
Abstract
Individuals surviving cancer and brain tumors may experience growth hormone (GH) deficiency as a result of tumor growth, surgical resection and/or radiotherapy involving the hypothalamic-pituitary region. Given the pro-mitogenic and anti-apoptotic properties of GH and insulin-like growth factor-I, the safety of GH replacement in this population has raised hypothetical safety concerns that have been debated for decades. Data from multicenter studies with extended follow-up have generally not found significant associations between GH replacement and cancer recurrence or mortality from cancer among childhood cancer survivors. Potential associations with secondary neoplasms, especially solid tumors, have been reported, although this risk appears to decline with longer follow-up. Data from survivors of pediatric or adult cancers who are treated with GH during adulthood are scarce, and the risk versus benefit profile of GH replacement of this population remains unclear. Studies pertaining to the safety of GH replacement in individuals treated for nonmalignant brain tumors, including craniopharyngioma and non-functioning pituitary adenoma, have generally been reassuring with regards to the risk of tumor recurrence. The present review offers a summary of the most current medical literature regarding GH treatment of patients who have survived cancer and brain tumors, with the emphasis on areas where active research is required and where consensus on clinical practice is lacking.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The large-scale production of biosynthetic growth hormone (GH) since 1985 has allowed the widespread treatment of children with different conditions associated with short stature and the treatment of adults with growth hormone deficiency (GHD), with approved indications varying among countries [1]. Some cancer survivors may require treatment with GH due to the development of GHD related to the malignancy and/or to adverse effects of its treatment, including chemotherapy, surgery, radiotherapy and biological therapy (antigen–specific monoclonal antibodies and cytotoxic T-cells) [2]. The treatment of malignancies has greatly improved over the past four decades, with a substantial increase in the number of cancer survivors. Poor longitudinal growth and GHD are often a consequence of cancer treatment during childhood [3]. In adult life, GH replacement therapy attenuates the clinical features and comorbities associated with GHD. This review presents a summary of the most current medical literature related to safety of GH treatment in children and adults who have survived cancer and brain tumors, with emphasis on the main questions where consensus on clinical practice is lacking (Table 1).
Cancer incidence
Approximately 360,000 documented cases of cancer occurred in children in 2015 [4], and it is a major cause of death worldwide. The cancer incidence rate among 0–19-year-old individuals has been reported at 155,8 per million person-years with numbers slightly higher in boys than in girls [5]. The most common cancers in childhood are leukemias (28.8%), central nervous system (CNS) tumors (24.0%) and lymphomas (11.2%) [4]. Acute lymphoblastic leukemia (ALL) accounts for approximately 80% of leukemia cases in childhood, with event-free survival rates approaching 90% with advanced multiagent chemotherapy [6]. Medulloblastoma, an embryonal tumor of the posterior fossa, is the most frequent malignant CNS neoplasm in children, and is most often diagnosed before 15 years of age. Craniopharyngioma accounts for about 5–10% of pediatric CNS tumors [7,8,9]. Lymphomas are the third most frequent childhood malignancy [4, 5, 10], including Hodgkin and non-Hodgkin lymphomas, with considerable risk for secondary malignancies [10].
Growth hormone treatment and cancer in different cohorts of patients
A possible association between GH treatment and malignancy emerged from cases of leukemia in GH treated patients reported in the 1980s. Until de year 1992, 31 cases were reported, including recipients of pituitary derived GH and patients with known risk factors for malignancy [11, 12]. This finding has led to the evaluation of malignancy risk in GH-treated subjects [13,14,15,16,17,18], especially in childhood cancer survivors (CCS) with GHD [16,17,18,19,20,21,22,23,24,25,26,27,28,29]. Table 2 shows studies addressing this issue including patients treated with GH due to different indications. Population-based cohorts, long-term surveillance studies from pharmaceutical companies and collaborative international cohort studies, including SAGhE (Safety and Appropriateness of Growth Hormone Treatments in Europe, with 396,344 person-years, averaging 16.5 years per patient), showed that the overall risk of primary cancer was not increased in patients who had received GH treatment and who did not have previous risk factors for malignancy. These cohorts included children with isolated GHD, idiopathic short stature and being born small for gestational age [17, 18, 28,29,30]. In contrast, an increased risk of malignancy was suggested in those children who received GH therapy and had underlying conditions that are associated with an increased predisposition to cancer, including RASopathies such as Noonan syndrome, chromosomal breakage syndromes or DNA-repair disorders, such as Fanconi’s anemia and Bloom syndrome [30, 31]. An increased risk for malignancy was also found in patients treated with CNS radiotherapy in some [18, 26, 27, 32, 33], but not all studies [2, 19,20,21, 25, 26]. Studies in patients with benign intracranial tumors, including craniopharyngioma, did not show an increased risk of recurrence of these tumors in those individuals treated with GH [25, 26, 34,35,36].
GH/IGF-I and cancer – background
Experimental evidence for the role of GH-IGF-I system in carcinogenesis
The effects of GH in stimulating mitosis, cell differentiation and growth has been known since the early years of the twentieth century [37, 38]. In the 1950s, it was shown that GH action in peripheral tissues could be mediated by insulin-like growth factor I (IGF-I) [39], and since then, novel components of the GH-IGF-I signaling system and their roles on normal and abnormal cell growth and metabolism have been progressively unraveled [37,38,39].
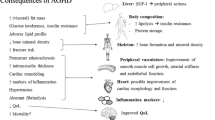
The mechanisms involved in the control of cell growth, differentiation and death are tightly regulated by a complex cascade of molecular events that, when disrupted, can lead to an increased risk of malignant transformation. Experimental models have shown the ability of endocrine and paracrine GH and IGF-I to promote cell proliferation and differentiation, angiogenesis, and inhibition of apoptosis, either directly or by synergy with other growth factors [30, 40,41,42,43,44]. In the multistep process of tumorigenesis, GH and IGF may potentially increase the number of mutations by reducing time for DNA repair during rapid progression of neoplastic cells [45]. In contrast, other players of the GH-IGF system, such as insulin-like growth factor binding protein 3 (IGFBP3) [46] and IGF-II receptors [47], inhibit mitogenesis, stimulate apoptosis and modulate IGF-I actions, thus acting as protective factors against tumor progression [30, 40,41,42, 46, 48] (Fig. 1). Additionally, various solid and hematologic malignancies have been associated with local production of GH, IGF-I, and IGFBPs, normal or altered expression of several receptors of the GH-IGF system, and deregulation of miRNAs induced by GH and IGF-I [30]. The final effect of these opposed endocrine and paracrine forces of the GH-IGF system in a tissue-specific environment might be critical for normal and abnormal cell growth, but how and when GH per se may participate in this process is largely unknown.

Diagrammatic representation of endocrine and paracrine effects of GH on mitogenesis, angiogenesis and apoptosis, either directly or by synergy with other growth factors. GH stimulates both IGF-I and IGBP3 [41]. While IGF-I and IGFBP proteases favors cell proliferation and inhibits apoptosis [43, 44, 47], IGFBP3 [46] and IGF-II receptors [47] act in the opposite direction, via an IGF-independent pathway. The result of these opposed forces in a tissue-specific environment might be critical for normal and abnormal cell growth [41]. Dotted lines: inhibition. Continuous line: stimulation.
The potential role of the GH-IGF-I system in the development of specific types of cancer has been comprehensively reviewed in several recent publications [30, 41]. It is important to note that GH-induced intracellular signaling pathways have been identified as the third most highly associated with breast cancer susceptibility among 421 pathways containing 3962 genes in a human genome-wide association study [49]. In breast cancer, pituitary and exogenous GH seems to be less involved, while local expression of GH has been shown to have profound autocrine/paracrine effects in breast tissue independent of IGF-I, leading to increased epithelial cell proliferation, and conferring an invasive phenotype on mammary carcinoma cells by affecting epithelial–mesenchymal transition [50, 51]. Moreover, a model to explain neoplastic colon growth has been proposed in which high endocrine or autocrine GH levels – for instance, as a result of acromegaly or colonic DNA damage and inflammation – inactivate tumor-suppressor genes, suppress apoptosis, and stimulates epithelial to mesenchymal transition, leading to changes in the intestinal mucosal field that favor malignant transformation [52]. Consequently, several components of the GH-IGF-I signaling cascades have been investigated as targets for the treatment of breast and colonic cancer, as well as for several other malignancies [53,54,55,56].
Cumulative data obtained from natural or genetically modified animals exhibiting normal or disrupted GH production or action, have also brought a wealth of evidence linking GH axis and carcinogenesis [56, 57]. The repression of the GH-IGF signaling system in many of these models has been associated with significant reductions in cancer rates and increased longevity. In contrast, transgenic mice with excessive circulating levels of GH or tissue overexpression of IGF-I exhibit an increased risk for hyperplasia and tumor formation [30, 58].
Epidemiological and clinical evidence in humans
From the 1990s onwards, a substantial amount of epidemiological data has found associations between serum IGF-I levels in the highest quartiles of the normal reference range with an increased incidence of several cancers in the general population [59,60,61,62,63]. Moreover, associations between normal-low IGFBP3 levels and prostate cancer and breast cancer in postmenopausal women were also observed, but this association was likely influenced by the concomitant association of normal-high IGF-I levels and the presence of these tumors in the general population [60]. In the last decade, data obtained from the European Prospective Investigation into Cancer and Nutrition (EPIC cohort) involving more than 500,000 healthy volunteers have confirmed the association between higher circulating IGF-I levels and the risk of breast cancer in receptor-positive tumors in women older than 50 years, thyroid cancer, low-grade gliomas and acoustic neuromas [64,65,66]. Nevertheless, there are many caveats and confounding factors in these epidemiological studies, and the associations are usually modest and without a threshold IGF-I level that would allow its use in cancer screening or monitoring. Therefore, a potentially causal relationship between elevated IGF-I and a higher risk for cancer lacks the certainty needed to apply these findings in routine clinical care.
In humans, congenital GHD or GH resistance (Laron dwarfism) have been associated with a lower incidence of malignancies, similarly to what is observed in animal models [67,68,69]. Cells treated with serum obtained from Ecuadorian patients with Laron syndrome have been shown to exhibit reduced DNA breaks and increased apoptosis [68]. In the Ecuadorian cohort, not a single case of cancer was noted during a 22 years follow-up, while in cohorts of GHD patients due to GHRH-R defect or congenital isolated or multiple GHD, in which small amounts of circulating GH can be demonstrated, few cases of cancer have been reported both in naïve and in patients treated with GH [67]. These data suggest that the protection against cancer in human GHD is not absolute and that GHR signaling via pituitary or exogenous GH might be mitogenic in the long term. In contrast, acquired hypopituitarism has been linked to either an increased risk of having cancer or an increased risk of dying from cancer in some, but not all, retrospective studies [70, 71]. Of note, in acquired hypopituitarism, factors other than GHD may play a role in these observed associations, such as the underlying pituitary disease, inherent elevated risk for tumors in patients with pituitary adenomas, radiotherapy, associated morbidities and inadequate or inappropriate pituitary hormone replacement [70, 71]. Studies investigating the risk of cancer in acromegaly patients who are chronically exposed to very high levels of GH-IGF-I for many years have produced inconsistent and controversial findings. Acromegaly is characterized by a prolonged and excessive secretion of GH which, in turn, induces both IGF-I and IGFBP3 production, resulting in a dysregulated, unpredictable balance of cell cycle regulation, characterized by signals for cell growth competing with signals for cell death [72]. The ultimate consequences of these antagonistic mechanisms are the basis for concerns and disputes on the cancer risks in patients with active acromegaly. More recent data suggest that other factors, such as age, comorbidities, inhered predisposition and regional differences of cancer prevalence are likely to contribute equally or more to the risk of cancer in acromegaly than the GH-IGF-I levels per se [30, 72].
GH treatment in cancer survivors in childhood
Many factors lead to reductions in longitudinal growth in CCS, including poor nutrition, low body mass index, long-term treatment with glucocorticoids, growth plate damage, background syndromic short stature and GHD. Tumors and surgeries that involve the hypothalamic-pituitary region are major risk factors for hypothalamic-pituitary axis (HPA) dysfunction, which often are present at baseline or soon after the procedure. On the other hand, CNS radiation frequently causes hypothalamic-pituitary dysfunctions several months or years after treatment [73]. GHD is the most frequent hypothalamic-pituitary disorder in CCS, with a prevalence of 22.2% in all groups and of 40.2% following radiation of hypothalamic-pituitary region [3, 74]. The average time between completion of tumor therapy and the onset of poor longitudinal growth has been reported as 37.7 months in children treated with radiotherapy [75], ranging from 3 months to 5 years [76]. The risk of short adult height is higher when radiotherapy is given at younger age and before puberty [73, 77]. GHD has been associated with radiotherapy doses ≥ 18 Gy to the hypothalamic-pituitary region area as well as with total body irradiation using a 10 Gy single dose or fractionated doses totaling 12 Gy [77]. In a study of 192 children with primary brain tumors, the authors predicted the occurrence of GHD on the basis of dose and time after irradiation: GHD appeared 12 months if the radiotherapy dose was greater than 60 Gy, in 36 months if the dose was between 25 and 30 Gy, and 60 months if the dose was between 15 to 20 Gy [78]. Morphological changes affecting the pituitary are associated with linear growth decline among patients surviving ALL [79] and nasopharyngeal carcinoma [80]. The pituitary height was significantly reduced after radiotherapy, possibly due to radiation-induced pituitary cell apoptosis, or because radiation may have affected vascular function and oxygen supply to the pituitary gland [80]. Proton based radiotherapy is increasingly used for the treatment of brain tumors, a development that carries a promise in reducing scatter to normal tissue, including the hypothalamic-pituitary area when lesions are located elsewhere in the head [81, 82].
Spinal irradiation is an independent risk factor for growth impairment; it may cause disproportionate height due to reduced growth of vertebral bodies and spinal deformities [83].The resulting skeletal dysplasia can be detected by measuring an increased sitting height to standing height ratio [84, 85]. CCS treated with higher doses of spinal radiation (> 20 Gy) at a younger age, and to a larger volume of the spine, are at increased risk of short adult height [77]. The disproportionate growth may be evident as early as one year following radiotherapy. It becomes progressively more evident during puberty [84] and GH replacement in GHD cancer survivors who also received spinal irradiation may result in an improvement in leg length but not in spinal length and total height [77]. GHD is unlikely after traditional chemotherapy, but some newer agents may interfere with normal growth by affecting growth plates or dysregulating GH-IGF-I signaling pathways (tyrosine kinase inhibitors) or causing an autoimmune hypophysitis (immune checkpoint inhibitors) [73]. Some alkylating agents, such as busulfan and lomustine, may increase the risk for short stature, particularly after leukemia or neuroblastoma, possibly by increasing the vulnerability of the hypothalamus-pituitary to damage from radiotherapy or by directly affecting the hypothalamic-pituitary axis [85]. They are also risk factors for gonadal dysfunction and hypothyroidism, which may further exacerbate growth impairment [85]. Other chemotherapy agents may directly damage the growth plates and cause severe short stature; these include cis-retinoic acid for the treatment of neuroblastoma and hedgehog pathway inhibitors [86, 87].
The growth of children who have undergone cancer treatment should be assessed every 6–12 months, and lifelong assessment may be necessary to screen for the development of GHD. It is recommended to measure standing and sitting height in children treated with spinal radiotherapy, i.e., total body irradiation, craniospinal irradiation, as well as radiation to the chest, abdomen, or pelvis [77]. GHD should be suspected when growth deceleration is observed with a deflection of at least 0.3 SDS/year or height deviates from the familial background [88]. The measurement of serum IGF-I levels is not recommended as a biomarker for the diagnosis of GHD in CCS, especially among those exposed to hypothalamic-pituitary irradiation [77]. Normal serum IGF-I levels have been observed in CCS who failed in GH dynamic testing, especially those treated with low doses of radiotherapy. In survivors of childhood ALL treated with cranial radiotherapy, IGF-I levels < −2 SDS showed a sensitivity of only 17.86%; sensitivity of IGF-I as a diagnostic tool was even lower (7.14%) in subjects treated with 14.4 Gy total body irradiation before bone marrow transplantation. The use of IGF-I as a screening tool should be restricted to patients treated with high cranial radiation doses [89]. Similarly, serum IGFBP3 do not aid in the diagnosis of GHD in CCS children [89,90,91].
In patients with a high probability of GHD (poor growth, other pituitary hormones deficiencies), one GH provocative test is sufficient to make the diagnosis, after excluding other growth failure etiologies (e.g., total body irradiation, imatinib mesylate and cis-retinoic acid treatment) [90]. The insulin tolerance test has been considered as the gold standard provocative stimulation test to make the diagnosis of GHD, but caution should be exerted and alternative secretagogues should be considered in patients at risk for seizures [90]. Growth hormone-releasing hormone (GHRH) alone or in combination with arginine is not recommended in CCS after hypothalamic-pituitary irradiation because radiation doses less than 40 Gy cause predominantly hypothalamic damage, and consequently, exogenous hypothalamic hormones administration may cause false-negative results [74, 77]. The Pediatric Endocrine Society guidelines suggest that one can make the diagnosis of GHD without using a GH dynamic test in patients with the following conditions: auxological criteria as mentioned before, hypothalamic-pituitary defect (such as major congenital malformation, tumor or irradiation), and deficiency of at least one additional pituitary hormone [92]. Provocative GH stimulation tests are also not needed in patients who have three other confirmed hypothalamic-pituitary hormone deficiencies [77]. According to Cattoni et al. [89], the peak GH level attained on a stimulation test decreases on average 0.1 μg/L for each additional Gy of pituitary exposure to radiation. It is important to avoid false-positive tests in the diagnosis of GHD, with special attention to obesity and considerations about GH assays and cutoff values [90]. Brain imaging should be performed prior to starting GH therapy to rule out a preexisting tumor and to serve as a baseline study in children whose cancer monitoring does not routinely include CNS imaging [25].
Treatment with GH is usually indicated in CCS with proven GHD after careful discussion with the patient, his/her family if appropriate, as well as with the treating oncologist or neurosurgeon. The most appropriate and safest time to start GH is still a topic of debate, primarily due to a paucity of data and lack of controlled trials [93]. The Endocrine Society and the Pediatric Endocrine Society guidelines suggest waiting for 12 months after completing cancer treatments [77, 92]. In children with residual tumor and stable disease, as is often the case with optic pathway tumors and low-grade gliomas, the safety and timing of the initiation of GH treatment should specifically be discussed with the oncologist [77]. In children with craniopharyngiomas, which are considered benign tumors, GH therapy may be safely initiated as early as 0.7 year from diagnosis [77]. The safety of GH replacement for children on treatment with tyrosine kinase inhibitors has not been established and therefore is not recommended at the present time [77]. GH doses are similar to those used for the treatment of GHD in non-CCS [77, 92], with a starting dose of 0.022–0.035 mg/kg/day (0.15–0.25 mg/kg/week) and individualization of subsequent dosing [92]. Serum IGF-I levels should be measured during the treatment and kept in the normal range for sex, age, and pubertal status [77, 88, 92]. In conditions associated with increased predisposition to malignancy, e.g., Down syndrome, Fanconi anemia, Noonan syndrome, Bloom syndrome, neurofibromatosis 1, among others, the decision of whether to start GH therapy or not is a very controversial issue [92, 93]. Table 3 summarizes the main recommendations for GH treatment in CCS.
The use of GH in CCS carries additional safety concerns. Scoliosis is common following either spinal surgery and/or spinal or craniospinal irradiation [77, 94], and GH therapy may exacerbate this condition due to rapid growth [92]. Slipped capital femoral epiphysis is more frequently seen during GH treatment in GHD following intracranial neoplasms, craniopharyngioma and after bone marrow transplantation compared with patients with idiopathic GHD. GH replacement is also associated with a slightly increased risk of intracranial hypertension [92]. Some chemotherapy agents, e.g., cisplatin, alkylating agents, anthracyclines, and camptothecins, may induce insulin resistance, hyperinsulinemia, and impaired glucose control by directly influencing insulin sensitivity [95,96,97]. Survivors of craniopharyngioma are at risk for hypothalamic obesity and insulin resistance [73]. Hence, cranial radiotherapy may induce GHD, which may promote the development of metabolic syndrome. Cranial radiotherapy is a major risk factor for obesity, dyslipidemia, and insulin resistance in CCS [95]. A higher body mass index before hematopoietic stem cell transplantation for childhood ALL and GHD were also associated with increased risk for metabolic syndrome [97].
GH treatment in cancer survivors during adulthood
Adults with hypopituitarism and documented GHD have distinct clinical features that have been demonstrated in many studies [98, 99]. These include overweight, abdominal obesity, reduced lean muscle mass, decreased extracellular fluid volume, raised cholesterol and triglycerides, and low bone mineral density with likely increased vertebral fracture rate [99]. Adults with hypopituitarism including GHD also describe reduced energy and well-being, possibly due to reduced muscle strength and exercise capacity. Many of these outcomes have been shown to improve with GH replacement, which has thus garnered increasing support by expert panels over the past two decades [99,100,101,102]. Excess mortality due to cardiovascular risk factors has also been reported in adults with multiple hypothalamic-pituitary deficits including GHD. However, the specific contribution of GHD to all causes of mortality in this population has yet to be established, especially in light of markedly higher mortality rates in adults with a history of cancer or craniopharyngioma when compared to those with non-functioning pituitary adenoma [103,104,105]. Despite the potential benefits, the perceived increased risk for tumor recurrence and the controversy surrounding a potential association with secondary neoplasia [24] may explain provider reluctance to offer at-risk adult CCS testing for and treatment of GHD. For instance, in the St. Jude Lifetime Cohort study, more than 99% of patients with GHD were not given GH replacement therapy [106].
GHD and hypopituitarism were commonly reported late effects in CCS, in particular those surviving CNS tumors, and older patients treated for childhood leukemia during the 1970s [106,107,108,109]. The proportion of individuals receiving radiation therapy for these tumors decreased during the 1990s [109]. In general, the same diagnostic approach for GHD can be used in adult cancer survivors as in patients with hypothalamic-pituitary disorders due to other causes [100], with adjustments made to testing approaches as summarized in Sect. 3, especially in relation to the GHRH and the combined GHRH-arginine stimulation tests after brain irradiation. Macimorelin, an oral ghrelin mimetic, has demonstrated a diagnostic accuracy similar to that of the ITT, is well-tolerated and safe. In adults without other pituitary hormonal deficiencies, it is good practice to perform two stimulation tests to diagnose GHD unless IGF-1 is also below the lower limit of the reference range [99, 110]. GH replacement in adult cancer survivors involves individualized dose titration that maintains serum IGF-I levels within the age-related reference range and achieves an appropriate clinical response, for example, an improvement in the AGHDA (Adult Growth Hormone Deficiency Assessment) score [100, 101, 111, 112]. Hypopituitary women treated with GH should receive estrogen replacement preferably by non-oral route, as oral estrogen reduces the serum IGF-I response to GH therapy [113]. The expected benefits of GH treatment are the same as those reported in adults with GHD due to other causes not related to cancer [2], including reduction of total body fat mass and abdominal fat mass, improvement in lipid profile, left ventricular systolic function, muscle strength, quality of life (QoL) and cognitive performance [114,115,116,117,118,119].
Hypopituitary adults treated with GH have been reported to have a similar subsequent cancer risk compared to individuals not treated with GH [120,121,122], whereas a meta-analysis even suggested that the cancer risk could be reduced [123], although this observation is likely due to selection bias. Data from the ongoing PATRO pharmaceutical sponsored post-marketing surveillance study of children (n = 136) and adults (n = 293) with GHD were analyzed over 10 years of real-life clinical experience. No increased risk was seen for neoplasia compared with other GH treatments [124]. Further analysis from the same database including 1293 adults, of which 637 (49.3%) were GH treatment-naïve at study entry and the majority having multiple pituitary hormone deficiency (n = 1128, 87.2%), demonstrated that GH treatment did not result in an increased cancer risk, although an increased risk of second new malignancies in patients with previous cancer could not be excluded [125]. Conversely, the SAGhE studies of GH therapy in young adults with childhood-onset GHD suggested an increased risk for certain cancer types or a trend towards increased risk of mortality with GH therapy [17, 126]. These divergent results could be explained by a variety of confounding factors, possibly unrelated to GHD, including differences in sampling size and study design, types of cancers, prevalence of specific cancer types in the control populations, underlying pituitary disease, surgery, radiation, associated co-morbidities, and sub-optimal pituitary hormone replacement therapies. Therefore, clinicians have to navigate challenging safety questions when considering GH replacement therapy, especially in newly diagnosed GHD adults with a previous history of cancer and those who developed cancer while previously receiving GH. Current Endocrine Society recommendations suggest starting GH therapy after 1 year of disease remission following childhood cancer treatment [77], but it remains unknown whether 1 year is sufficient or deferring beyond 1 year is safer as there are no hard data to support this recommendation. In the case of chronic or not totally eradicable oncological disease, the choice of whether to start GH or not should be tailored according to cancer type and patient comorbidities. If GH therapy is considered, the decision should be individualized after a thorough discussion with the patient and clearance from the oncologist [77, 127]. Published guidelines do not recommend treatment and the label contains a “black box” warning against using the GH in patients with active cancer. For these patients, it may be reasonable to start GH at least two years after cancer remission if the patient has expressed a keen desire to start GH replacement therapy; however, these patients should be counseled that conclusive data on the effects of GH replacement and cancer risk are still lacking [127]. The benefits of GH replacement should be carefully balanced against the possible, yet unsubstantiated, increased cancer risk. A prospective surveillance study of a large cohort of GH-treated patients using optimal dosing and untreated GHD hypopituitary patients with a history of cancer in remission is an unmet need; however, conducting such a large and long-term study will be challenging due to the high cost and adjustments needed to be made for the differences in demographics between GH-treated and untreated subjects [128].
Adherence to daily subcutaneous GH injection is often challenging. Numerous studies have shown that most children and adults are non-adherent to daily GH injections [129, 130] leading to high treatment discontinuation rates [131, 132]. Thus, there has been a push to develop long-acting GH formulations to allow reductions of injection frequency in order to improve treatment adherence [133,134,135]. At present, two long-acting GH formulations have been introduced to the market in Asia, several long-acting GH formulations are in the latter stages of development, and one has been approved in the United States and Europe but is not commercially available yet [133, 135]. Current evidence indicates that long-acting GH formulations are safe in non-cancer GH-deficient patients [133, 135], although it may be argued that these studies are too short of a duration to address this question. Whether they are equally safe in GH-deficient cancer survivors especially given their differences in pharmacokinetic and pharmacodynamic profiles compared to daily GH injections requires further long-term studies.
GH treatment and second neoplasm
The risk of developing a second neoplasm is an important consideration during follow-up of cancer survivors, as CCS are at a significantly higher risk during their lifetime. The most frequently reported second neoplasms are non-melanoma skin cancer, breast cancer, meningiomas, thyroid cancer, soft tissue sarcomas, and CNS tumors, by order of frequency [136, 137]. Genetic factors and adverse effects from cancer treatment may synergize in raising the risk among survivors [106]. Cranial radiotherapy is known to increase the risk for developing meningioma [109, 137]. While a nonsignificant association was reported between GH replacement therapy and recurrence of the primary cancer, studies have reported that treatment with GH may increase the risk for secondary neoplasia in CCS [17, 22, 23, 138,139,140,141]. In the SAGhE cohort, of 10,403 patients treated with GH, 38 were diagnosed with meningioma. Thirty of them had been treated with cranio-spinal radiotherapy, given a standardized incidence ratio (SIR) for meningioma in the overall cohort of 75.4 [18]. However, the risk of meningioma was not increased in patients whose diagnosis before GH treatment was not cancer (SIR = 2.4). There were no significant associations between this risk for meningioma and age of GH start, time since starting treatment, mean daily GH dose, duration of treatment, or cumulative dose of GH [18]. Sklar and colleagues [23] reported an increased risk of second solid neoplasms after starting GH (RR, 3.21), mainly in survivors of acute leukemia/lymphoma (RR, 4.98), with no secondary leukemias. In a control-matched study with patients treated with CNS irradiation with a follow-up of 14.5 years, the incidence of recurrent or secondary tumors did not differ significantly between GH-treated and controls [25]. The risk of GH-dependent effects on secondary tumor was 10% in both GH-treated and untreated patients with a mean latency time of 22.5 years [25]. Again, meningiomas were the most frequent second tumor in the GH-treated groups [24,25,26]. Female sex, young age at primary cancer diagnosis, a long time period since cranial irradiation [2], CNS radiation dose ≥ 20 Gy and cumulative doses of multiple alkylating agents [142] were associated with meningioma development. These reports reinforce the need to interpret cautiously the risk of secondary tumors in GH treated patients. It is advisable to consider the relatively small number of events, high confidence intervals and low levels of significance. In addition, the risk of outcome bias due to the fact that CCS are already at increased risk of second neoplasm [18].
GH treatment in non-malignant sellar tumors
Craniopharyngioma
Craniopharyngioma represents about 5–10% of pediatric CNS tumors and 3% of intracranial tumors for all age groups, with peak incidences in the age categories of 5 to 9 and 40 to 44 years old. Approximately 40–87% of children will have at least one deficient hypothalamic-pituitary hormone at time of diagnosis [7,8,9, 143, 144]. Craniopharyngioma is primarily treated surgically, with or without adjuvant radiotherapy [7, 8, 143]. Patients who are not GHD at diagnosis will often develop GHD post resection or following focal radiotherapy [143]. Progression rates range from 71–90% after surgery alone to 21% when radiotherapy is used after partial resection [8]. The safety concerns related to GH treatment in patients with craniopharyngioma are the potential effects of GH on growth of known residual disease or recurrence of radiologically “cured” disease following surgical intervention ± radiotherapy.
Carefully conducted case control studies provide most of the information on this tumor. The majority of these studies report on a mix of childhood and adult-onset disease. In the study of Olsson et al. [145] that included all age categories, 29% of the patients in the GH treated group had residual tumor compared to 47% in the untreated group. However, there was no difference in tumor progression between the two groups up to 15 years after treatment. Other similar studies where patients with craniopharyngiomas were studied alone or included in larger cohorts of sellar tumors, have also failed to demonstrate any evidence of craniopharyngioma recurrence with GH therapy [19, 146,147,148,149,150]. A recent well conducted, single centre, retrospective analysis included 89 patients with adult onset craniopharyngioma with a median duration of treatment follow-up of greater than 7 years, again demonstrated no increased risk of craniopharyngioma recurrence following initial neurosurgery in those treated with GH therapy [151].
Safety data have also been derived from pharmaceutical sponsored post marketing surveillance studies in both children and adults with craniopharyngioma. The report from the KIMS (Pharmacia & Upjohn International Metabolic Database) with 1000 adult patients registered at the time of the analysis, recorded 12% with a craniopharyngioma. Only 6 patients overall were reported as having tumor recurrence on GH therapy, none of whom had a craniopharyngioma [152]. More recently, the HypoCCS study of 1058 adults with craniopharyngioma found no association of GH treatment with risk of recurrence (RR 1.32; range 0.53–3.31, p = 0.55) with a mean follow-up of 4.8yrs [121]. Other open-label post marketing surveillance registries have similarly failed to show any increased risk of craniopharyngioma recurrence with GH treatment in childhood onset craniopharyngiomas [141, 153,154,155]. A recent meta-analysis of 10 studies investigating GH treatment in children with craniopharyngiomas compared 3436 patients who received GH with 51 who did not. Their results suggested that recurrence rates of craniopharyngioma were reduced in children receiving GH, although this may reflect selection bias within the individual studies towards favoring GH treatment in those with less aggressive tumors [36].
Despite the reassuring clinical data, in vitro studies have demonstrated growth of craniopharyngioma cells in culture in the presence of exogenous GH [156]. GH receptors have been identified on craniopharyngiomas [157], and increased GH receptors expression may reflect tumor aggressiveness with potential prognostic implications in some patients [158]. A diagnosis of craniopharyngioma is associated with an excess mortality rate compared to the general population, with most recent analyses demonstrating a SMR of between 2–3 [159, 160]. There is also an increased risk of metabolic complications, with data from KIMS demonstrating that patients with craniopharyngioma had a ninefold increased risk for developing diabetes mellitus compared to a background Swedish population [160]. Use of GH has not been shown to increase the risk of diabetes mellitus in this population, [159] although one study showed a decline in insulin sensitivity markers over a longer duration of GH treatment [161].
Pituitary adenomas
Non-Functioning Pituitary Adenomas (NFPAs) are the most frequent pituitary tumor leading to adult GHD. Alongside craniopharyngiomas, NFPAs make up the majority of patients represented in the largest registries of GH replacement therapy [162, 163]. The data with regard to growth or recurrence of NFPAs with GH therapy are reassuring. The most recent and largest, single centre study, of adult patients treated surgically for NFPAs demonstrated no evidence of a difference in NFPA recurrence in 74 patients treated with GH compared to 120 patients who chose not to receive GH [151]. A retrospective case-controlled study from 2009 provided 10-year tumor progression free data in over 200 patients with NFPA and GHD, again demonstrating no difference in NFPA progression between those treated or not treated with GH [164]. These reassuring data reflect a number of other carefully conducted case-controlled studies and post-marketing surveillance studies [121, 152, 165,166,167,168,169] with up to 14 years of follow-up analyzed.
The safety of GH in the management of GHD in patients with an original diagnosis of acromegaly (acroGHD) is less well elucidated. A randomized placebo-controlled trial of GH in 30 patients [170] showed improved QoL and body composition with no detrimental effects or safety concerns over 6 months although an earlier open label study in 20 patients had shown increased cardiovascular events in the GH treated group [171]. Retrospective analysis of the KIMS data set, comparing acroGHD and NFPA GHD patients treated with GH to a background reference population demonstrated an increased cardiovascular mortality in the acroGHD compared to background population and the NFPA group. It was not possible to determine if this was related to the use of GH or to the previous acromegaly, but caution was advised in treating this group of patients with GH. Of note, markers of glucose tolerance showed a small but significant decline over time in both the GH treated acroGHD and NFPA group [172].
Although the data are reassuring with regard to overall risk of recurrence/regrowth of the original tumor not being increased with GH therapy, the majority of craniopharyngiomas that do recur will recur within 7 years, and those non-functioning adenomas that recur will grow within 5 years, with growth becoming less common after 10 years [173]. Many of the studies on the safety of GH treatment in these patients have a shorter duration of follow-up and therefore may underestimate recurrence and regrowth rates.
Summary
GH replacement has been shown to improve linear growth during childhood as well as to enhance body composition, bone health, quality of life and well-being parameters in adults. Many studies and several expert panels have attempted to assess whether these benefits override potential safety concerns in individuals who have experienced a malignancy or CNS benign neoplasia. Currently published data regarding survivors of childhood and adult cancer do not suggest that GH replacement increases future cancer risk, although it remains difficult to identify factors that may modulate cancer risk in older patients, individuals with increased predisposition to malignancy and those with a strong family history of cancer. Despite the growing availability of data from cohorts with long-term follow-up, consensus on clinical practice is lacking in several areas. These include whether in-vitro pro-neoplastic properties can truly provide the basis for safety concerns related to GH replacement, whether treatment with GH with increased serum concentrations of IGF-I could independently contribute to worse tumor or mortality outcomes in at-risk populations, how to manage potential safety concerns in individuals who are GHD and are predisposed to cancer and finally, whether best practices in the management of GHD could reduce some of the risk potentially conferred by GH replacement, and whether long-term use of long-acting GH preparations may exacerbate the risk of cancer in CCS.
References
Deodati A, Cianfarani S (2017) The rationale for growth hormone therapy in children with short stature. J Clin Res Pediatr Endocrinol 9(Suppl 2):23–32. https://doi.org/10.4274/jcrpe.2017.S003
Tamhane S, Sfeir JG, Kittah NEN, Jasim S, Chemaitilly W, Cohen LE et al (2018) GH therapy in childhood cancer survivors: a systematic review and meta-analysis. J Clin Endocrinol Metab 103(8):2794–2801. https://doi.org/10.1210/jc.2018-01205
van Iersel L, Li Z, Srivastava DK, Brinkman TM, Bjornard KL, Wilson CL et al (2019) Hypothalamic-pituitary disorders in childhood cancer survivors: prevalence, risk factors and long-term health outcomes. J Clin Endocrinol Metab 104(12):6101–6115. https://doi.org/10.1210/jc.2019-00834
Johnston WT, Erdmann F, Newton R, Steliarova-Foucher E, Schuz J, Roman E (2021) Childhood cancer: estimating regional and global incidence. Cancer Epidemiol 71(Pt B):101662. https://doi.org/10.1016/j.canep.2019.101662
Steliarova-Foucher E, Colombet M, Ries LAG, Moreno F, Dolya A, Bray F et al (2017) International incidence of childhood cancer, 2001–10: a population-based registry study. Lancet Oncol 18(6):719–731. https://doi.org/10.1016/S1470-2045(17)30186-9
Kaplan JA (2019) Leukemia in children. Pediatr Rev 40(7):319–331. https://doi.org/10.1542/pir.2018-0192
Garre ML, Cama A (2007) Craniopharyngioma: modern concepts in pathogenesis and treatment. Curr Opin Pediatr 19(4):471–479. https://doi.org/10.1097/MOP.0b013e3282495a22
Bogusz A, Muller HL (2018) Childhood-onset craniopharyngioma: latest insights into pathology, diagnostics, treatment, and follow-up. Expert Rev Neurother 18(10):793–806. https://doi.org/10.1080/14737175.2018.1528874
Udaka YT, Packer RJ (2018) Pediatric brain tumors. Neurol Clin 36(3):533–556. https://doi.org/10.1016/j.ncl.2018.04.009
Buhtoiarov IN (2017) Pediatric Lymphoma. Pediatr Rev 38(9):410–423. https://doi.org/10.1542/pir.2016-0152
Stahnke K, Ritter J, Schellong G, Beck JD, Kabisch H, Lampert F et al (1992) Treatment of recurrence of acute myeloid leukemia in childhood. A retrospective analysis of recurrence in the AML-BFM-83 study. Klin Padiatr 204(4):253–257. https://doi.org/10.1055/s-2007-1025356
Watanabe S, Mizuno S, Oshima LH, Tsunematsu Y, Fujimoto J, Komiyama A (1993) Leukemia and other malignancies among GH users. J Pediatr Endocrinol 6(1):99–108. https://doi.org/10.1515/jpem.1993.6.1.99
Nishi Y, Tanaka T, Takano K, Fujieda K, Igarashi Y, Hanew K et al (1999) Recent status in the occurrence of leukemia in growth hormone-treated patients in Japan. GH treatment study committee of the foundation for growth science. Japan. J Clin Endocrinol Metab. 84(6):1961–1965. https://doi.org/10.1210/jcem.84.6.5716
Wilton P, Mattsson AF, Darendeliler F (2010) Growth hormone treatment in children is not associated with an increase in the incidence of cancer: experience from KIGS (Pfizer International Growth Database). J Pediatr 157(2):265–270. https://doi.org/10.1016/j.jpeds.2010.02.028
Libruder C, Blumenfeld O, Dichtiar R, Laron Z, Zadik Z, Shohat T et al (2016) Mortality and cancer incidence among patients treated with recombinant growth hormone during childhood in Israel. Clin Endocrinol (Oxf) 85(5):813–818. https://doi.org/10.1111/cen.13131
Swerdlow AJ, Cooke R, Albertsson-Wikland K, Borgstrom B, Butler G, Cianfarani S et al (2015) Description of the SAGhE cohort: A large European study of mortality and cancer incidence risks after childhood treatment with recombinant growth hormone. Horm Res Paediatr 84(3):172–183. https://doi.org/10.1159/000435856
Swerdlow AJ, Cooke R, Beckers D, Borgstrom B, Butler G, Carel JC et al (2017) Cancer risks in patients treated with growth hormone in childhood: The SAGhE European cohort study. J Clin Endocrinol Metab 102(5):1661–1672. https://doi.org/10.1210/jc.2016-2046
Swerdlow AJ, Cooke R, Beckers D, Butler G, Carel JC, Cianfarani S et al (2019) Risk of meningioma in European patients treated with growth hormone in childhood: results from the SAGhE cohort. J Clin Endocrinol Metab 104(3):658–664. https://doi.org/10.1210/jc.2018-01133
Arslanian SA, Becker DJ, Lee PA, Drash AL, Foley TP Jr (1985) Growth hormone therapy and tumor recurrence. Findings in children with brain neoplasms and hypopituitarism. Am J Dis Child 139(4):347–350. https://doi.org/10.1001/archpedi.1985.02140060029020
Clayton PE, Shalet SM, Gattamaneni HR, Price DA (1987) Does growth hormone cause relapse of brain tumours? Lancet 1(8535):711–713. https://doi.org/10.1016/s0140-6736(87)90355-2
Corrias A, Picco P, Einaudi S, de Sanctis L, Besenzon L, Garre ML et al (1997) Growth hormone treatment in irradiated children with brain tumors. J Pediatr Endocrinol Metab 10(1):41–49. https://doi.org/10.1515/jpem.1997.10.1.41
Leung W, Rose SR, Zhou Y, Hancock ML, Burstein S, Schriock EA et al (2002) Outcomes of growth hormone replacement therapy in survivors of childhood acute lymphoblastic leukemia. J Clin Oncol 20(13):2959–2964. https://doi.org/10.1200/JCO.2002.09.142
Sklar CA, Mertens AC, Mitby P, Occhiogrosso G, Qin J, Heller G et al (2002) Risk of disease recurrence and second neoplasms in survivors of childhood cancer treated with growth hormone: a report from the childhood cancer survivor study. J Clin Endocrinol Metab 87(7):3136–3141. https://doi.org/10.1210/jcem.87.7.8606
Ergun-Longmire B, Mertens AC, Mitby P, Qin J, Heller G, Shi W et al (2006) Growth hormone treatment and risk of second neoplasms in the childhood cancer survivor. J Clin Endocrinol Metab 91(9):3494–3498. https://doi.org/10.1210/jc.2006-0656
Mackenzie S, Craven T, Gattamaneni HR, Swindell R, Shalet SM, Brabant G (2011) Long-term safety of growth hormone replacement after CNS irradiation. J Clin Endocrinol Metab 96(9):2756–2761. https://doi.org/10.1210/jc.2011-0112
Patterson BC, Chen Y, Sklar CA, Neglia J, Yasui Y, Mertens A et al (2014) Growth hormone exposure as a risk factor for the development of subsequent neoplasms of the central nervous system: a report from the childhood cancer survivor study. J Clin Endocrinol Metab 99(6):2030–2037. https://doi.org/10.1210/jc.2013-4159
Brignardello E, Felicetti F, Castiglione A, Fortunati N, Matarazzo P, Biasin E et al (2015) GH replacement therapy and second neoplasms in adult survivors of childhood cancer: a retrospective study from a single institution. J Endocrinol Invest 38(2):171–176. https://doi.org/10.1007/s40618-014-0179-1
Raman S, Grimberg A, Waguespack SG, Miller BS, Sklar CA, Meacham LR et al (2015) Risk of neoplasia in pediatric patients receiving growth hormone therapy–a report from the pediatric endocrine society drug and therapeutics committee. J Clin Endocrinol Metab 100(6):2192–2203. https://doi.org/10.1210/jc.2015-1002
Child CJ, Zimmermann AG, Chrousos GP, Cummings E, Deal CL, Hasegawa T et al (2019) Safety outcomes during pediatric gh therapy: final results from the prospective GeNeSIS observational program. J Clin Endocrinol Metab 104(2):379–389. https://doi.org/10.1210/jc.2018-01189
Boguszewski CL, Boguszewski M (2019) Growth hormone’s links to cancer. Endocr Rev 40(2):558–574. https://doi.org/10.1210/er.2018-00166
Villani A, Greer MC, Kalish JM, Nakagawara A, Nathanson KL, Pajtler KW et al (2017) Recommendations for cancer surveillance in individuals with RASopathies and other rare genetic conditions with increased cancer risk. Clin Cancer Res 23(12):e83–e90. https://doi.org/10.1158/1078-0432.CCR-17-0631
Bell J, Parker KL, Swinford RD, Hoffman AR, Maneatis T, Lippe B (2010) Long-term safety of recombinant human growth hormone in children. J Clin Endocrinol Metab 95(1):167–177. https://doi.org/10.1210/jc.2009-0178
Woodmansee WW, Zimmermann AG, Child CJ, Rong Q, Erfurth EM, Beck-Peccoz P et al (2013) Incidence of second neoplasm in childhood cancer survivors treated with GH: an analysis of GeNeSIS and HypoCCS. Eur J Endocrinol 168(4):565–573. https://doi.org/10.1530/EJE-12-0967
Shen L, Sun CM, Li XT, Liu CJ, Zhou YX (2015) Growth hormone therapy and risk of recurrence/progression in intracranial tumors: a meta-analysis. Neurol Sci 36(10):1859–1867. https://doi.org/10.1007/s10072-015-2269-z
Wang ZF, Chen HL (2014) Growth hormone treatment and risk of recurrence or development of secondary neoplasms in survivors of pediatric brain tumors. J Clin Neurosci 21(12):2155–2159. https://doi.org/10.1016/j.jocn.2014.04.016
Alotaibi NM, Noormohamed N, Cote DJ, Alharthi S, Doucette J, Zaidi HA et al (2018) Physiologic growth hormone-replacement therapy and craniopharyngioma recurrence in pediatric patients: a meta-analysis. World Neurosurg 109487–496:e481. https://doi.org/10.1016/j.wneu.2017.09.164
Lindholm J (2006) Growth hormone: historical notes. Pituitary 9(1):5–10. https://doi.org/10.1007/s11102-006-7557-4
Ranke MB, Wit JM (2018) Growth hormone - past, present and future. Nat Rev Endocrinol 14(5):285–300. https://doi.org/10.1038/nrendo.2018.22
Salmon, W.D., Jr., Daughaday, W.H. A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro. J Lab Clin Med. 49(6), 825–836 (1957). https://www.ncbi.nlm.nih.gov/pubmed/13429201
Samani AA, Yakar S, LeRoith D, Brodt P (2007) The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev 28(1):20–47. https://doi.org/10.1210/er.2006-0001
Boguszewski CL, Boguszewski MC, Kopchick JJ (2016) Growth hormone, insulin-like growth factor system and carcinogenesis. Endokrynol Pol 67(4):414–426. https://doi.org/10.5603/EP.a2016.0053
Pollak M (2012) The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer 12(3):159–169. https://doi.org/10.1038/nrc3215
Resnicoff, M., Abraham, D., Yutanawiboonchai, W., Rotman, H.L., Kajstura, J., Rubin, R., et al. The insulin-like growth factor I receptor protects tumor cells from apoptosis in vivo. Cancer Res. 55(11): 2463–2469 (1995). https://www.ncbi.nlm.nih.gov/pubmed/7758000
Resnicoff, M., Burgaud, J.L., Rotman, H.L., Abraham, D., Baserga, R. Correlation between apoptosis, tumorigenesis, and levels of insulin-like growth factor I receptors. Cancer Res. 55(17), 3739–3741 (1995). https://www.ncbi.nlm.nih.gov/pubmed/7641185
Podlutsky A, Valcarcel-Ares MN, Yancey K, Podlutskaya V, Nagykaldi E, Gautam T et al (2017) The GH/IGF-1 axis in a critical period early in life determines cellular DNA repair capacity by altering transcriptional regulation of DNA repair-related genes: implications for the developmental origins of cancer. Geroscience 39(2):147–160. https://doi.org/10.1007/s11357-017-9966-x
Rajah R, Valentinis B, Cohen P (1997) Insulin-like growth factor (IGF)-binding protein-3 induces apoptosis and mediates the effects of transforming growth factor-beta1 on programmed cell death through a p53- and IGF-independent mechanism. J Biol Chem 272(18):12181–12188. https://doi.org/10.1074/jbc.272.18.12181
Brahmkhatri VP, Prasanna C, Atreya HS (2015) Insulin-like growth factor system in cancer: novel targeted therapies. Biomed Res Int. https://doi.org/10.1155/2015/538019
Melmed S (2001) Acromegaly and cancer: not a problem? J Clin Endocrinol Metab 86(7):2929–2934. https://doi.org/10.1210/jcem.86.7.7635
Menashe I, Maeder D, Garcia-Closas M, Figueroa JD, Bhattacharjee S, Rotunno M et al (2010) Pathway analysis of breast cancer genome-wide association study highlights three pathways and one canonical signaling cascade. Cancer Res 70(11):4453–4459. https://doi.org/10.1158/0008-5472.CAN-09-4502
Mukhina S, Mertani HC, Guo K, Lee KO, Gluckman PD, Lobie PE (2004) Phenotypic conversion of human mammary carcinoma cells by autocrine human growth hormone. Proc Natl Acad Sci U S A 101(42):15166–15171. https://doi.org/10.1073/pnas.0405881101
Waters MJ, Conway-Campbell BL (2004) The oncogenic potential of autocrine human growth hormone in breast cancer. Proc Natl Acad Sci U S A 101(42):14992–14993. https://doi.org/10.1073/pnas.0406396101
Chesnokova V, Zonis S, Zhou C, Recouvreux MV, Ben-Shlomo A, Araki T et al (2016) Growth hormone is permissive for neoplastic colon growth. Proc Natl Acad Sci U S A 113(23):E3250-3259. https://doi.org/10.1073/pnas.1600561113
Perry JK, Wu ZS, Mertani HC, Zhu T, Lobie PE (2017) Tumour-derived human growth hormone as a therapeutic target in oncology. Trends Endocrinol Metab 28(8):587–596. https://doi.org/10.1016/j.tem.2017.05.003
Harvey S, Martinez-Moreno CG, Luna M, Aramburo C (2015) Autocrine/paracrine roles of extrapituitary growth hormone and prolactin in health and disease: An overview. Gen Comp Endocrinol. https://doi.org/10.1016/j.ygcen.2014.11.004
Iams WT, Lovly CM (2015) Molecular pathways: clinical applications and future direction of insulin-like growth factor-1 receptor pathway blockade. Clin Cancer Res 21(19):4270–4277. https://doi.org/10.1158/1078-0432.CCR-14-2518
Beckwith H, Yee D (2015) Minireview: were the IGF signaling inhibitors all bad? Mol Endocrinol 29(11):1549–1557. https://doi.org/10.1210/me.2015-1157
Berryman DE, Christiansen JS, Johannsson G, Thorner MO, Kopchick JJ (2008) Role of the GH/IGF-1 axis in lifespan and healthspan: lessons from animal models. Growth Horm IGF Res 18(6):455–471. https://doi.org/10.1016/j.ghir.2008.05.005
Bartke A, Chandrashekar V, Bailey B, Zaczek D, Turyn D (2002) Consequences of growth hormone (GH) overexpression and GH resistance. Neuropeptides 36(2–3):201–208. https://doi.org/10.1054/npep.2002.0889
Endogenous Hormones, Breast Cancer Collaborative Group, Key, T.J., Appleby, P.N., Reeves, G.K., Roddam, A.W (2010) Insulin-like growth factor 1 (IGF1), IGF binding protein 3 (IGFBP3), and breast cancer risk: pooled individual data analysis of 17 prospective studies. Lancet Oncol 11(6):530–542. https://doi.org/10.1016/S1470-2045(10)70095-4
Clayton PE, Banerjee I, Murray PG, Renehan AG (2011) Growth hormone, the insulin-like growth factor axis, insulin and cancer risk. Nat Rev Endocrinol 7(1):11–24. https://doi.org/10.1038/nrendo.2010.171
Roddam AW, Allen NE, Appleby P, Key TJ, Ferrucci L, Carter HB et al (2008) Insulin-like growth factors, their binding proteins, and prostate cancer risk: analysis of individual patient data from 12 prospective studies. Ann Intern Med 149(7):461–471. https://doi.org/10.7326/0003-4819-149-7-200810070-00006
Jenkins PJ, Mukherjee A, Shalet SM (2006) Does growth hormone cause cancer? Clin Endocrinol (Oxf) 64(2):115–121. https://doi.org/10.1111/j.1365-2265.2005.02404.x
Wang J, Li YC, Deng M, Jiang HY, Guo LH, Zhou WJ et al (2017) Serum insulin-like growth factor-1 and its binding protein 3 as prognostic factors for the incidence, progression, and outcome of hepatocellular carcinoma: a systematic review and meta-analysis. Oncotarget 8(46):81098–81108. https://doi.org/10.18632/oncotarget.19186
Kaaks R, Johnson T, Tikk K, Sookthai D, Tjonneland A, Roswall N et al (2014) Insulin-like growth factor I and risk of breast cancer by age and hormone receptor status-A prospective study within the EPIC cohort. Int J Cancer 134(11):2683–2690. https://doi.org/10.1002/ijc.28589
Schmidt JA, Allen NE, Almquist M, Franceschi S, Rinaldi S, Tipper SJ et al (2014) Insulin-like growth factor-I and risk of differentiated thyroid carcinoma in the European Prospective Investigation into Cancer and Nutrition. Cancer Epidemiol Biomarkers Prev 23(6):976–985. https://doi.org/10.1158/1055-9965.EPI-13-1210-T
Rohrmann S, Linseisen J, Becker S, Allen N, Schlehofer B, Overvad K et al (2011) Concentrations of IGF-I and IGFBP-3 and brain tumor risk in the European Prospective Investigation into Cancer and Nutrition. Cancer Epidemiol Biomarkers Prev 20(10):2174–2182. https://doi.org/10.1158/1055-9965.EPI-11-0179
Steuerman R, Shevah O, Laron Z (2011) Congenital IGF1 deficiency tends to confer protection against post-natal development of malignancies. Eur J Endocrinol 164(4):485–489. https://doi.org/10.1530/EJE-10-0859
Guevara-Aguirre J, Balasubramanian P, Guevara-Aguirre M, Wei M, Madia F, Cheng CW et al (2011) Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med. 3(70):70ra13. https://doi.org/10.1126/scitranslmed.3001845
Marinho CG, Mermejo LM, Salvatori R, Assirati JAJ, Oliveira CRP, Santos EG et al (2018) Occurrence of neoplasms in individuals with congenital, severe GH deficiency from the Itabaianinha kindred. Growth Horm IGF Res 41:71–74. https://doi.org/10.1016/j.ghir.2018.03.004
Pekic S, Popovic V (2013) GH therapy and cancer risk in hypopituitarism: what we know from human studies. Eur J Endocrinol 169(5):R89-97. https://doi.org/10.1530/EJE-13-0389
Sherlock M, Ayuk J, Tomlinson JW, Toogood AA, Aragon-Alonso A, Sheppard MC et al (2010) Mortality in patients with pituitary disease. Endocr Rev 31(3):301–342. https://doi.org/10.1210/er.2009-0033
Boguszewski CL, Ayuk J (2016) Management OF endocrine DISEASE: acromegaly and cancer: an old debate revisited. Eur J Endocrinol 175(4):R147-156. https://doi.org/10.1530/EJE-16-0178
van Santen HM, Chemaitilly W, Meacham LR, Tonorezos ES, Mostoufi-Moab S (2020) Endocrine health in childhood cancer survivors. Pediatr Clin North Am 67(6):1171–1186. https://doi.org/10.1016/j.pcl.2020.08.002
Darzy KH, Shalet SM (2009) Hypopituitarism following radiotherapy. Pituitary 12(1):40–50. https://doi.org/10.1007/s11102-008-0088-4
Hidalgo Santos AD, de Mingo Alemany MDC, Moreno Macian F, Leon Carinena S, Collado Ballesteros E, Canete Nieto A (2019) Endocrinological late effects of oncologic treatment on survivors of medulloblastoma. Rev Chil Pediatr 90(6):598–605. https://doi.org/10.32641/rchped.v90i6.994
Heikens J, Michiels EM, Behrendt H, Endert E, Bakker PJ, Fliers E (1998) Long-term neuro-endocrine sequelae after treatment for childhood medulloblastoma. Eur J Cancer 34(10):1592–1597. https://doi.org/10.1016/s0959-8049(98)00212-3
Sklar CA, Antal Z, Chemaitilly W, Cohen LE, Follin C, Meacham LR et al (2018) Hypothalamic-pituitary and growth disorders in survivors of childhood cancer: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 103(8):2761–2784. https://doi.org/10.1210/jc.2018-01175
Merchant TE, Rose SR, Bosley C, Wu S, Xiong X, Lustig RH (2011) Growth hormone secretion after conformal radiation therapy in pediatric patients with localized brain tumors. J Clin Oncol 29(36):4776–4780. https://doi.org/10.1200/JCO.2011.37.9453
Paakko, E., Talvensaari, K., Pyhtinen, J., Lanning, M. Decreased pituitary gland height after radiation treatment to the hypothalamic-pituitary axis evaluated by MR. AJNR Am J Neuroradiol. 15(3), 537–541 (1994). https://www.ncbi.nlm.nih.gov/pubmed/8197954
Xie C, Li J, Weng Z, He LJ, Yin S, Zhang J et al (2018) Decreased pituitary height and stunted linear growth after radiotherapy in survivors of childhood nasopharyngeal carcinoma cases. Front Endocrinol (Lausanne). https://doi.org/10.3389/fendo.2018.00643
Viswanathan V, Pradhan KR, Eugster EA (2011) Pituitary hormone dysfunction after proton beam radiation therapy in children with brain tumors. Endocr Pract 17(6):891–896. https://doi.org/10.4158/EP10391.OR
Mailhot Vega R, Kim J, Hollander A, Hattangadi-Gluth J, Michalski J, Tarbell NJ et al (2015) Cost effectiveness of proton versus photon radiation therapy with respect to the risk of growth hormone deficiency in children. Cancer 121(10):1694–1702. https://doi.org/10.1002/cncr.29209
Oshiro Y, Mizumoto M, Pan H, Kaste SC, Gajjar A, Merchant TE (2020) Spinal changes after craniospinal irradiation in pediatric patients. Pediatr Blood Cancer 67(12):e28728. https://doi.org/10.1002/pbc.28728
Clayton PE, Shalet SM (1991) The evolution of spinal growth after irradiation. Clin Oncol (R Coll Radiol) 3(4):220–222. https://doi.org/10.1016/s0936-6555(05)80744-7
Demoor-Goldschmidt C, Allodji RS, Journy N, Rubino C, Zrafi WS, Debiche G et al (2020) Risk factors for small adult height in childhood cancer survivors. J Clin Oncol 38(16):1785–1796. https://doi.org/10.1200/JCO.19.02361
Mostoufi-Moab S (2016) Skeletal impact of retinoid therapy in childhood cancer survivors. Pediatr Blood Cancer 63(11):1884–1885. https://doi.org/10.1002/pbc.26180
Robinson GW, Kaste SC, Chemaitilly W, Bowers DC, Laughton S, Smith A et al (2017) Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget 8(41):69295–69302. https://doi.org/10.18632/oncotarget.20619
Collett-Solberg PF, Ambler G, Backeljauw PF, Bidlingmaier M, Biller BMK, Boguszewski MCS et al (2019) Diagnosis, genetics, and therapy of short stature in children: a growth hormone research society international perspective. Horm Res Paediatr 92(1):1–14. https://doi.org/10.1159/000502231
Cattoni A, Clarke E, Albanese A (2018) The predictive value of insulin-like growth factor 1 in irradiation-dependent growth hormone deficiency in childhood cancer survivors. Horm Res Paediatr 90(5):314–325. https://doi.org/10.1159/000495760
Sfeir JG, Kittah NEN, Tamhane SU, Jasim S, Chemaitilly W, Cohen LE et al (2018) Diagnosis of GH deficiency as a late effect of radiotherapy in survivors of childhood cancers. J Clin Endocrinol Metab 103(8):2785–2793. https://doi.org/10.1210/jc.2018-01204
Tillmann V, Shalet SM, Price DA, Wales JK, Pennells L, Soden J et al (1998) Serum insulin-like growth factor-I, IGF binding protein-3 and IGFBP-3 protease activity after cranial irradiation. Horm Res 50(2):71–77. https://doi.org/10.1159/000023237
Grimberg A, DiVall SA, Polychronakos C, Allen DB, Cohen LE, Quintos JB et al (2016) Guidelines for growth hormone and insulin-like growth factor-I treatment in children and adolescents: growth hormone deficiency, idiopathic short stature, and primary insulin-like growth factor-I deficiency. Horm Res Paediatr 86(6):361–397. https://doi.org/10.1159/000452150
Allen DB, Backeljauw P, Bidlingmaier M, Biller BM, Boguszewski M, Burman P et al (2016) GH safety workshop position paper: a critical appraisal of recombinant human GH therapy in children and adults. Eur J Endocrinol 174(2):P1-9. https://doi.org/10.1530/EJE-15-0873
Paulino AC, Suzawa HS, Dreyer ZE, Hanania AN, Chintagumpala M, Okcu MF (2021) Scoliosis in children treated with photon craniospinal irradiation for medulloblastoma. Int J Radiat Oncol Biol Phys 109(3):712–717. https://doi.org/10.1016/j.ijrobp.2020.09.055
Chueh HW, Yoo JH (2017) Metabolic syndrome induced by anticancer treatment in childhood cancer survivors. Ann Pediatr Endocrinol Metab 22(2):82–89. https://doi.org/10.6065/apem.2017.22.2.82
Faienza MF, Delvecchio M, Giordano P, Cavallo L, Grano M, Brunetti G et al (2015) Metabolic syndrome in childhood leukemia survivors: a meta-analysis. Endocrine 49(2):353–360. https://doi.org/10.1007/s12020-014-0395-7
Oudin C, Auquier P, Bertrand Y, Contet A, Kanold J, Sirvent N et al (2015) Metabolic syndrome in adults who received hematopoietic stem cell transplantation for acute childhood leukemia: an LEA study. Bone Marrow Transplant 50(11):1438–1444. https://doi.org/10.1038/bmt.2015.167
Higham CE, Johannsson G, Shalet SM (2016) Hypopituitarism Lancet 388(10058):2403–2415. https://doi.org/10.1016/S0140-6736(16)30053-8
Melmed S (2019) Pathogenesis and diagnosis of growth hormone deficiency in adults. N Engl J Med 380(26):2551–2562. https://doi.org/10.1056/NEJMra1817346
Fleseriu M, Hashim IA, Karavitaki N, Melmed S, Murad MH, Salvatori R et al (2016) Hormonal replacement in hypopituitarism in adults: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 101(11):3888–3921. https://doi.org/10.1210/jc.2016-2118
Ho KK (2007) GH deficiency consensus workshop participants. consensus guidelines for the diagnosis and treatment of adults with GH deficiency II: a statement of the GH Research society in association with the European society for pediatric endocrinology, lawson wilkins society, European society of endocrinology, Japan endocrine society, and endocrine society of Australia. Eur J Endocrinol 157(6):695–700. https://doi.org/10.1530/EJE-07-0631
Yuen KCJ, Biller BMK, Radovick S, Carmichael JD, Jasim S, Pantalone KM et al (2019) American association of clinical endocrinologists and American college of endocrinology guidelines for management of growth hormone deficiency in adults and patients transitioning from pediatric to adult care. Endocr Pract 25(11):1191–1232. https://doi.org/10.4158/GL-2019-0405
Krzyzanowska-Mittermayer K, Mattsson AF, Maiter D, Feldt-Rasmussen U, Camacho-Hubner C, Luger A et al (2018) New Neoplasm during GH replacement in adults with pituitary deficiency following malignancy: a kims analysis. J Clin Endocrinol Metab 103(2):523–531. https://doi.org/10.1210/jc.2017-01899
Olsson DS, Andersson E, Bryngelsson IL, Nilsson AG, Johannsson G (2015) Excess mortality and morbidity in patients with craniopharyngioma, especially in patients with childhood onset: a population-based study in Sweden. J Clin Endocrinol Metab 100(2):467–474. https://doi.org/10.1210/jc.2014-3525
Olsson DS, Nilsson AG, Bryngelsson IL, Trimpou P, Johannsson G, Andersson E (2015) Excess mortality in women and young adults with nonfunctioning pituitary adenoma: a Swedish nationwide study. J Clin Endocrinol Metab 100(7):2651–2658. https://doi.org/10.1210/jc.2015-1475
Chemaitilly W, Li Z, Huang S, Ness KK, Clark KL, Green DM et al (2015) Anterior hypopituitarism in adult survivors of childhood cancers treated with cranial radiotherapy: a report from the St Jude Lifetime Cohort study. J Clin Oncol 33(5):492–500. https://doi.org/10.1200/jco.2014.56.7933
de Fine Licht S, Winther JF, Gudmundsdottir T, Holmqvist AS, Bonnesen TG, Asdahl PH et al (2014) Hospital contacts for endocrine disorders in adult life after childhood cancer in scandinavia (ALiCCS): a population-based cohort study. Lancet 383(9933):1981–1989. https://doi.org/10.1016/s0140-6736(13)62564-7
Rose SR, Horne VE, Howell J, Lawson SA, Rutter MM, Trotman GE et al (2016) Late endocrine effects of childhood cancer. Nat Rev Endocrinol 12(6):319–336. https://doi.org/10.1038/nrendo.2016.45
Turcotte LM, Liu Q, Yasui Y, Arnold MA, Hammond S, Howell RM et al (2017) Temporal trends in treatment and subsequent neoplasm risk among 5-year survivors of childhood cancer, 1970–2015. JAMA 317(8):814–824. https://doi.org/10.1001/jama.2017.0693
Garcia JM, Biller BMK, Korbonits M, Popovic V, Luger A, Strasburger CJ et al (2018) Macimorelin as a diagnostic test for adult GH deficiency. J Clin Endocrinol Metab 103(8):3083–3093. https://doi.org/10.1210/jc.2018-00665
Hoybye C, Beck-Peccoz P, Simsek S, Zabransky M, Zouater H, Stalla G et al (2020) Safety of current recombinant human growth hormone treatments for adults with growth hormone deficiency and unmet needs. Expert Opin Drug Saf 19(12):1539–1548. https://doi.org/10.1080/14740338.2020.1839410
Molitch ME, Clemmons DR, Malozowski S, Merriam GR, Vance ML, Endocrine S (2011) Evaluation and treatment of adult growth hormone deficiency: an endocrine Society clinical practice guideline. J Clin Endocrinol Metab 96(6):1587–1609. https://doi.org/10.1210/jc.2011-0179
Birzniece V, Ho KKY (2017) Sex steroids and the GH axis: Implications for the management of hypopituitarism. Best Pract Res Clin Endocrinol Metab 31(1):59–69. https://doi.org/10.1016/j.beem.2017.03.003
Follin C, Thilén U, Ahrén B, Erfurth EM (2006) Improvement in cardiac systolic function and reduced prevalence of metabolic syndrome after two years of growth hormone (GH) treatment in GH-deficient adult survivors of childhood-onset acute lymphoblastic leukemia. J Clin Endocrinol Metab 91(5):1872–1875. https://doi.org/10.1210/jc.2005-2298
Follin C, Thilén U, Osterberg K, Björk J, Erfurth EM (2010) Cardiovascular risk, cardiac function, physical activity, and quality of life with and without long-term growth hormone therapy in adult survivors of childhood acute lymphoblastic leukemia. J Clin Endocrinol Metab 95(8):3726–3735. https://doi.org/10.1210/jc.2010-0117
Huisman J, Aukema EJ, Deijen JB, van Coeverden SC, Kaspers GJ, van der Pal HJ et al (2008) The usefulness of growth hormone treatment for psychological status in young adult survivors of childhood leukaemia: an open-label study. BMC Pediatr. https://doi.org/10.1186/1471-2431-8-25
Mukherjee A, Tolhurst-Cleaver S, Ryder WD, Smethurst L, Shalet SM (2005) The characteristics of quality of life impairment in adult growth hormone (GH)-deficient survivors of cancer and their response to GH replacement therapy. J Clin Endocrinol Metab 90(3):1542–1549. https://doi.org/10.1210/jc.2004-0832
Murray RD, Darzy KH, Gleeson HK, Shalet SM (2002) GH-deficient survivors of childhood cancer: GH replacement during adult life. J Clin Endocrinol Metab 87(1):129–135
van den Heijkant S, Hoorweg-Nijman G, Huisman J, Drent M, van der Pal H, Kaspers GJ et al (2011) Effects of growth hormone therapy on bone mass, metabolic balance, and well-being in young adult survivors of childhood acute lymphoblastic leukemia. J Pediatr Hematol Oncol 33(6):e231-238. https://doi.org/10.1097/MPH.0b013e31821bbe7a
Child CJ, Zimmermann AG, Woodmansee WW, Green DM, Li JJ, Jung H et al (2011) Assessment of primary cancers in GH-treated adult hypopituitary patients: an analysis from the Hypopituitary Control and Complications Study. Eur J Endocrinol 165(2):217–223. https://doi.org/10.1530/EJE-11-0286
Child CJ, Conroy D, Zimmermann AG, Woodmansee WW, Erfurth EM, Robison LL (2015) Incidence of primary cancers and intracranial tumour recurrences in GH-treated and untreated adult hypopituitary patients: analyses from the Hypopituitary Control and Complications Study. Eur J Endocrinol 172(6):779–790. https://doi.org/10.1530/EJE-14-1123
Popovic V, Mattsson AF, Gaillard RC, Wilton P, Koltowska-Haggstrom M, Ranke MB (2010) Serum insulin-like growth factor I (IGF-I), IGF-binding proteins 2 and 3, and the risk for development of malignancies in adults with growth hormone (GH) deficiency treated with GH: data from KIMS (Pfizer International Metabolic Database). J Clin Endocrinol Metab 95(9):4449–4454. https://doi.org/10.1210/jc.2010-0287
Li Z, Zhou Q, Li Y, Fu J, Huang X, Shen L (2016) Growth hormone replacement therapy reduces risk of cancer in adult with growth hormone deficiency: A meta-analysis. Oncotarget 7(49):81862–81869. https://doi.org/10.18632/oncotarget.13251
Lundberg E, Kristrom B, Zouater H, Deleskog A, Hoybye CT (2020) years with biosimilar rhGH in clinical practice in Sweden - experience from the prospective PATRO children and adult studies. BMC Endocr Disord 20(1):55. https://doi.org/10.1186/s12902-020-0535-4
Beck-Peccoz P, Hoybye C, Murray RD, Simsek S, Zabransky M, Zouater H et al (2020) Malignancy risk in adults with growth hormone deficiency undergoing long-term treatment with biosimilar somatropin (Omnitrope((R))): data from the PATRO Adults study. Ther Adv Endocrinol Metab. https://doi.org/10.1177/2042018820943377
Savendahl L, Cooke R, Tidblad A, Beckers D, Butler G, Cianfarani S et al (2020) Long-term mortality after childhood growth hormone treatment: the SAGhE cohort study. Lancet Diabetes Endocrinol 8(8):683–692. https://doi.org/10.1016/S2213-8587(20)30163-7
Yuen KC, Popovic V (2015) Growth hormone replacement in patients with a history of malignancy: a review of the literature and best practice for offering treatment. Expert Rev Endocrinol Metab 10(3):319–326. https://doi.org/10.1586/17446651.2015.996130
Hartman ML, Xu R, Crowe BJ, Robison LL, Erfurth EM, Kleinberg DL et al (2013) Prospective safety surveillance of GH-deficient adults: comparison of GH-treated vs untreated patients. J Clin Endocrinol Metab 98(3):980–988. https://doi.org/10.1210/jc.2012-2684
Holdaway IM, Hunt P, Manning P, Cutfield W, Gamble G, Ninow N et al (2015) Three-year experience with access to nationally funded growth hormone (GH) replacement for GH-deficient adults. Clin Endocrinol (Oxf) 83(1):85–90. https://doi.org/10.1111/cen.12691
Kremidas D, Wisniewski T, Divino VM, Bala K, Olsen M, Germak J et al (2013) Administration burden associated with recombinant human growth hormone treatment: perspectives of patients and caregivers. J Pediatr Nurs 28(1):55–63. https://doi.org/10.1016/j.pedn.2011.12.006
Auer MK, Stieg MR, Hoffmann J, Stalla GK (2016) Is insulin-like growth factor-I a good marker for treatment adherence in growth hormone deficiency in adulthood? Clin Endocrinol (Oxf) 84(6):862–869. https://doi.org/10.1111/cen.13030
Mancini A, Vergani E, Bruno C, Palladino A, Brunetti A (2019) Relevance of adherence monitoring in adult patients with growth hormone deficiency under replacement therapy: preliminary monocentric data with easypod(TM) connect. Front Endocrinol (Lausanne). https://doi.org/10.3389/fendo.2019.00416
Miller BS, Velazquez E, Yuen KCJ (2020) Long-acting growth hormone preparations - current status and future considerations. J Clin Endocrinol Metab 105(6):e2121–e2133. https://doi.org/10.1210/clinem/dgz149
Yuen KCJ, Miller BS, Biller BMK (2018) The current state of long-acting growth hormone preparations for growth hormone therapy. Curr Opin Endocrinol Diabetes Obes 25(4):267–273. https://doi.org/10.1097/MED.0000000000000416
Yuen KCJ, Miller BS, Boguszewski CL, Hoffman AR (2021) Usefulness and potential pitfalls of long-acting growth hormone analogs. Front Endocrinol (Lausanne). https://doi.org/10.3389/fendo.2021.637209
Armstrong GT, Liu W, Leisenring W, Yasui Y, Hammond S, Bhatia S et al (2011) Occurrence of multiple subsequent neoplasms in long-term survivors of childhood cancer: a report from the childhood cancer survivor study. J Clin Oncol 29(22):3056–3064. https://doi.org/10.1200/JCO.2011.34.6585
Bowers DC, Nathan PC, Constine L, Woodman C, Bhatia S, Keller K et al (2013) Subsequent neoplasms of the CNS among survivors of childhood cancer: a systematic review. Lancet Oncol 14(8):e321-328. https://doi.org/10.1016/S1470-2045(13)70107-4
Chemaitilly W, Robison LL (2012) Safety of growth hormone treatment in patients previously treated for cancer. Endocrinol Metab Clin North Am 41(4):785–792. https://doi.org/10.1016/j.ecl.2012.07.002
Nishio S, Morioka T, Inamura T, Takeshita I, Fukui M, Sasaki M et al (1998) Radiation-induced brain tumours: potential late complications of radiation therapy for brain tumours. Acta Neurochir (Wien) 140(8):763–770. https://doi.org/10.1007/s007010050177
Bogarin R, Steinbok P (2009) Growth hormone treatment and risk of recurrence or progression of brain tumors in children: a review. Childs Nerv Syst 25(3):273–279. https://doi.org/10.1007/s00381-008-0790-6
Darendeliler F, Karagiannis G, Wilton P, Ranke MB, Albertsson-Wikland K, Anthony Price D et al (2006) Recurrence of brain tumours in patients treated with growth hormone: analysis of KIGS (Pfizer International Growth Database). Acta Paediatr 95(10):1284–1290. https://doi.org/10.1080/08035250600577889
Journy NMY, Zrafi WS, Bolle S, Fresneau B, Alapetite C, Allodji RS et al (2021) Risk factors of subsequent central nervous system tumors after childhood and adolescent cancers: findings from the french childhood cancer survivor study. Cancer Epidemiol Biomarkers Prev 30(1):133–141. https://doi.org/10.1158/1055-9965.EPI-20-0735
Drapeau A, Walz PC, Eide JG, Rugino AJ, Shaikhouni A, Mohyeldin A et al (2019) Pediatric craniopharyngioma. Childs Nerv Syst 35(11):2133–2145. https://doi.org/10.1007/s00381-019-04300-2
Cossu G, Jouanneau E, Cavallo LM, Elbabaa SK, Giammattei L, Starnoni D et al (2020) Surgical management of craniopharyngiomas in adult patients: a systematic review and consensus statement on behalf of the EANS skull base section. Acta Neurochir (Wien) 162(5):1159–1177. https://doi.org/10.1007/s00701-020-04265-1
Olsson DS, Buchfelder M, Wiendieck K, Kremenevskaja N, Bengtsson BA, Jakobsson KE et al (2012) Tumour recurrence and enlargement in patients with craniopharyngioma with and without GH replacement therapy during more than 10 years of follow-up. Eur J Endocrinol 166(6):1061–1068. https://doi.org/10.1530/EJE-12-0077
Kanev PM, Lefebvre JF, Mauseth RS, Berger MS (1991) Growth hormone deficiency following radiation therapy of primary brain tumors in children. J Neurosurg 74(5):743–748. https://doi.org/10.3171/jns.1991.74.5.0743
Moshang T Jr (1995) Is brain tumor recurrence increased following growth hormone treatment? Trends Endocrinol Metab 6(6):205–209. https://doi.org/10.1016/1043-2760(95)00090-5
Karavitaki N, Warner JT, Marland A, Shine B, Ryan F, Arnold J et al (2006) GH replacement does not increase the risk of recurrence in patients with craniopharyngioma. Clin Endocrinol (Oxf) 64(5):556–560. https://doi.org/10.1111/j.1365-2265.2006.02508.x
Alotaibi NM, Zaidi HA, Noormohamed N, Cote DJ, Crocker E, Doucette J et al (2017) Comparison of physiologic growth hormone replacement therapy to no replacement on craniopharyngioma recurrence in pediatric patients. J Neurol Surg B Skull Base. 78:1–156. https://doi.org/10.1055/s-0037-1600744
Rohrer TR, Langer T, Grabenbauer GG, Buchfelder M, Glowatzki M, Dorr HG (2010) Growth hormone therapy and the risk of tumor recurrence after brain tumor treatment in children. J Pediatr Endocrinol Metab 23(9):935–942. https://doi.org/10.1515/jpem.2010.150
Losa M, Castellino L, Pagnano A, Rossini A, Mortini P, Lanzi R (2020) Growth hormone therapy does not increase the risk of craniopharyngioma and nonfunctioning pituitary adenoma recurrence. J Clin Endocrinol Metab 105(5):1573–1580. https://doi.org/10.1210/clinem/dgaa089
Abs R, Bengtsson BA, Hernberg-Stahl E, Monson JP, Tauber JP, Wilton P et al (1999) GH replacement in 1034 growth hormone deficient hypopituitary adults: demographic and clinical characteristics, dosing and safety. Clin Endocrinol (Oxf) 50(6):703–713. https://doi.org/10.1046/j.1365-2265.1999.00695.x
Moshang T Jr, Rundle AC, Graves DA, Nickas J, Johanson A, Meadows A (1996) Brain tumor recurrence in children treated with growth hormone: the National Cooperative Growth Study experience. J Pediatr 128(5 Pt 2):S4-7. https://doi.org/10.1016/s0022-3476(96)70002-1
Price DA, Wilton P, Jonsson P, Albertsson-Wikland K, Chatelain P, Cutfield W et al (1998) Efficacy and safety of growth hormone treatment in children with prior craniopharyngioma: an analysis of the Pharmacia and Upjohn International Growth Database (KIGS) from 1988 to 1996. Horm Res 49(2):91–97. https://doi.org/10.1159/000023133
Smith TR, Cote DJ, Jane JA Jr, Laws ER Jr (2016) Physiological growth hormone replacement and rate of recurrence of craniopharyngioma: the genentech national cooperative growth study. J Neurosurg Pediatr 18(4):408–412. https://doi.org/10.3171/2016.4.PEDS16112
Li Q, You C, Liu L, Rao Z, Sima X, Zhou L et al (2013) Craniopharyngioma cell growth is promoted by growth hormone (GH) and is inhibited by tamoxifen: involvement of growth hormone receptor (GHR) and IGF-1 receptor (IGF-1R). J Clin Neurosci 20(1):153–157. https://doi.org/10.1016/j.jocn.2012.04.014
Hofmann BM, Hoelsken A, Fahlbusch R, Blumcke I, Buslei R (2010) Hormone receptor expression in craniopharyngiomas: a clinicopathological correlation. Neurosurgery 67(3):617–625. https://doi.org/10.1227/01.NEU.0000372918.68453.5B
Ogawa Y, Kudo M, Watanabe M, Tominaga T (2020) Heterogeneity of growth hormone receptor expression in craniopharyngioma-implications for surgical strategy. World Neurosurg. https://doi.org/10.1016/j.wneu.2020.02.022
Wijnen M, Olsson DS, van den Heuvel-Eibrink MM, Hammarstrand C, Janssen J, van der Lely AJ et al (2018) Excess morbidity and mortality in patients with craniopharyngioma: a hospital-based retrospective cohort study. Eur J Endocrinol 178(1):93–102. https://doi.org/10.1530/EJE-17-0707
Yuen KCJ, Mattsson AF, Burman P, Erfurth EM, Camacho-Hubner C, Fox JL et al (2018) Relative risks of contributing factors to morbidity and mortality in adults with craniopharyngioma on growth hormone replacement. J Clin Endocrinol Metab 103(2):768–777. https://doi.org/10.1210/jc.2017-01542
Profka E, Giavoli C, Bergamaschi S, Ferrante E, Malchiodi E, Sala E et al (2015) Analysis of short- and long-term metabolic effects of growth hormone replacement therapy in adult patients with craniopharyngioma and non-functioning pituitary adenoma. J Endocrinol Invest 38(4):413–420. https://doi.org/10.1007/s40618-014-0196-0
Webb SM, Strasburger CJ, Mo D, Hartman ML, Melmed S, Jung H et al (2009) Changing patterns of the adult growth hormone deficiency diagnosis documented in a decade-long global surveillance database. J Clin Endocrinol Metab 94(2):392–399. https://doi.org/10.1210/jc.2008-0713
Abs R, Mattsson AF, Thunander M, Verhelst J, Goth MI, Wilton P et al (2013) Prevalence of diabetes mellitus in 6050 hypopituitary patients with adult-onset GH deficiency before GH replacement: a KIMS analysis. Eur J Endocrinol 168(3):297–305. https://doi.org/10.1530/EJE-12-0807
Olsson DS, Buchfelder M, Schlaffer S, Bengtsson BA, Jakobsson KE, Johannsson G et al (2009) Comparing progression of non-functioning pituitary adenomas in hypopituitarism patients with and without long-term GH replacement therapy. Eur J Endocrinol 161(5):663–669. https://doi.org/10.1530/EJE-09-0572
Hatrick AG, Boghalo P, Bingham JB, Ayres AB, Sonksen PH, Russell-Jones DL (2002) Does GH replacement therapy in adult GH-deficient patients result in recurrence or increase in size of pituitary tumours? Eur J Endocrinol 146(6):807–811. https://doi.org/10.1530/eje.0.1460807
Buchfelder M, Kann PH, Wuster C, Tuschy U, Saller B, Brabant G et al (2007) Influence of GH substitution therapy in deficient adults on the recurrence rate of hormonally inactive pituitary adenomas: a case control study. Eur J Endocrinol 157(2):149–156. https://doi.org/10.1530/EJE-07-0164
Arnold JR, Arnold DF, Marland A, Karavitaki N, Wass JA (2009) GH replacement in patients with non-functioning pituitary adenoma (NFA) treated solely by surgery is not associated with increased risk of tumour recurrence. Clin Endocrinol (Oxf) 70(3):435–438. https://doi.org/10.1111/j.1365-2265.2008.03391.x
van Varsseveld NC, van Bunderen CC, Franken AA, Koppeschaar HP, van der Lely AJ, Drent ML (2015) tumor recurrence or regrowth in adults with nonfunctioning pituitary adenomas using GH replacement therapy. J Clin Endocrinol Metab 100(8):3132–3139. https://doi.org/10.1210/jc.2015-1764
Gasco V, Caputo M, Cambria V, Beccuti G, Caprino MP, Ghigo E et al (2019) Progression of pituitary tumours: impact of GH secretory status and long-term GH replacement therapy. Endocrine 63(2):341–347. https://doi.org/10.1007/s12020-018-1787-x
Miller KK, Wexler T, Fazeli P, Gunnell L, Graham GJ, Beauregard C et al (2010) Growth hormone deficiency after treatment of acromegaly: a randomized, placebo-controlled study of growth hormone replacement. J Clin Endocrinol Metab 95(2):567–577. https://doi.org/10.1210/jc.2009-1611
Norrman LL, Johannsson G, Sunnerhagen KS, Svensson J (2008) Baseline characteristics and the effects of two years of growth hormone (GH) replacement therapy in adults with GH deficiency previously treated for acromegaly. J Clin Endocrinol Metab 93(7):2531–2538. https://doi.org/10.1210/jc.2007-2673
Tritos NA, Johannsson G, Korbonits M, Miller KK, Feldt-Rasmussen U, Yuen KC et al (2014) Effects of long-term growth hormone replacement in adults with growth hormone deficiency following cure of acromegaly: a KIMS analysis. J Clin Endocrinol Metab 99(6):2018–2029. https://doi.org/10.1210/jc.2014-1013
Roelfsema F, Biermasz NR, Pereira AM (2012) Clinical factors involved in the recurrence of pituitary adenomas after surgical remission: a structured review and meta-analysis. Pituitary 15(1):71–83. https://doi.org/10.1007/s11102-011-0347-7
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Conflict of interest
MCSB received speaker honorarium from Pfizer. CLB has served as consultant for Pfizer. GJ has served as consultant for Shire and Astra Zeneca, and has received lecture fees from Eli Lilly, Ipsen, Novartis, Novo Nordisk, Merck Serono, Otsuka, and Pfizer. KCJY is an investigator on research grants from Pfizer, Novo Nordisk, and OPKO Biologics, and has consulted for Pfizer, Novo Nordisk, Sandoz, and Ascendis. AACD, WC and CEH have nothing to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Boguszewski, M.C.S., Cardoso-Demartini, A.A., Boguszewski, C.L. et al. Safety of growth hormone (GH) treatment in GH deficient children and adults treated for cancer and non-malignant intracranial tumors—a review of research and clinical practice. Pituitary 24, 810–827 (2021). https://doi.org/10.1007/s11102-021-01173-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11102-021-01173-0