
Abstract
Cushing’s disease is a rare, but devastating condition, caused by corticotroph tumors. It rarely manifests as syndrome and very few isolated cases present with germline mutations. Instead, the vast majority of corticotroph tumors are sporadic monoclonal neoplasms. At present, the major recurrent somatic driver mutations are found in the USP8 gene, which encodes for a deubiquitinase that rescues proteins regulating ACTH synthesis. Almost half of functional corticotroph tumors carry somatic USP8 mutations that associate with a distinct transcriptomic and clinical profile. Other genes mutated in a small fraction of corticotroph tumors include the deubiquitinase encoding gene USP48 and the glucocorticoid receptor expressing NR3C1. Recent reports on somatic TP53 and ATRX mutations in corticotroph macroadenomas and carcinomas indicate that within specific patient subpopulations they are not as rare as assumed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the early 1930’s, “basophil adenomas of the anterior lobe of the pituitary” were established as the cause of Cushing’s disease [1]. Flash forward 50 years, advancements in molecular techniques established their monoclonality, but hypothesis based analyses failed to identify driver gene alterations (reviewed in [2]). The advent of next generation sequencing led to the discovery of the USP8 mutational hotspot and prompted us to revisit genes that were previously studied but considered to be rarely mutated, such as NR3C1 encoding for the glucocorticoid receptor and the tumor suppressor gene TP53. This short review summarizes our current knowledge on mutational events found in functional corticotroph tumors.
Germline mutations
Cushing’s disease is very rare in genetic syndromes, such as, multiple endocrine neoplasia 1 (MEN1 encoding for menin), MEN4 (CDKN1B encoding for the cell cycle inhibitor p27/Kip1) and Carney complex (PRKAR1A encoding for type 1 alpha regulatory subunit of the cAMP-dependent protein kinase A) (reviewed in [2, 3]). Similarly, it is very rarely found in patients with familial isolated pituitary adenomas (FIPA). Screening pediatric and adult patients with evidently sporadic corticotroph tumors revealed very few cases with germline mutations in CDKN1B or AIP (aryl hydrocarbon receptor-interacting protein; mutated in ~ 10% of FIPA).
DICER1 syndrome (inactivating mutations in the gene encoding for the type III endoribonuclease Dicer involved in small non-coding RNA processing) is a rare pediatric condition that predisposes to pleuropulmonary blastoma and other dysplasias. Cushing’s disease often manifests in the rare cases (< 1%) of DICER1 patients with unique pituitary blastoma [4].
Lynch syndrome predisposes to several cancers and is caused by germline mutations in DNA mismatch repair genes (e.g. MSH2, MSH6 and PMS2). Although pituitary tumors are seldom, when present they are often invasive corticotroph tumors or carcinomas (reviewed in [3]).
Screening of 182 patients with corticotroph tumors (including 116 pediatric) identified germline missense variants in CABLES1 (CDK5 and ABL1 enzyme substrate 1; mediates the antiproliferative action of glucocorticoids in corticotroph tumor cells) in 4 patients (2 pediatric) with macroadenomas (> 10 mm) and difficult to manage disease [5].
Sporadic
Cushing’s disease occurs mainly in a sporadic setting. Somatic mutations in genes involved in the trophic hypothalamic signaling are rare (guanine nucleotide binding protein alpha stimulating, GNAS) or nonexistent (corticotrophin releasing hormone receptor 1, CRHR1; vasopressin V3 receptor, V3R). Regarding the negative glucocorticoid feedback, somatic mutations in the NR3C1 (nuclear receptor subfamily 3 group C member 1) gene encoding for glucocorticoid receptor were initially considered as extremely rare, but recent sequencing efforts identified them in 0–10% of corticotroph tumors (reviewed in [2]). No distinguishing clinical parameters were attributed to NR3C1 mutants, probably due to low numbers [6].
At present, the genetic basis of Cushing’s disease is recurrent somatic mutations in the ubiquitin specific protease 8 (USP8) gene found in almost half of functional corticotroph tumors, including pediatric and Nelson’s syndrome [7,8,9,10; reviewed in 2, 3]. All USP8 mutations are somatic, with one germline mutation reported in a pediatric patient with severe Cushing’s disease (reviewed in [3]). The USP8 mutational hotspot disrupts the 14-3-3 binding motif resulting in enhanced catalytic activity. In corticotroph tumor cells, these highly active USP8 mutants rescue epidermal growth factor receptor (EGFR) from ligand-induced ubiquitination and lysosomal degradation and potentiate its signaling resulting in enhanced ACTH synthesis [6]. USP8 mutant corticotroph tumors show female predominance and are smaller, less invasive, but more likely to recur after surgery. They also show increased somatostatin receptor 5 (SSTR5) expression, suggesting favorable response to the SSTR5 ligand pasireotide [10]. In vitro studies demonstrated a better response to the antisecretory action of pasireotide in human USP8 mutant corticotroph tumors [11]. USP8 wild type tumors, on the other hand, are found in both female and male patients and are usually macroadenomas and more likely to be aggressive [7,8,9,10].
A second mutational hotspot was found in ubiquitin specific protease 48 (USP48) in a small percentage (up to 23%) of USP8 wild type tumors [12, 13]. Similar to USP8, USP48 mutant tumors are more frequently found in female patients and are smaller.
An unexpected gene picked by whole exome sequencing is the BRAF proto-oncogene [12]. The BRAF V600E mutation was found in 16.5% of USP8 wild type corticotroph tumors in a Chinese patient cohort, where it correlated with higher midnight ACTH and serum cortisol, but in only 1/91 Caucasian cases (female patient with macroadenoma) and none in other Asiatic patient cohorts [12,13,14].
Activating somatic mutations in the PIK3CA gene that encodes for the p110α catalytic subunit and constitutively activate the PI3K/AKT survival pathway were found in 1 out of 6 invasive corticotroph tumors, which also harbored an oncogenic HRAS mutation, and in none of the 16 noninvasive corticotroph tumors [15]. A separate study reported PIK3CA mutations in 1 out of 6 corticotroph microadenomas [16].
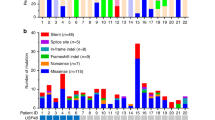
Finally, there are increasing reports of somatic mutations in TP53 and ATXR (alpha thalassemia/ mental retardation syndrome X-linked) in aggressive corticotroph tumors and corticotroph carcinomas. In selected populations of functional corticotroph tumors (i.e. USP8 wild type macroadenomas), TP53 mutations were found in up to ~ 35% of cases [13, 17]. Similarly, ATXR variants were found in 7/25 aggressive corticotroph tumors and carcinomas that were ATXR immunonegative [18]. Few cases carried mutations in both TP53 and ATXR or its interaction partner DAXX (death domain associated protein) [13, 18].
Multi-omics
Epigenetic profiling revealed that corticotroph tumors group in distinct miRNome and methylome clusters [10, 19]. The methylome of functional corticotroph tumors shows few areas of hypomethylation that include the POMC promoter [10, 19]. Notably corticotroph tumors from Cushing’s disease patients cluster separately from silent corticotroph tumors in terms of methylome and transcriptome [10]. Enrichment analysis revealed high expression of cell cycle related genes in functional corticotroph tumors [10]. Interestingly, USP8 wild type tumors show distinct transcriptomic profile with enrichment in epithelial-mesenchymal transition signatures, which may explain their more invasive nature [10].
Conclusion
During the last five years, we have gained a vast amount of information on the mutational landscape of Cushing’s disease. The discovery of the driver USP8 mutational hotspot highlighted distinct clinical profiles of Cushing’s disease patients: those with USP8 mutant tumors are predominantly female and less likely to go into remission after surgery, but have increased SSTR5 expression, which may indicate certain treatment strategies for the management of these tumors. In contrast, USP8 wild type tumors form a heterogeneous group that include tumors with no identified driver mutations, but also cases with TP53 mutations and ATXR mutations, which may allude to a more aggressive tumor behavior that may require intense management and long-term follow up. Although not of relevance in the general context of Cushing’s disease, isolated reports of somatic TP53 and ATXR mutations in aggressive corticotroph tumors and carcinomas, suggest that genetic TP53 screening and/or ATXR immunohistochemistry in selected patient subpopulations may indicate those more likely to require increased surveillance. Finally, ongoing multi-omics analyses are expected to provide with additional tools for the improved diagnosis and monitoring of patients with Cushing’s disease.
References
Cushing H (1932) The basophilic adenomas of the pituitary body and their clinical manifestations (pituitary basophilism) British Medical Journal. Bull Johns Hopkins Hosp 50:138–195
Simon J, Theodoropoulou M (2022) Genetics of Cushing’s disease. J Neuroendocrinol 25:e13148. https://doi.org/10.1111/cen.13457
Coopmans EC, Korbonits M (2022) Molecular genetic testing in the management of pituitary disease. Clin Endocrinol. https://doi.org/10.1111/cen.14706
Liu APY, Kelsey MM, Sabbaghian N, Park SH, Deal CL, Esbenshade AJ, Ploner O, Peet A, Traunecker H, Ahmed YHE, Zacharin M, Tiulpakov A, Lapshina AM, Walter AW, Dutta P, Rai A, Korbonits M, de Kock L, Nichols KE, Foulkes WD, Priest JR (2021) Clinical outcomes and complications of pituitary blastoma. J Clin Endocrinol Metab 106(2):351–363. https://doi.org/10.1210/clinem/dgaa857
Hernández-Ramírez LC, Gam R, Valdés N, Lodish MB, Pankratz N, Balsalobre A, Gauthier Y, Faucz FR, Trivellin G, Chittiboina P, Lane J, Kay DM, Dimopoulos A, Gaillard S, Neou M, Bertherat J, Assié G, Villa C, Mills JL, Drouin J, Stratakis CA (2017) Loss-of-function mutations in the CABLES1 gene are a novel cause of Cushing’s disease. Endocr Relat Cancer 24(8):379–392. https://doi.org/10.1530/ERC-17-0131
Miao H, Liu Y, Lu L, Gong F, Wang L, Duan L, Yao Y, Wang R, Chen S, Mao X, Zhang D, Heaney AP, Zhu H (2021) Effect of 3 NR3C1 mutations in the pathogenesis of pituitary ACTH adenoma. Endocrinology 162(11):bqab167. https://doi.org/10.1210/endocr/bqab167
Reincke M, Sbiera S, Hayakawa A, Theodoropoulou M, Osswald A, Beuschlein F, Meitinger T, Mizuno-Yamasaki E, Kawaguchi K, Saeki Y, Tanaka K, Wieland T, Graf E, Saeger W, Ronchi CL, Allolio B, Buchfelder M, Strom TM, Fassnacht M, Komada M (2015) Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat Genet 47(1):31–38. https://doi.org/10.1038/ng.3166
Pérez-Rivas LG, Theodoropoulou M, Puar TH, Fazel J, Stieg MR, Ferraù F, Assié G, Gadelha MR, Deutschbein T, Fragoso MC, Kusters B, Saeger W, Honegger J, Buchfelder M, Korbonits M, Bertherat J, Stalla GK, Hermus AR, Beuschlein F, Reincke M (2018) Somatic USP8 mutations are frequent events in corticotroph tumor progression causing Nelson’s tumor. Eur J Endocrinol 178(1):57–63. https://doi.org/10.1530/EJE-17-0634
Faucz FR, Tirosh A, Tatsi C, Berthon A, Hernández-Ramírez LC, Settas N, Angelousi A, Correa R, Papadakis GZ, Chittiboina P, Quezado M, Pankratz N, Lane J, Dimopoulos A, Mills JL, Lodish M, Stratakis CA (2017) Somatic USP8 gene mutations are a common cause of pediatric cushing disease. J Clin Endocrinol Metab 102(8):2836–2843. https://doi.org/10.1210/jc.2017-00161
Neou M, Villa C, Armignacco R, Jouinot A, Raffin-Sanson ML, Septier A, Letourneur F, Diry S, Diedisheim M, Izac B, Gaspar C, Perlemoine K, Verjus V, Bernier M, Boulin A, Emile JF, Bertagna X, Jaffrezic F, Laloe D, Baussart B, Bertherat J, Gaillard S, Assié G (2020) Pangenomic classification of pituitary neuroendocrine tumors. Cancer Cell 37(1):123-134.e5. https://doi.org/10.1016/j.ccell.2019.11.002
Albani A, Perez-Rivas LG, Tang S, Simon J, Lucia KE, Colón Bolea P, Schopohl J, Roeber S, Buchfelder M, Rotermund R, Flitsch J, Thorsteinsdottir J, Herms J, Stalla G, Reincke M, Theodoropoulou M (2022) Improved pasireotide response in USP8 mutant corticotroph tumours in vitro. Endocr Relat Cancer 29(8):503–511
Chen J, Jian X, Deng S, Ma Z, Shou X, Shen Y, Zhang Q, Song Z, Li Z, Peng H, Peng C, Chen M, Luo C, Zhao D, Ye Z, Shen M, Zhang Y, Zhou J, Fahira A, Wang Y, Li S, Zhang Z, Ye H, Li Y, Shen J, Chen H, Tang F, Yao Z, Shi Z, Chen C, Xie L, Wang Y, Fu C, Mao Y, Zhou L, Gao D, Yan H, Zhao Y, Huang C, Shi Y (2018) Identification of recurrent USP48 and BRAF mutations in Cushing’s disease. Nat Commun 9(1):3171. https://doi.org/10.1038/s41467-018-05275-5
Sbiera S, Perez-Rivas LG, Taranets L, Weigand I, Flitsch J, Graf E, Monoranu CM, Saeger W, Hagel C, Honegger J, Assie G, Hermus AR, Stalla GK, Herterich S, Ronchi CL, Deutschbein T, Reincke M, Strom TM, Popov N, Theodoropoulou M, Fassnacht M (2019) Driver mutations in USP8 wild-type Cushing’s disease. Neuro Oncol 21(10):1273–1283. https://doi.org/10.1093/neuonc/noz109
Abraham AP, Pai R, Beno DL, Chacko G, Asha HS, Rajaratnam S, Kapoor N, Thomas N, Chacko AG (2022) USP8, USP48, and BRAF mutations differ in their genotype-phenotype correlation in Asian Indian patients with Cushing’s disease. Endocrine 75(2):549–559. https://doi.org/10.1007/s12020-021-02903-x
Lin Y, Jiang X, Shen Y, Li M, Ma H, Xing M, Lu Y (2009) Frequent mutations and amplifications of the PIK3CA gene in pituitary tumors. Endocr Relat Cancer 16(1):301–310. https://doi.org/10.1677/ERC-08-0167
Murat CB, Braga PB, Fortes MA, Bronstein MD, Correa-Giannella ML, Giorgi RR (2012) Mutation and genomic amplification of the PIK3CA proto-oncogene in pituitary adenomas. Braz J Med Biol Res 45(9):851–855
Uzilov AV, Taik P, Cheesman KC, Javanmard P, Ying K, Roehnelt A, Wang H, Fink MY, Lau CY, Moe AS, Villar J, Bederson JB, Stewart AF, Donovan MJ, Mahajan M, Sebra R, Post KD, Chen R, Geer EB (2021) USP8 and TP53 drivers are associated with CNV in a corticotroph adenoma cohort enriched for aggressive tumors. J Clin Endocrinol Metab 106(3):826–842. https://doi.org/10.1210/clinem/dgaa853
Casar-Borota O, Boldt HB, Engström BE, Andersen MS, Baussart B, Bengtsson D, Berinder K, Ekman B, Feldt-Rasmussen U, Höybye C, Jørgensen JOL, Kolnes AJ, Korbonits M, Rasmussen ÅK, Lindsay JR, Loughrey PB, Maiter D, Manojlovic-Gacic E, Pahnke J, Poliani PL, Popovic V, Ragnarsson O, Schalin-Jäntti C, Scheie D, Tóth M, Villa C, Wirenfeldt M, Kunicki J, Burman P (2021) Corticotroph aggressive pituitary tumors and carcinomas frequently harbor ATRX mutations. J Clin Endocrinol Metab 106(4):1183–1194. https://doi.org/10.1210/clinem/dgaa749
Salomon MP, Wang X, Marzese DM, Hsu SC, Nelson N, Zhang X, Matsuba C, Takasumi Y, Ballesteros-Merino C, Fox BA, Barkhoudarian G, Kelly DF, Hoon DSB (2018) The epigenomic landscape of pituitary adenomas reveals specific alterations and differentiates among acromegaly, Cushing’s disease and endocrine-inactive subtypes. Clin Cancer Res 24(17):4126–4136. https://doi.org/10.1158/1078-0432.CCR-17-2206
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the Deutsche Forschungsgemeinschaft (DFG) (Project Number: 314061271-TRR 205) to MT and MR, by the Deutsche Forschungsgemeinschaft (DFG) (Project Number RE 752/30-1) to MR, and by a grant from the Else Kröner-Fresenius Stiftung (2012_A103 and 2015_A228) to MR.
Author information
Authors and Affiliations
Contributions
M.T. and M.R. wrote the main manuscript text. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The authors have no conflicts of interest to declare that are relevant to the content of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Theodoropoulou, M., Reincke, M. Genetics of Cushing’s disease: from the lab to clinical practice. Pituitary 25, 689–692 (2022). https://doi.org/10.1007/s11102-022-01253-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11102-022-01253-9