and common taxa across the different soil compartments (Fig. 2A and B). The majority of bacterial and fungal taxa were specific to the different soil compartments. A total of 5584 ASVs of bacterial taxa were shared across all three compartments (Fig. 2A). The bulk soil, rhizosphere, and endosphere each harboured 3215, 5833, and 2899 unique ASVs, respectively. The rhizosphere had the richest diversity of bacterial taxa compared to other compartments and the rhizosphere and bulk soil shared 2751 ASVs. Similarly, with the fungal communities, the rhizosphere had greater unique ASVs (699) compared to other compartments (Fig. 2B). The shared ASVs across all compartments, and between the rhizosphere and endosphere were the same (22). However, the fungal bulk and rhizosphere soil shared higher ASVs (132) relative to other shared ASVs between compartments. Furthermore, the bulk soil and endosphere showed lower overlap in bacterial (275) and fungal (4) ASVs. There was a high overlap of bacterial and fungal taxa between bulk soil and rhizosphere, and the endosphere had the least unique ASVs.

The Venn diagram shows the number of A) bacterial and B) fungal amplicon sequence variants (ASVs) that are unique and shared across the three soil compartments. Alpha-diversity of C) bacterial and D) fungal communities in the three compartments. A Shannon diversity index was used to measure species richness (number of different ASVs in the sample). Simpson diversity index measures the evenness of the microbial community (abundance of ASVs relative to each other) within each compartment
Alpha and Beta-diversity
To understand the community diversity of M. flabellifolia’s root microbiome, we characterised the alpha (within the community) and beta (across the communities) diversity of the bulk soil, rhizosphere, and endosphere compartments. Rarefaction curves at the ASV level show the sequencing depth of both bacterial and fungal community diversity (Fig. S2A and S2B). Alpha-diversity, both in terms of species richness using Shannon’s index and species evenness using Simpson was measured. In bacterial communities, species richness was significantly higher in bulk and rhizosphere soil compared to the endosphere (Wilcoxon test, P = 0.05) (Fig. 2C). Similarly, fungal richness was significantly higher (P = 0.02) in bulk and rhizosphere soil than in the endosphere (Fig. 2D). There was no significant difference between bulk and rhizosphere soil for bacteria (P = 0.39) or fungi (P = 0.25). Moreover, the bulk and rhizosphere soil exhibited a high degree of evenness in their bacterial and fungal communities, whereas there was more variability in the endosphere (Fig. 2C-D). The bulk and rhizosphere soil had a greater diversity of bacteria and fungi compared to the endosphere compared.
Next, we calculated beta-diversity–a measure of variation in species diversity between compartments (Fig. 3A and B). The PERMANOVA indicated significant variation in bacterial and fungal communities across compartments. The separation between the groups was statistically significant (PERMANOVA, P = 0.01), providing strong evidence for a difference in bacterial composition between soil compartments and endosphere. (Fig. 3A). There was an overlap in bacterial ASVs between the rhizosphere and bulk soil compartments, which was further observed in hierarchical clustering. The fungal community showed substantial variation in species diversity between compartments with a high statistical difference (PERMANOVA, P = 0.001) in variation between three compartments (Fig. 3B). Moreover, hierarchical cluster analysis further confirmed the clustering of compartments with Bray-Curtis dissimilarity distances ranging from 0.1 to 1 distance across compartments for both bacterial (Fig. 3C) and fungal ASVs (Fig. 3D). However, the fungal dissimilarity distance between the compartments was one, indicating that observations were well clustered and had distinct taxa compared to bacterial hierarchy. Taken together, the endosphere microbiome was more distinct compared to the bulk and rhizosphere soil for both bacteria and fungi clustering.

Differences in microbial composition (beta-diversity) between endosphere, bulk, and rhizosphere soil were analysed using non-metric multidimensional scaling (NMDS) based on the PERMANOVA test. A) Bacterial and B) fungal community composition in different compartments were analysed at the amplicon sequence variant (ASV) level. C) Bacterial and D) fungal hierarchical cluster analysis of taxa using Bray-Curtis dissimilarity
Relative and differential abundance
To better understand the possible functional impact of the root microbiome, relative abundances of different taxa were evaluated (Fig. 4). Not only were different taxa present in different compartments, but the abundance of these bacterial and fungal taxa differed significantly. The top six most abundant bacterial phyla across all compartments were Acidobacteriota, Actinobacteriota, Chloroflexota, Planctomycetota, Pseudomonadota, and WPS-2 (Fig. 4A). The relative abundance of Chloroflexota, Crenarchaeota, Cyanophyta, Gemmatimonadota, RCP2–54, and WPS-2 was more enriched in bulk soil (Fig. 4A). In contrast, the rhizosphere had a high abundance of Bdellovibrionota, Dependentiae, Bacillota, GAL-15, Spirochaetota, and Verrucomicrobiota. Furthermore, we observed an enrichment of Acidobacteriota, Actinobacteriota, Elusimicrobiota, Myxococcota, Patescibacteria, Planctomycetota, and Pseudomonadota in the endosphere. Interestingly, the relative abundance of Chloroflexota decreased from bulk-rhizosphere soil to endosphere, whereas Actinobacteriota increased from bulk-rhizosphere to endosphere. Interestingly there was no significant difference in the relative abundance of bacterial taxa between the rhizosphere and endosphere, but we found that the bacterial microbiome composition was significantly different between the soil compartments (bulk and rhizosphere soil) and endosphere (P = 0.00054). The most dominant fungal phyla found across all compartments were Ascomycota and Basidiomycota (Fig. 4B), but the fungal microbiome differed significantly between the three compartments (P = 0.01). The rhizosphere harboured a more diverse fungal microbiome than the endosphere and bulk soil, and the significantly enriched phyla were Chytridiomycota, Mortierellomycota, Mucoromycota, Rozellomycota, and Zoopagomycota. The endosphere and rhizosphere had a similar composition of the dominant bacterial phyla, while fungal composition across compartments was dominated by Ascomycota.

Relative abundance of A) Bacterial B) Fungal microbial community composition of bulk soil, rhizosphere, and endosphere compartments at phylum level during extreme drought stress. Significant differences (P < 0.01) in the Dirichlet-multinomial distribution of bacterial and fungal phyla between bulk and rhizosphere soil, and bulk soil and endosphere. C) Bacterial and D) fungal heatmap plots of the abundance of taxa differing significantly between three soil compartments. Presented data are centred and scaled to the average of each taxon abundance, and each vertical bar corresponds to a sample. The log2 fold change shows the abundance of different taxa in the sample
To identify the bacterial and fungal lineages enriched within the root relative to the rhizosphere and bulk soil, a negative binomial model was used to test for the differential abundance of ASVs across compartments. Many bacterial and fungal genera were more abundant in the rhizosphere compared to other compartments. A total of 84 bacterial ASVs were significantly enriched (nbGLM, P < 0.01) across all compartments (Fig. 4C). The most significantly enriched taxa in bulk soil were Leptolyngbya, Flavisolibacter, Cyanophyta, and unclassified genera (B12-WMSP1, AD3, TK10, Coleofasciculaceae, Gemmatimonadaceae). There was high variability within rhizosphere samples for both bacterial and fungal communities (Fig. 4C and D). The enriched taxa in the rhizosphere were Asticcacaulis, Nitrolancea, Pedomicrobium, Hyphomicrobium, Cytophaga, Micropepsis, Nocardioides, Planctopirus, Novosphingobium, Tailbaiella, Flavobacterium, Edaphobaculum, Parvibaculum, Chitinophaga, Parafilimonas, Clostridium, Tistrella, Caulobacter, Candidatus, Mucilaginibacter, Devosia, Streptomyces and unclassified genera (Sphingobacteriaceae, Rickettsiaceae, Cytophagales, CPIa-3 termite group, Saccharimonadaceae, Micropepsaceae, Bacterioidia, Lachnospiraceae, Rhizobiaceae, Devosiaceae and Microscillaceae). In the endosphere, we detected only Pirellulaceae, TM7a, Mucilagnibacter, Pajaroellobacter, and an unclassified genus (B12-WMSP1). A total of 71 fungal ASVs were significantly different (P = 0.01) across the three compartments. The most significantly enriched fungal genera in bulk soil were Chrysosporium, Epicoccum, Leucucuprinus, Pseudophaeomoniella, Westerdykella, Metarhizium, Tolyposporium, Paracamarosporium, and Sphaeropsis (Fig. 4D). Some fungal genera were significantly enriched in the rhizosphere including Apiotrichum sporotrichoides, Clitopilus hobsonii, Nematoctonus pachysporus, Meyerozyma caribbica, Mortierella capitata, Penicillin kongi, and Talaromyces diversus. In contrast, the endosphere compartment hosts only a small number of fungal genera namely Cladosporium and Penicillium. Taken together, the most significantly different phyla in the bulk soil were Cyanophyta and Chloroflexota for bacteria, Ascomycota, and Basidiomycota for fungi relative to other compartments. Although the rhizosphere showed high variability, this compartment had a higher number of significantly enriched taxa compared to other compartments.
Environmental factors influencing the microbial composition
To explore the effects of edaphic factors on the microbial composition of bulk and rhizosphere soil of M. flabellifolia, we performed a redundancy analysis (RDA) to relate elemental soil variables to microbiome composition for both bacteria (Fig. 5A and C) and fungi (Fig. 5B and D). Bulk soil samples clustered together more than the rhizosphere samples, indicating a higher variability within the rhizosphere compared to the bulk soil compartment. The soil physicochemical factors pH, moisture, organic carbon and matter, and soil elements positively correlated with a high abundance of bacterial and fungal species in the rhizosphere compartment. Furthermore, the soil elements Ca, K, Mg, Na, P, and Zn were associated with the enrichment of bacterial genera Gordonia, Taonella, and Rhodobacter (Fig. S3A), whereas Ca, Fe, and Mn were significantly associated with the enrichment of fungal genera Spegazzinia, Dothideales and Vishniacozyma in both bulk and rhizosphere soil (Fig. S3B). There was a strong correlation between the rhizosphere microbiome. and soil physicochemical factors.

Redundancy analysis of soil physicochemical parameters of bulk and rhizosphere soil and their influence on bacterial (A and C) and fungal communities (B and D)
Discussion
Plant-microbe interactions play an important role in plant growth, health, and productivity. Despite the importance of plant growth-promoting microbes in plants, particularly in the rhizosphere, microbial species associated with resurrection plants have not been explored (Tebele et al. 2021). Our study provides one of the first high-resolution characterisations of the root microbiome of a resurrection plant. We described the bacterial and fungal microbiomes associated with the resurrection plant M. flabellifolia under air-dry conditions (≤ 10% RWC) and investigated how the microbial community composition varied across compartments from bulk soil to the rhizosphere, to the endosphere. While this provided only a snapshot of communities present under extreme conditions, precluding a full understanding of progressive changes in community dynamics during dehydration, this study does allow speculation on the role of these communities and their contribution to the unique resilience of M. flabellifolia. Our analyses revealed substantial differences in the microbiome of the bulk soil, rhizosphere, and endosphere compartments. Species richness and microbial dynamism were observed in the bulk soil and, to a higher degree, in the rhizosphere compartment. In contrast, the endosphere microbiome was notably reduced in such properties, with only a few select species being present, suggesting that strong filtering occurs at the soil-to-root interface. These findings suggest that regardless of extreme drought conditions, M. flabellifolia harbors a diverse microbial community and may actively regulate microbial assemblages in different compartments.
The most dominant bacterial phyla across all compartments were Acidobacteriota, Actinobacteriota, Chloroflexota, Planctomycetota, Pseudomonadota, and WPS-2, whereas the most dominant fungal phyla were Ascomycota, Basidiomycota, Mortierellomycota and Mucoromycota. These results are parallel to previous studies that also reported the presence of Actinobacteriota, Chloroflexota, and Planctomycetota in the resurrection plant Ramonda as well as in chickpea, sorghum, and other plant hosts (Ahkami et al. 2017; Đokić et al. 2010; Xu et al. 2018; Yadav et al. 2021). However, there are notable differences between the findings on Ramonda spp and the current study. Most importantly, the presence of Flavobacterium, Mucilaginibacter, and Nocardioides in M. flabellifolia but absent in the Ramonda species’ rhizosphere, suggests that these microbes could be environmentally or geographically determined (M. flabellifolia occurs in southern Africa whereas Ramonda spp. occurs in Eurasia). In desiccation sensitive species, such as sorghum, Xu et al. (2018) have a low abundance of Acidobacteriota in roots under drought stress. In contrast, our study shows a high enrichment of Acidobacteriota in the endosphere of M. flabellifolia suggesting a possible role for these taxa in coping with extreme environmental stresses. The abundance of these distinctive microbial strains in M. flabellifolia demonstrates that extreme environmental stress may restructure the plant microbiome. Drought stress undoubtedly introduces physiological, metabolic, and genetic responses while also impacting nutrient availability in both microbiome and host plants. Bulk soil samples were clustered together more strongly compared to the rhizosphere compartment (Fig. 5). There was variability in the rhizosphere samples, suggesting that the rhizosphere of each plant hosts a slightly different microbial composition and this could be due to the landscape topography of the natural environment.
We identified differences in soil physicochemical factors that were associated with changing microbiome composition across soil compartments. The bioavailability of micronutrients such as Ca, Cu, Mg, Mn, Na, and Zn in the rhizosphere soil of M. flabellifolia under drought stress seems to impact the relative microbiome abundance and hints at an intriguing plant-microbe-soil interaction. Previous studies have conclusively shown that arbuscular mycorrhizal enhances plant growth by increasing root access to immobile mineral ions and binding to heavy metals and transporting them from roots to shoot tissues (Srinivasagam et al. 2013). These findings showed that rhizospheric fungal species have synergistic interaction with bacterial species in enhancing nutrient uptake and heavy metals resistance in M. flabellifolia. The content of soil moisture and exchangeable H+ were significantly correlated with bacterial and fungal communities in the rhizosphere compartment. The exchangeable H+ is the predominant factor that influences soil pH (Zheng et al. 2020), therefore, indicating that rhizosphere soil is less acidic relative to bulk soil and this enhances soil nutrients. Taken together, the rhizosphere soil had higher moisture content and micronutrients and it was less acidic compared to bulk soil, which could attract diverse and beneficial microbial communities.
The enrichment of particular bacteria and fungi under extreme drought could indicate a role for those microbes in supporting drought tolerance through close linkage with the host plant. We identified numerous taxa that were uniquely enriched in different compartments of M. flabellifolia. The enriched taxa could point towards a fundamental role of microbes in enhancing the drought tolerance of M. flabellifolia. The M. flabellifolia microbiome is extremely complex consisting of over 900 unique bacterial and fungal taxa in each compartment. To summarize this complexity, we highlight selected taxa from each compartment and discuss their possible role in mitigating drought stress.
This study found a significant enrichment of monoderm (Actinobacteriota, Chloroflexota, Bacillota) and diderm (Acidobacteriota, Bacteriodota, and Pseudomonadota) lineages in rhizosphere and endosphere compared to the bulk soil, except for Chloroflexota, which showed high enrichment in bulk soil. The bulk soil compartment of the M. flabellifolia microbiome hosted an abundance of bacterial taxa in Coleofascicus, Flavisolibacter, and Leptolyngbya. These bacterial genera are known to be resistant to drought stress and can improve the physical and biological conditions of rhizosheath soil (Liu et al. 2021; Moreira et al. 2021). The finding of Cyanophyta in bulk soil is significant, as these species are normally restricted to the upper surface of soil crusts (due to their photosynthetic ability). Such species have been shown to play a significant role in soil stabilisation and the addition of organic carbon (Gao et al. 2020b) and their role in the bulk soil could be significant. Interestingly, our study revealed that the bulk soil compartment hosts unique, beneficial bacterial and fungal taxa, and this might be due to the ecological niche of M. flabellifolia.
The significantly enriched fungal taxa in bulk soil were Epicoccum dendrobii, Metarhizium lepidiotae, and Mycenastrum. These enriched fungal genera also have multiple functions related to resilience. For instance, Epicoccum dendrobii is an antifungal agent that enters the plant tissue through stomatal cells and secretes compounds that inhibit anthracnose lesion development caused by pathogens (Bian et al. 2021). Metarhizium spp. is an endophyte that proliferates propagule levels in the rhizosphere (Steinwender et al. 2015). Remarkably, Metarhizium spp was found in the bulk soil of M. flabellifolia, instead of the endosphere compared to previous studies. These findings indicate that both bacteria and fungi have synergic effects in M. flabellifolia, which is in accordance with the study by Sheteiwy et al. (2021b) reported the co-inoculation of soybean seedlings with AMF and Bradyrhizobium improved root length and dry weight of the plant as compared to the controls. The microbial diversity in the bulk soil depends on the plant species and habitat, and the bulk soil compartment of M. flabellifolia hosted a high relative abundance of Chloroflexota and Cyanophyta compared to other compartments.
A plethora of bacterial and fungal taxa were significantly enriched in the rhizosphere compared to other compartments. The bacterial genera include Flavobacterium, Nocardioides, and Streptomyces. These bacterial genera produce indoleacetic acid (IAA) which improves xylem and phloem formation in roots and further increases the formation of lateral and adventitious roots to mitigate drought effects (Khan et al. 2014). This suggests that these species play a vital role during drought stress in M. flabellifolia because a large root system plays a significant role in sourcing water in the deeper soil profiles. A recent study reported the upregulation of the TaIAA15-1A gene is enhanced by exogenous abscisic acid (ABA) treatment and overexpression of TaIAA15-1A in Brachypodium improved drought tolerance through the regulation of antioxidant pathways and ABA signalling, which further reduces ROS and decreased malondialdehyde content (Su et al. 2023). Moreover, high amounts of iron in the rhizosphere soil might be associated with the abundance of Acidicapsa species (Fig. S3A), which produces IAA (Kielak et al. 2016). Interestingly maize primed with Pseudomonas species enhanced the upregulation of protein kinases (SnRKs), which are positive regulators of ABA receptors (SkZ et al. 2018). This may suggest that numerous diverse protein kinases involved in the desiccation tolerance of M. flabellifolia (Ma et al. 2015) might be enhanced by Pseudomonatota to regulate extreme drought stress. Water limitation and nutrient starvation in plants stimulate the transcription of multiple drought-responsive genes that ameliorate drought stress effects.
The enriched fungal taxa in the rhizosphere included Actinomucor, Aspergillus, and Penicillium spp. Actinomucor sp. has been shown to produce abundant quantities of proteases, lipases, and amylases that hydrolyse the components of sorghum seeds (Gao et al. 2020a). Therefore, the presence of hydrolytic-enzyme-producing species under drought conditions in the rhizosphere suggests that they might be involved in the rearrangement of metabolism through the selective degradation of short-lived proteins (Vaseva et al. 2012). A recent study by Sheteiwy et al. (2021b) reported high antioxidant activity and upregulation of catalase and peroxidase gene expression in the soybean seedling under drought stress was enhanced by Bradyrhizobium and mycorrhizal co-inoculant. The presence of trehalose in M. flabellifolia’s leaves has been related to fungal species, and trehalose is a desiccation-induced osmoticum in resurrection plants (Farrant 2000; Moore et al. 2011). Although there was variation in the rhizosphere microbiome, this compartment hosted diverse microbial communities that may confer drought tolerance and promote plant growth to the host.
The endosphere of M. flabellifolia was significantly less diverse in microbial taxa than the other two compartments, suggesting that stringent filtering occurs at the soil-root interface. Bacterial genera significantly enriched in the endosphere were Mucilaginibacter, TMa7, and unclassified Pirellulaceae, and fungal taxa were Cladosporium and Penicillium. The Mucilaginibacter and Cladosporium genera are known to produce exopolysaccharides which form biofilms and adhere to root surfaces (Fan et al. 2018; Kielak et al. 2016; Mahapatra and Banerjee 2013). Exopolysaccharides also act as defense mechanisms against drought by sustaining a hydrated microenvironment and reducing water loss (Morcillo and Manzanera 2021). As the rhizosphere soil moisture was significantly greater than bulk soil, it is possible that the presence of exopolysaccharide-producing microbes may slow water loss and alleviate drought stress. These results provide further support for the hypothesis that root-associated microorganisms confer drought tolerance to their host plant. In addition, bacterial (Mucilaginibacter) and fungal genera (Penicillium and Aspergillus) are known to be resistant to heavy metals and remove or absorb Zn2+, Cd2+, and Cu2+ (Anahid et al. 2011; Fan et al. 2018). This suggests that these species do not only tolerate drought stress but also may play a role in minimising heavy metal toxicity in M. flabellifolia.
Conclusion
Myrothamnus flabellifolia hosts a diverse microbiome that may assist the plant in surviving extreme drought stress. Understanding the microbial composition and the distribution across three compartments and characterising the core taxa in the bulk soil, rhizosphere, and endosphere provides a great opportunity to identify the microbes involved in enhancing M. flabellifolia drought tolerance. Taken together, this study demonstrated that the most dominant bacterial phyla across all compartments were Acidobacteriota, Actinobacteriota, Chloroflexota, Planctomycetota, Pseudomonadota, and WPS-2, and fungal phyla were Ascomycota, Basidiomycota, Mortierellomycota and Mucoromycota. Additionally, the rhizosphere hosted a high level of bacterial and fungal diversity compared to other compartments. Future studies should consider investigating M. flabellifolia under rehydration and dehydration conditions using meta-transcriptomic and metabolomic approaches to explore the expressed microbial genes and responsive metabolites under extreme drought conditions. The identified microbial communities associated with M. flabellifolia roots could have the potential to improve plant resilience and well-being.
Data availability
All raw sequencing data have been submitted to the European Nucleotide Archive (ENA) database under the accession number PRJEB58375.
References
Ahkami AH, White RA III, Handakumbura PP, Jansson C (2017) Rhizosphere engineering: enhancing sustainable plant ecosystem productivity. Rhizosphere 3:233–243
Al-Harahsheh M, Kingman S, Somerfield C, Ababneh F (2009) Microwave-assisted total digestion of sulphide ores for multi-element analysis. Anal Chim Acta 638:101–105
Anahid S, Yaghmaei S, Ghobadinejad Z (2011) Heavy metal tolerance of fungi. Scientia Iranica 18:502–508
Bentley J, Moore JP, Farrant JM (2019) Metabolomics as a complement to phylogenetics for assessing intraspecific boundaries in the desiccation-tolerant medicinal shrub Myrothamnus flabellifolia (Myrothamnaceae). Phytochemistry 159:127–136
Bewley JD (1979) Physiological aspects of desiccation tolerance. Annu Rev Plant Physiol 30:195–238
Bian J-Y, Fang Y-L, Song Q, Sun M-L, Yang J-Y, Ju Y-W, Li D-W, Huang L (2021) The fungal endophyte Epicoccum dendrobii as a potential biocontrol agent against Colletotrichum gloeosporioides. Phytopathology® 111:293–303. https://doi.org/10.1094/phyto-05-20-0170-r
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP (2016) DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583
Đokić L, Savić M, Narančić T, Vasiljević B (2010) Metagenomic analysis of soil microbial communities. Archives of Biological Sciences 62:559–564
Edwards J, Santos-Medellín C, Sundaresan V (2018) Extraction and 16S rRNA sequence analysis of microbiomes associated with rice roots. Bio-protocol 8(12):e2884–e2884
Erhabor J, Komakech R, Kang Y, Tang M, Matsabisa M (2020) Ethnopharmacological importance and medical applications of Myrothamnus flabellifolius Welw.(Myrothamnaceae)-a review. J Ethnopharmacol 252:112576
Fan X, Tang J, Nie L, Huang J, Wang G (2018) High-quality-draft genome sequence of the heavy metal resistant and exopolysaccharides producing bacterium Mucilaginibacter pedocola TBZ30T. Stand Genomic Sci 13:1–8
Farrant JM (2000) A comparison of mechanisms of desiccation tolerance among three angiosperm resurrection plant species. Plant Ecol 151:29–39
Farrant JM, Kruger L (2001) Longevity of dry Myrothamnus flabellifolius in simulated field conditions. Plant Growth Regul 35:109–120
Fitzpatrick CR, Copeland J, Wang PW, Guttman DS, Kotanen PM, Johnson MT (2018a) Assembly and ecological function of the root microbiome across angiosperm plant species. Proc Natl Acad Sci 115:E1157–E1165
Fitzpatrick CR, Lu-Irving P, Copeland J, Guttman DS, Wang PW, Baltrus DA, Dlugosch KM, Johnson MT (2018b) Chloroplast sequence variation and the efficacy of peptide nucleic acids for blocking host amplification in plant microbiome studies. Microbiome 6:1–10
Gaff DF (1989) Responses of desiccation tolerant ‘resurrection’plants to water stress. Structural and functional responses to environmental stresses:255–268
Gao C, Montoya L, Xu L, Madera M, Hollingsworth J, Purdom E, Singan V, Vogel J, Hutmacher RB, Dahlberg JA (2020a) Fungal community assembly in drought-stressed sorghum shows stochasticity, selection, and universal ecological dynamics. Nat Commun 11:1–14
Gao L, Sun H, Xu M, Zhao Y (2020b) Biocrusts resist runoff erosion through direct physical protection and indirect modification of soil properties. J Soils Sediments 20:133–142. https://doi.org/10.1007/s11368-019-02372-w
Hartman K, Tringe SG (2019) Interactions between plants and soil shaping the root microbiome under abiotic stress. Biochem J 476:2705–2724. https://doi.org/10.1042/bcj20180615
Hilhorst HW, Farrant JM (2018) Plant desiccation tolerance: a survival strategy with exceptional prospects for climate-smart agriculture. Annual plant reviews. Online:327–354
Kato Y, Okami M (2011) Root morphology, hydraulic conductivity and plant water relations of high-yielding rice grown under aerobic conditions. Ann Bot 108:575–583. https://doi.org/10.1093/aob/mcr184
Kennedy AC, de Luna LZ (2005) Rhizosphere. In: Hillel D (ed) Encyclopedia of soils in the environment. Elsevier, Oxford
Khan AL, Waqas M, Kang S-M, Al-Harrasi A, Hussain J, Al-Rawahi A, Al-Khiziri S, Ullah I, Ali L, Jung H-Y (2014) Bacterial endophyte Sphingomonas sp. LK11 produces gibberellins and IAA and promotes tomato plant growth. J Microbiol 52:689–695
Kielak AM, Cipriano MA, Kuramae EE (2016) Acidobacteria strains from subdivision 1 act as plant growth-promoting bacteria. Arch Microbiol 198:987–993
Kranner I, Beckett RP, Wornik S, Zorn M, Pfeifhofer HW (2002) Revival of a resurrection plant correlates with its antioxidant status. Plant J 31:13–24
Leborgne-Castel N, Adam T, Bouhidel K (2010) Endocytosis in plant–microbe interactions. Protoplasma 247:177–193. https://doi.org/10.1007/s00709-010-0195-8
Liu T-Y, Ye N, Wang X, Das D, Tan Y, You X, Long M, Hu T, Dai L, Zhang J, Chen M-X (2021) Drought stress and plant ecotype drive microbiome recruitment in switchgrass rhizosheath. J Integr Plant Biol 63:1753–1774. https://doi.org/10.1111/jipb.13154
Lizumi T, Ramankutty N (2016) Changes in yield variability of major crops for 1981–2010 explained by climate change. Environ Res Lett 11:034003
Ma C, Wang H, Macnish AJ, Estrada-Melo AC, Lin J, Chang Y, Reid MS, Jiang C-Z (2015) Transcriptomic analysis reveals numerous diverse protein kinases and transcription factors involved in desiccation tolerance in the resurrection plant Myrothamnus flabellifolia. Horticulture Research 2:15034. https://doi.org/10.1038/hortres.2015.34
Mahapatra S, Banerjee D (2013) Fungal exopolysaccharide: production, composition and applications. Microbiol Insights 6:1–16. https://doi.org/10.4137/mbi.S10957
Marks R, Mbobe M, Greyling M, Pretorius J, McLetchie D, Vanburen R, Farrant J (2022) Variability in functional traits along an environmental gradient in the south African resurrection plant Myrothamnus flabellifolia. Plants 11:1332. https://doi.org/10.3390/plants11101332
Martin KJ, Rygiewicz PT (2005) Fungal-specific PCR primers developed for analysis of the ITS region of environmental DNA extracts. BMC Microbiol 5:1–11
Moore J, Waldron M, Lindsey G, Farrant J, Brandt W (2011) An ultrastructural investigation of the surface microbiota present on the leaves and reproductive structures of the resurrection plant Myrothamnus flabellifolia. S Afr J Bot 77:485–491
Moore JP, Nguema-Ona E, Chevalier L, Lindsey GG, Brandt WF, Lerouge P, Farrant JM, Driouich A (2006) Response of the leaf cell wall to desiccation in the resurrection plant Myrothamnus flabellifolius. Plant Physiol 141:651–662
Moore JP, Westall KL, Ravenscroft N, Farrant JM, Lindsey GG, Brandt WF (2005) The predominant polyphenol in the leaves of the resurrection plant Myrothamnus flabellifolius, 3, 4, 5 tri-O-galloylquinic acid, protects membranes against desiccation and free radical-induced oxidation. Biochem J 385:301–308
Morcillo RJ, Manzanera M (2021) The effects of plant-associated bacterial exopolysaccharides on plant abiotic stress tolerance. Metabolites 11:337
Moreira F, Giraldo-Silva A, Roush D, Garcia-Pichel F (2021) Coleofasciculaceae, a monophyletic home for the Microcoleus steenstrupii complex and other desiccation-tolerant filamentous cyanobacteria. J Phycol 57:1563–1579
Nilsson RH, Larsson KH, Taylor AFS, Bengtsson-Palme J, Jeppesen TS, Schigel D, Kennedy P, Picard K, Glöckner FO, Tedersoo L, Saar I, Kõljalg U, Abarenkov K (2019) The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res 47:D259–d264. https://doi.org/10.1093/nar/gky1022
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2012) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. https://doi.org/10.1093/nar/gks1219
Robertson GP, Sollins P, Ellis BG, Lajtha K (1999) Exchangeable ions, pH, and cation exchange capacity. Standard soil methods for long-term ecological research 2:462
Sheibani-Tezerji R, Rattei T, Sessitsch A, Trognitz F, Mitter B (2015) Transcriptome profiling of the endophyte Burkholderia phytofirmans PsJN indicates sensing of the plant environment and drought stress. MBio 6:e00621–e00615
Sherwin HW, Pammenter N, February E, Vander Willigen C, Farrant JM (1998) Xylem hydraulic characteristics, water relations and wood anatomy of the resurrection plant Myrothamnus flabellifolius Welw. Ann Bot 81:567–575
Sheteiwy MS, Abd Elgawad H, Xiong YC, Macovei A, Brestic M, Skalicky M, Shaghaleh H, Alhaj Hamoud Y, El-Sawah AM (2021a) Inoculation with bacillus amyloliquefaciens and mycorrhiza confers tolerance to drought stress and improve seed yield and quality of soybean plant. Physiol Plant 172:2153–2169
Sheteiwy MS, Ali DFI, Xiong Y-C, Brestic M, Skalicky M, Hamoud YA, Ulhassan Z, Shaghaleh H, AbdElgawad H, Farooq M (2021b) Physiological and biochemical responses of soybean plants inoculated with arbuscular mycorrhizal fungi and Bradyrhizobium under drought stress. BMC Plant Biol 21:1–21
SkZ A, Vardharajula S, Vurukonda SSKP (2018) Transcriptomic profiling of maize (Zea mays L.) seedlings in response to Pseudomonas putida stain FBKV2 inoculation under drought stress. Ann Microbiol 68:331–349. https://doi.org/10.1007/s13213-018-1341-3
Srinivasagam K, Natarajan B, Raju M, Selvan RK (2013) Myth and mystery of soil mycorrhiza: a review. Afr J Agric Res 8:4706–4717
Steinwender BM, Enkerli J, Widmer F, Eilenberg J, Kristensen HL, Bidochka MJ, Meyling NV (2015) Root isolations of Metarhizium spp. from crops reflect diversity in the soil and indicate no plant specificity. J Invertebr Pathol 132:142–148
Su P, Sui C, Li J, Wan K, Sun H, Wang S, Liu X, Guo S (2023) The aux/IAA protein TaIAA15-1A confers drought tolerance in Brachypodium by regulating abscisic acid signal pathway. Plant Cell Rep 42:385–394. https://doi.org/10.1007/s00299-022-02965-9
Tebele SM, Marks RA, Farrant JM (2021) Two decades of desiccation biology: a systematic review of the best studied angiosperm resurrection plants. Plants 10: 2784
Timmusk S, Kim S-B, Nevo E, Abd El Daim I, Ek B, Bergquist J, Behers L (2015) Sfp-type PPTase inactivation promotes bacterial biofilm formation and ability to enhance wheat drought tolerance. Front Microbiol 6:387
Tomer S, Suyal DC, Goel R (2016) Biofertilizers: a timely approach for sustainable agriculture. Plant-microbe interaction: an approach to sustainable agriculture. Springer, Berlin
Urbanek S (2022) R. for macOS. Cran.R-project
Vaseva I, Sabotič J, Šuštar-Vozlič J, Meglič V, Kidrič M, Demirevska K, Simova-Stoilova L (2012) The response of plants to drought stress: the role of dehydrins, chaperones, proteases and protease inhibitors in maintaining cellular protein function. Droughts: new research 1:1–45
Walker-Black I (1934) An examination of the Degtajareff method for soil organic matter determination and a proposed modification of the chronic acid titration. Soil Sci 37:29–38
Xu L, Naylor D, Dong Z, Simmons T, Pierroz G, Hixson KK, Kim Y-M, Zink EM, Engbrecht KM, Wang Y (2018) Drought delays development of the sorghum root microbiome and enriches for monoderm bacteria. Proc Natl Acad Sci 115:E4284–E4293
Yadav AN, Kour D, Kaur T, Devi R, Yadav A, Dikilitas M, Abdel-Azeem AM, Ahluwalia AS, Saxena AK (2021) Biodiversity, and biotechnological contribution of beneficial soil microbiomes for nutrient cycling, plant growth improvement and nutrient uptake. Biocatalysis and Agricultural Biotechnology 33:102009
Zheng X, Song W, Guan E, Wang Y, Hu X, Liang H, Dong J (2020) Response in physicochemical properties of tobacco-growing soils and N/P/K accumulation in tobacco plant to tobacco straw biochar. J Soil Sci Plant Nutr 20:293–305
Acknowledgements
We would like to thank the Swebeswebe Nature reserve owners and personnel for providing access to the plant materials. We are grateful for using the University of Cape Town’s ICTS High-Performance Computing for data analysis. The authors thank Dr. Jeanne Korsman for reviewing this article. SMT acknowledges National Research Foundation Scholarship (PMDS22052313807) for financial support. RAM was supported by NSF Postdoctoral Research Fellowship in Biology (IOS-1906094) and by the Plant Resiliency Institute at Michigan State University and JMF acknowledges funding from the Department of Science and Innovation, National Research Foundation South African Research Chair grant number 98406.
Funding
Open access funding provided by University of Cape Town.
Author information
Authors and Affiliations
Contributions
SMT and JMF designed the study. JMF collected the field samples and SMT conducted the laboratory analysis. SMT performed data processes and wrote the manuscript. SMT and RAM performed data visualisation. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no potential or apparent conflict of interest that could have appeared to influence the work reported in this article.
Additional information
Responsible Editor: Manuel T. Oliveira.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(DOCX 1907 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tebele, S.M., Marks, R.A. & Farrant, J.M. Exploring the root-associated microbiome of the resurrection plant Myrothamnus flabellifolia. Plant Soil (2023). https://doi.org/10.1007/s11104-023-06019-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11104-023-06019-1