
Abstract
Pure nickel and four binary Ni–Cr (5, 10, 20 and 30 wt%) alloys were exposed to Ar–20O2, Ar–20O2–20H2O and Ar–20H2O (vol.%) at 700°C. In dry and wet O2, three Ni–Cr alloys (5, 10 and 20 wt%) formed a three-layered structure: an external NiO layer, an inner oxide layer of NiO + Cr2O3 and an internal oxidation zone (IOZ) with Cr2O3 dispersed. The Ni–30Cr alloy formed a continuous chromia layer, with locally outer NiO islands. In pure water vapor, all alloys underwent internal oxidation, nickel metal expulsion and NiO formation. Compared with the results at 650°C, increasing the temperature to 700°C did not alter the morphology of the oxide scale, but enhanced the scale growth kinetics. A significant difference between these two temperatures was that the strong retarding effect of water vapor on NiO formation in oxygen at 650°C became insignificant at 700°C. However, in water vapor only condition, NiO formation remained much slower at both temperatures. The effects of water vapor and oxygen partial pressure on NiO formation of different alloys and the effect of temperature on scale growth kinetics and morphology were discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Thermal power is widely used for energy generation in the world. In order to reach higher efficiency, the temperatures of inlet steam can reach 700 ~ 720°C in northern Europe and even higher at 760°C in the USA [1]. Nickel-based alloys are considered as likely construction materials for high temperature components in the next generation of advanced ultra-supercritical power plants, due to a combination of superior corrosion resistance and high creep strength at elevated temperatures [1,2,3]. A slow growing, continuous and compact oxide scale, commonly Cr2O3, is formed to protect the underlying alloys as a barrier from attack in aggressive environments [4, 5].
In our previous study [6, 7], we found that the corrosion rates of Ni–Cr alloys were generally lower in wet O2/CO2 or pure water vapor than in dry O2/CO2. However, the effect of water vapor on oxidation behavior is complex and depends on the oxygen partial pressure. The addition of H2O–CO2 gas not only changes the growth kinetics, but also scale morphology. The continuous NiO layers formed in dry CO2 were replaced by loose NiO growths and/or scattered Ni metal nodules in wet gas. In contrast, the addition of water vapor to oxygen only slows overall alloy oxidation rates but does not change the scale morphology. Similarly, reduced chromia scale thicknesses in wet O2 and air compared with those grown in the corresponding dry gases were also found in other investigations [8, 9]. In addition, Ni–Cr alloys developed similar scale morphologies in steam and in wet CO2: internal oxidation, nickel metal expulsion and external NiO growth, which differed significantly from those frequently observed in oxygen [10, 11].
In spite of these past studies, the effect of water vapor on Ni–Cr alloy oxidation at higher temperatures in the varied pO2 atmospheres is still not clear. This work is focused on Ni and Ni–Cr alloy oxidation at 700°C in O2, O2 + H2O and H2O only gases. The results are compared with those at 650°C reported before [6, 7] to deduce the effect of temperature on corrosion of these materials.
Experimental Procedures
Pure nickel and four model Ni–Cr alloys (Ni–5Cr, 10Cr, 20Cr, and 30Cr) (nominal composition, all in wt%), were made from high purity metals of Ni (99.95%) and Cr (99.995%) by arc melting using a non-consumable tungsten electrode under an Ar–5%H2 atmosphere. The cast metal and alloy ingots were annealed under a flowing Ar–5%H2 gas at 1050°C for 50 h for homogenization, and then slowly cooled to room temperature in the furnace. The annealed alloys were cut into small rectangular plates with the dimensions (7.5–11.5) mm × (5–8) mm × (0.9–2.5) mm. All surfaces of the sample coupons were ground to 1200-grit finish, polished to a 3 µm finish and then further electro-polished in a hydrochloric solution (17 ml 32 wt% HCl and 23 ml H2O). The purpose of electro-polishing is to remove the surface work hardened zone created during the sample preparation. All samples were ultrasonically cleaned in ethanol after each step and dried for the reaction. The grain sizes of samples were measured and estimated by using the samples after electro-polishing. The grains exhibit elongated shapes, and the grain sizes are large, around hundreds of microns to several millimeters.
All corrosion experiments were proceeded in a horizontal furnace with an alumina tubular reactor. All samples were exposed to Ar–20O2, Ar–20H2O and Ar–20O2–20H2O atmospheres with a linear flow rate of 2 cm/s and a total pressure of 1 atm at 700°C. The reaction time was set at 10, 100 and 300 h, respectively. The gas flow rates of Ar and O2 were regulated by rotameters, and water vapor was generated by Ar gas or the mixture of Ar and O2 passing through a thermostatted water saturator containing demineralized water. The temperature of demineralized water was set to generate excess water vapor which was then condensed inside a condenser with a setting temperature corresponding to the required water vapor content. All samples were fixed on a sample holder in the cold zone of the alumina tubular reactor. Before reaction, the alumina tubular reactor was purged with pure argon for 1 h to expel air. Then the furnace was heated at a rate of 10°C/min to 700°C and stabilized for 20 min. After that, the sample holder was pushed into the hot zone and the reaction gas was introduced subsequently. After the exposure for the desired time, the flowing gas in the tubular reactor was changed from reaction gas to pure argon and the sample was pulled out to the cold zone for cooling.
Before and after the reaction, each sample was measured by an analytical balance (Mettler Toledo XP205) with an accuracy of 0.01 mg. The samples after 100 h of reaction in three different atmospheres were selected to identify the phases of the oxide scales by using X-ray diffractometry (XRD; Empyrean II, Co source). The setting voltage and current were 45 kV and 40 mA, respectively, and the scan angle (2θ) was from 20° to 100°. Cross sections of reacted samples were characterized by optical microscopy (OM, Nikon 200) and scanning electron microscopy (SEM; FEI Nova NanoSEM 450) equipped with energy dispersive spectroscopy (EDS; Bruker).
Results
Weight Gain Kinetics
Figure 1 shows the weight gain kinetics of pure nickel and the Ni–Cr alloys exposed to three different atmospheres at 700°C. In general, weight gain kinetics in dry (Fig. 1a) and wet oxygen (Fig. 1b) were comparable, with the highest rate for Ni–5Cr and the lowest one for Ni–30Cr. In pure water vapor, however, the weight gain kinetics were much lower than those in both oxygen-containing gases (Fig. 1c), and the highest weight gains were for Ni–20Cr and 30Cr in this case. For pure nickel, the weight gain kinetics were the lowest in the water vapor only condition but much higher than those of Ni–30Cr in both wet and dry O2 conditions. It should be mentioned that the appearance of weight decrease with reaction time for the dry O2 and H2O only conditions could be related to possible scale spallation.

Weight gain kinetics of pure Ni and Ni–Cr alloys exposed to a Ar–20O2, b Ar–20O2–20H2O and c Ar–20H2O at 700°C
Assuming diffusion is the controlling step for all situations, the parabolic rate constants of weight gain kinetics, \({k}_{w}\), were calculated by the following equation.
where \(\Delta W\) is the weight gain, \({\text{A}}\) is the surface area of tested samples, and \({\text{t}}\) is the reaction time. The calculated results are summarized in Table 1. The parabolic rate constants for Ni–10Cr in dry oxygen and Ni–30Cr in all three atmospheres were not determined due to the irregular weight change behavior. The results showed clearly that the parabolic rate constants of Cr alloys (5–20Cr alloys) in wet oxygen were similar to those in dry oxygen. In pure water vapor, the parabolic rate constants were the lowest in all three environments.
Reaction Product Morphology
Figure 2a–c show metallographic cross sections of pure Ni after exposure at 700°C in the three atmospheres for 100 h. In dry oxygen, a continuous and dense NiO layer with the thickness of approximate 10 μm was formed (Fig. 2a). In wet oxygen, the NiO scale was porous, with numerous pores distributed near the scale-alloy interface. The thickness was similar to that in dry oxygen (Fig. 2b), in line with the weight gain kinetics (Fig. 1b). In pure water vapor, the surface of pure nickel formed a very thin layer after 100 h reaction (Fig. 2c).

Metallographic cross sections of pure Ni after reaction in a Ar–20O2, b Ar–20O2–20H2O, c Ar–20H2O and d XRD analysis results at 700°C for 100 h
Further analysis using BSE-SEM is shown in Fig. 3, which provides more information about the morphology of the scale. The scales formed in both dry and wet oxygen were similar, with numerous pores mainly distributed in the inner part of the scale (Fig. 3a, b). In dry O2, the matrix and the scale were separated by a gap, while in wet oxygen, no such a gap was observed, and the oxide scale kept in good contact with the alloy. In pure water vapor, however, there was a very thin dense and continuous NiO layer about 1 μm in thickness (Fig. 3c). These oxides were confirmed by XRD analysis (shown in Fig. 2d) to be NiO. After 300 h reaction, the morphologies of pure Ni exposed to the three atmospheres remained the same but the thickness increased. The measured thickness of oxide scale in different atmospheres is shown in Table 2.

BSE-SEM cross sections of pure Ni after reaction in a Ar–20O2, b Ar–20O2–20H2O and c Ar–20H2O at 700°C for 100 h
Figure 4a–c show the metallographic cross sections of Ni–5Cr alloy after reaction for 100 h at 700°C in the three atmospheres. Compared with pure nickel in dry oxygen, the oxide morphology was changed significantly when adding 5 wt% Cr. The scale contained a three-layered structure: a top dense and continuous gray layer; an inner layer with some pores; and an internal oxidation zone (IOZ). There were gaps between the outer layer and the inner layer, and the inner layer and the IOZ. Surface X-ray diffraction (Fig. 4d) indicated that the oxide scale was composed of NiO and Cr2O3. The presence of NiCr2O4 spinel was expected based on Ni–Cr–O phase diagram but was not detected in this work, probably because of very slow rate of spinel formation at a low temperature and the amount of this spinel too low to be detected.

Metallographic cross sections of Ni–5Cr alloy after reaction in a Ar–20O2, b Ar–20O2–20H2O, c Ar–20H2O and d XRD analysis results at 700°C for 100 h
Further analysis by the BSE-SEM of the scale and EDS mapping (Fig. 5) indicated that the outer layer was mainly Ni-rich oxide and Cr was detected in the inner layer. According to the SEM image, the IOZ consisted of the needle-like precipitates which were scatteredly distributed in the alloy near the alloy-scale interface. These needle-like precipitates were identified by TEM and EDS to be Cr2O3 in dry oxygen at 650°C [6]. Therefore, the top layer was NiO, followed by NiO and Cr2O3 mixture in the inner layer and Cr2O3 precipitates in the IOZ. After 300 h reaction, the cross section morphology did not change much (not shown), and only the scale and IOZ became thicker.

a BSE-SEM cross section and b-d corresponding EDS mappings of Ni–5Cr alloy after reaction in Ar–20O2 for 100 h
By adding water in oxygen, the morphology did not change too much. The oxide scale (Fig. 4b) consisted of three layers which resembled that in dry oxygen. However, more pores were observed in the inner layer than those in dry oxygen. There were a dense NiO layer on the top, a porous NiO + Cr2O3 mixed inner layer, and the needle-like Cr2O3 internal precipitates inside the matrix. The XRD results (Fig. 4d) indicated that the phases in the scale were the same as those in dry oxygen. After 300 h reaction, the cross-section morphology had no change except the increased thickness (not shown).
When Ni–5Cr alloy was reacted in water vapor only condition, the morphology was changed significantly compared with those in dry and wet oxygen. The scale was much thinner than those in other two atmospheres. Figure 4c shows the metallographic cross-section image of the alloy after 100 h reaction where an IOZ can be observed. Further analysis by XRD indicated that the scale consisted of NiO and Cr2O3 (Fig. 4d). High magnification BSE-SEM image shown in Fig. 6a revealed a very thin external layer on the top of IOZ. The EDS point analysis results showed that this thin layer mainly consisted of nickel oxide. Therefore, the scale was composed of an external thin NiO layer and internal Cr2O3 precipitates. After 300 h reaction, the morphology was changed compared with that after 100 h reaction (shown in Fig. 6b). The NiO layer was much thicker (about 3 µm) than that after 100 h reaction (0.3 µm). In addition, some metallic Ni islands were visible beneath the NiO layer. Whisker-shaped NiO formed on the top of NiO layer, and the IOZ contained both needle-like and fine precipitates. The thickness of IOZ increased significantly.

BSE-SEM cross-section images and corresponding EDS points analysis results of Ni–5Cr alloy after reaction in Ar–20H2O for a 100 h and b 300 h
Figure 7a–c show the metallographic cross sections of Ni–10Cr after 100 h reaction in the three atmospheres. Similar to Ni–5Cr, in dry and wet O2, the scale also had three layers which were the outer dense layer, the inner layer with few pores inside, and IOZ. The XRD analysis is shown in Fig. 7d, which consisted of NiO and Cr2O3, that is, the outer layer was composed of NiO, the inner layer NiO and Cr2O3 mixture, and the IOZ with Cr2O3 precipitates. The scale thickness in dry and wet oxygen was more or less the same. The only difference was the inner layer formed in wet oxygen had a higher porosity than that in dry oxygen. After 300 h reaction, the morphology in both conditions remained the same (not shown). In pure water vapor, there was an IOZ near the surface in the matrix. Additionally, there were some light-color nodules covered by a dark layer formed on the top of the scale. The oxide scale contained Ni, NiO and Cr2O3 identified by XRD analysis (Fig. 7d). The thin top layer covering Ni nodules was NiO and the IOZ was with Cr2O3 precipitates. After 300 h reaction, the morphology was changed to a similar structure to that in Ni–5Cr after 300 h reaction. A thin NiO layer was observed with whisker-shaped NiO on the top of the scale, and the IOZ was separated by a metallic Ni layer.

Metallographic cross sections of Ni–10Cr alloy after reaction in a Ar–20O2, b Ar–20O2–20H2O, c Ar–20H2O and d XRD analysis results at 700°C for 100 h
Figures 8a–c show metallographic cross sections of Ni–20Cr alloy after reaction in the three atmospheres for 100 h. In both dry and wet oxygen, the oxide scale structure was similar to that formed on the surface of Ni–10Cr in the same reaction condition, i.e., the outer light gray NiO layer, inner gray NiO and Cr2O3 mixture and internal Cr2O3 precipitates. In pure water vapor, the scale structure was also similar to that formed on the surface of Ni–10Cr in the same condition, i.e., the external light-color nodules wrapped by a thin dark oxide layer, and a rather thick internal oxidation zone. The oxide composition of the scale was similar to that of Ni–10Cr, which were NiO and Cr2O3 confirmed by the XRD (shown in Fig. 8d).

Metallographic cross sections of Ni–20Cr alloy after reaction in a Ar–20O2, b Ar–20O2–20H2O, c Ar–20H2O and d XRD analysis results at 700°C for 100 h
Metallographic cross sections of Ni–30Cr alloy in the three conditions are shown in Figs. 9a–c. In dry and wet O2, the scale consisted of two different contrasts of thin dense layers without any internal oxidation zone observed. The scale was comprised of NiO and Cr2O3 layers which were confirmed by XRD analysis (Fig. 9d).

Metallographic cross sections of Ni–30Cr alloy after reaction in a Ar–20O2, b Ar–20O2–20H2O, c Ar–20H2O and d XRD analysis results at 700°C for 100 h
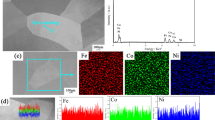
A high magnification BSE-SEM image of the scale with EDS mapping is shown in Fig. 10. The mapping results indicated that the outer and inner layers were Ni-rich layer and Cr-rich layer, respectively. Combining these results, the external outer layer was NiO followed by a Cr2O3 layer. After 300 h reaction, the morphology was similar to that in 100 h reaction.

a BSE-SEM image and b-d corresponding EDS mappings of Ni–30Cr after reaction in dry oxygen for 100 h
A metallographic cross section of the scale in water vapor only condition is shown in Fig. 9c, which consisted of the outer NiO nodules and the IOZ. There was a dense internal Cr2O3 band formed beneath the IOZ in contact with the matrix. The scale structure was the same as that of Ni–20Cr alloy in the same condition.
The measured external NiO thicknesses of different alloys after reaction in three atmospheres for 300 h are listed in Table 2. There was not much difference between dry and wet oxygen. The external NiO thickness in water vapor only condition was significantly lower than those in other two atmospheres.
Discussion
Effects of Water Vapor and Oxygen Partial Pressure on NiO Formation of Pure Nickel
As indicated in Figs. 2 and 3, only a NiO scale is formed after reaction of pure nickel in all atmospheres, but its morphology is different. The weight gain kinetics (Fig. 1) and scale thicknesses (Table 2) in both dry and wet oxygen gases are similar. The calculated parabolic rate constants in Table 1 further confirms that there is no significant difference between the oxidation rates of pure Ni in dry and wet oxygen. However, in water vapor only condition, the weight gain kinetics and the scale thickness are significantly lower than those of oxygen-containing gases. Besides, in both oxygen-containing gases, there are some fine pores distributed in the scale (Fig. 3a, b). The scale and metal are separated by a gap in dry oxygen (Fig. 3a), but a better scale-metal adherence (no gap) is observed in wet gases (Fig. 3b, c). In pure water vapor, the resulting scale is relatively thin and dense (Fig. 3c).
These observations clearly demonstrate the effects of water vapor and oxygen partial pressure on the oxidation of pure Ni. The effect of water vapor on pure Ni oxidation has been reported previously by several authors [13, 13, 14]. The presence of water vapor has been postulated to slow down the oxidation rate due to the preferential absorption tendency of H2O on NiO [15], leading to the non-acidity of NiO [13]. This non-acidity effect of NiO could be explained by the fact that the bond in OH− is difficult to be broken on the surface of NiO scale which results in the low acidity or non-acidity of the scale surface and less oxygen atoms to oxidize the substrate. In agreement with this, Galerie et al. [13] showed that NiO formation rate is lower in Ar–H2O than that in Ar–O2 at 1100°C when these two gases have the same low oxygen partial pressure. This explains well the observation of much slower NiO formation in water vapor only condition in this work.
By comparing the kinetic results in dry and wet oxygen (Fig. 1, Table 2), the oxidation rates were not affected much by the presence of water vapor in oxygen-containing gases in this work. This result is different from what has been reported in wet O2 and wet CO2 at 650°C. Xie et al. [13] observed that the weight gain of pure nickel is 0.76 mg/cm2 in dry CO2, but decreases to 0.06 mg/cm2 in wet CO2 at 650°C for 150 h. Also at this temperature, similar water vapor effects have been observed for pure Ni in oxygen [6]. The rate of NiO formation is substantially decreased in the presence of water vapor, with 0.55 mg/cm2 in dry O2 but very low 0.04 mg/cm2 in wet O2 after 150 h reaction [6]. Clearly increasing temperature to 700°C significantly changes the effect of water vapor in the presence of oxygen which will be discussed in the next section.
Although water vapor does not have much effect on the oxidation rate of pure Ni in the presence of oxygen at 700°C, it does change the ion diffusion process and mass transfer mechanism. The NiO scale grows in dry oxygen by the dominant outward diffusion of nickel ions via lattice and grain boundaries [16,17,18]. This could result in the cation vacancies after nickel ions diffuse outward which eventually condense as pores and even gaps (Fig. 3a). The inward oxygen diffusion may also be included in diffusion mechanisms [19, 20]; however, it is much slower than the outward diffusion of Ni ions [4, 16, 21]. In wet gases, the diffusion of ions changes from predominant cation outward diffusion in dry gas to oxygen inward diffusion mixed with the outward diffusion of cations. The presence of water vapor promotes the oxygen inward diffusion and suppresses the outward diffusion of nickel ions [6]. Compared with oxygen, water vapor has a higher priority of adsorption tendency [15] which could preferencially occupy grain boundaries and other defects, reducing outward diffusion of nickel ions along the grain boundaries. The faster oxygen inward diffusion results in the inward growth of NiO which increases the adherence of the metal-scale interface as shown in Figs. 3b, c. The presence of water vapor was also reported to increase oxide scale plasticity which could also contribute to the good contact of oxide scale with the metal substrate [4]. The formation of some pores in this condition could also be attributed to the coalescence of condensed vacancies due to the injection of vacancies and their coalescence. The presence of water vapor was reported to accelerate the pore transport toward the scale surface by the Rahmel-Tobolski mechanism [22] (Fig. 3b).
The oxidation of pure Ni is sensitive to the oxygen partial pressure. In this work, the oxidation kinetics and scale thickness in pure water vapor are significantly lower than that in wet oxygen. This result is different from what was reported in [19] where no clear oxygen pressure dependence was found at 700°C. This difference could be due to the presence of water vapor whose dissociation may control oxidation kinetics. Clearly this surface reaction is affected by temperature, as apparent different behaviors of nickel oxide formation in wet oxygen at 650 and 700°C mentioned above.
Effect of Temperature on Scale Growth Kinetics and Morphology
As revealed above, the pure water vapor condition reduces significantly NiO growth kinetics at both 650 [6] and 700°C. However, water vapor effect in the presence of oxygen on NiO formation is quite different with a very slow rate at 650°C but almost no affection at 700°C. This finding indicates that a strong temperature effect on the role of water vapor in the process of nickel oxidation. Nickel oxidation starts from adsorption and dissociation of gaseous oxidants which are water vapor and oxygen in this case. Therefore, there is competitive adsorption and dissociation in the mixed gases. Adsorption and dissociation of water vapor were reported to be more rapid at lower temperatures than for example oxygen, but this difference was less pronounced at higher temperatures [23]. As a result, oxygen is the dominant diffusing species, overwhelming any water vapor effect. This analysis explains well the quite different NiO forming kinetics observed in this work, slow kinetics at a lower temperature 650°C due to dominant water adsorption and dissociation, but high kinetics at a higher temperature 700°C due to dominant oxygen dissolution. The negligible effect of water vapor on NiO formation is further supported by the work of Rahmel [24] at even higher temperatures at 1000°C and 1100°C where water vapor has little effect on the oxidation of pure Ni.
Further kinetic analysis of the external NiO formation of Ni–Cr alloys in both wet and dry oxygen at 650 and 700°C is shown in Tables 2 and 3, respectively. Similar to that for pure Ni, the external NiO formation has been retarded significantly at 650°C but only slightly reduced at 700°C in wet oxygen gas. Similar observation was also reported by Rahmel [24] in his experiment on oxidation of Nimonic 75 (Ni–20Cr) alloy at even higher temperatures at 1000°C and 1100°C. At high temperatures, absorption and dissociation of water vapor are significantly reduced and water vapor effect is not obvious and does not produce a significant change in the oxidation rate. Although this temperature effect is clear, the detailed mechanism of this effect needs further investigation.
In spite of this difference, the addition of water vapor to oxygen, in general, did not change the morphology of oxides formed at 650 and 700°C. In dry and wet O2, for alloys containing 5, 10 and 20 wt% Cr, the scale produced an external NiO layer, internal chromium oxidation and an intermediate zone of NiO + Cr2O3 (Figs. 4, 7 and 8, [6]), while the Ni–30Cr alloy formed a continuous chromia layer, with local outer NiO islands (Fig. 9a, b). In contrast, in pure water vapor, a thin film of NiO was developed on Ni nodules/layers protruded from the IOZ under the surface of the alloy (Figs. 7c, 8c, 9c). The temperature difference did not change the morphology, but changed the scale growth kinetics, which could be well attributed to the different effect of water vapor on NiO forming kinetics. In summary, in water vapor only condition, water vapor substantially reduces the rate of nickel oxidation; however, in water vapor and oxygen mixed gases, this retarding effect due to water vapor can only be observed at lower temperatures, e.g., at 650°C, but not at higher temperatures, e.g., at 700°C.
Conclusions
Pure nickel and four Ni–Cr alloys with different Cr concentrations were reacted in Ar–20O2, Ar–20O2–20H2O and Ar–20H2O at 700°C, and the effects of water vapor and oxygen partial pressure on NiO formation of different alloys were investigated and the effect of temperature on scale growth kinetics and morphology was also discussed by comparing with the results at 650°C reported before.
For pure nickel, the weight gain kinetics and scale thicknesses in both dry and wet oxygen gases are similar, but significantly higher than that in water vapor only condition. Water vapor reduced the rate of NiO formation in H2O-only condition, and led to the formation of a relatively dense NiO scale. Besides, the presence of water vapor accelerated the oxygen inward diffusion rate by diffusing water molecules and hydroxyl ions via grain boundaries. This leads to the inward growth of NiO and better scale-metal adherence in wet gases.
The corrosion performance of different alloys at 700°C is similar with that corroded in the corresponding atmosphere at 650°C in terms of the scale morphology. In both dry and wet oxygen, a typical structure of an external NiO layer, an inner oxide of NiO + Cr2O3 and an IOZ was observed for alloys containing 5, 10 and 20 wt% Cr. For Ni–30Cr alloy, a continuous chromia layer, with local outer NiO islands, was formed. In contrast, in pure water vapor, a thin NiO layer was developed on Ni nodules/layers together with an IOZ. The temperature difference did not change the oxide morphology, but changed the scale growth kinetics. At 650°C, NiO formation kinetics were significantly suppressed by water vapor in the presence of oxygen. However, at 700°C, these kinetics were only slightly reduced, probably due to the reduced adsorption and dissolution of water vapor at a higher temperature.
References
F. Abe, Engineering. 1, 211 (2015).
R. Viswanathan and W. Bakker, Journal of Materials Engineering and Performance 10, 81 (2001).
R. Viswanathan and W. Bakker, Journal of Materials Engineering and Performance 10, 96 (2001).
N. Birks, G. H. Meier, F. S. Pettit, Introduction to the high temperature oxidation of metals. Cambridge university press, 2006.
D. J. Young, High Temperature Oxidation and Corrosion of Metals, 2nd edn. (Elsevier, 2016), p. 549.
C. Jiang, Y. Xie, C. Kong, J. Zhang, and D. J. Young, Corrosion Science 174, 108801 (2020).
C. Jiang, J. Zhang, and D. J. Young, Oxidation of Metals 96, 57 (2021).
D. Simon, B. Gorr, and H. J. Christ, Oxidation of Metals 87, 417 (2017).
P. Berthod, L. Aranda, S. Mathieu, and M. Vilasi, Oxidation of metals 79, 517 (2013).
G. C. Wood, I. G. Wright, T. Hodgkiess, and D. P. Whittle, Materials and Corrosion 21, 900 (1970).
L. Latu-Romain, Y. Parsa, S. Mathieu, M. Vilasi, A. Galerie, and Y. Wouters, Corrosion Science. 126, 238 (2017).
A. Galerie, Y. Wouters, M. Caillet, Materials Science Forum, City; Trans Tech Publ, 2001, p. 231
Y. Xie, J. Zhang, and D. J. Young, Corrosion Science 136, 311 (2018).
S. R. J. Saunders, M. Monteiro, and F. Rizzo, Progress in Materials Science. 53, 775. (2008)
J. P. Abellán, T. Olszewski, G. H. Meier, L. Singheiser, and W. J. Quadakkers, International Journal of Materials Research 101, 287 (2010) .
M. Auchi, S. Hayashi, K. Toyota, and S. Ukai, Oxidation of Metals. 78, 51 (2012).
R. Peraldi, D. Monceau, and B. Pieraggi, Kinetics Oxidation of Metals. 58, 275 (2002).
C. K. Kim and L. W. Hobbs, Oxidation of Metals. 45, 247 (1996).
R. Haugsrud, Corrosion Science. 45, 211 (2003).
S. Chevalier, F. Desserrey, and J. P. Larpin, Oxidation of Metal. 64, 219 (2005).
A. Atkinson and R. I. Taylor, Philosophical Magazine A 43, 979 (1981).
A. Rahmel and J. Tobolski, Corrosion Science 5, 333 (1965).
K. Larsson, H. Björkman, and K. Hjort, Journal of Applied Physics. 90, 1026 (2001).
A. Rahmel, Corrosion Science. 5, 815 (1965).
Acknowledgments
Financial support by Australian Research Council under the Discovery Project is highly acknowledged.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Author information
Authors and Affiliations
Contributions
JC conducted experiments and analysis. JC and Dr. CJ wrote the main manuscript text. Prof. JZ reviewed and edited the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare that relevant to the content of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, J., Jiang, C. & Zhang, J. Corrosion Behaviors of Ni–Cr Alloys in O2, H2O and H2O + O2 Gases at 700°C and the Effect of Temperature. High Temperature Corrosion of mater. 100, 775–789 (2023). https://doi.org/10.1007/s11085-023-10190-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-023-10190-2