
Abstract
To understand chiral symmetry breaking on the molecular level, we developed a method to efficiently investigate reaction kinetics of single molecules. The model systems include autocatalysis as well as a reaction cascade to gain further insight into the prebiotic origin of homochirality. The simulated reactions start with a substrate and only a single catalyst molecule, and the occurrence of symmetry breaking was examined for its degree of dependence on randomness. The results demonstrate that interlocking processes, which e.g., form catalysts, autocatalytic systems, or reaction cascades that build on each other and lead to a kinetic acceleration, can very well amplify a statistically occurring symmetry breaking. These results suggest a promising direction for the experimental implementation and identification of such processes, which could have led to a shift out of thermodynamic equilibrium in the emergence of life.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A major incentive in the research concerning the origin of life is the elucidation of chiral symmetry breaking and the occurrence of homochirality. Spontaneous mirror symmetry breaking (SMSB) was first postulated in a theoretical model by Frank (1953), that is based on reaction rate equations, and has recently been part of a comprehensive review by Sallembien et al. (2022). These non-equilibrium systems, which may be closely related to the origin of the selected chirality of life are of great interest in current investigations and are summarized in a review by Ribó et al. (2017). Positive non-linear effects (NLE) can occur for systems in which the use of enantiomerically enriched reagents or catalysts lead to a significant increase in enantiomeric excess (ee) in the product. Although they are rarely observed, they were intensively studied over the last decades (Guillaneux et al. 1994; Girard and Kagan 1998, Kagan 2009; Blackmond 2010; Geiger et al. 2020). External influences are hypothesized to lead to symmetry breaking which can be induced via circularly polarized luminescence (CPL) (Bailey et al. 1998; Meinert et al. 2014; Bailey 2001), template surfaces (Haq et al. 2009, Kurata and Yoshizawa 2020) and autocatalysis (Bissette and Fletcher 2013). Self-amplification of symmetry breaking by catalyst—reaction product interaction (Storch and Trapp 2017, 2018), catalyst self-recognition (Scholtes and Trapp 2021), and in particular asymmetric autocatalysis—occur in the Soai reaction, which is one of the most intriguing and exceptional examples to date. Recent findings about the mechanism of the Soai reaction were published by Blackmond (2019) Buhse (2003, 2005) Buhse et al. (2021) Brown (2012), Denmark (2020), and Trapp (2020, 2022) and Trapp et al. (2020). The mechanistic investigations by Denmark (2020) and Trapp (2020) were recently reviewed and discussed by Geiger (2021). Hemiacetalate complexes were identified as transient catalysts, formed by reaction of aldehydes and the product alcoholate. To investigate possible scenarios leading to spontaneous symmetry breaking and amplification of the initial imbalance in the enantiomer ratio by autocatalysis theoretical approaches were developed. A stochastic kinetic approach for chiral autocatalysis was chosen by Lente (2004, 2005). Lente demonstrated that in particular autocatalytic reaction systems of higher reaction order result in symmetry breaking. Gillespie (2007) performed stochastic kinetic simulations of molecular systems to analyse for example biological cellular systems. Such systems are typically characterized by small molecular populations, which leads to deviations from the expected reaction progress using deterministic differential equations of classical chemical kinetics.
Our goal was to break down kinetic simulations at the molecular level to obtain an effective single molecule simulation and to gain further insight into systems that exhibit symmetry breaking. Therefore, we investigated reaction scenarios where two independent processes lead to the same reaction product. In such scenarios one reaction channel is controlled by a stochastic process, while the other is controlled by the reaction product, or an activator formed from the reaction product and another reactant. This represents a combination of a related stochastic and an autocatalytic process. Furthermore, we probed the idea that kinetic acceleration in consecutive reactions by autocatalytic processes or reaction cascades can be an efficient mechanism to amplify initial statistical imbalances in formed product enantiomers.
Materials and Methods
In order to conceptually understand symmetry breaking on a molecular scale, we developed a reaction network analysis tool based on a Runge–Kutta algorithm employing a system of non-linear differential equations (SMK; see Fig. 1).

Algorithm of the SMK program for statistical sampling on a molecular level. A: Exemplary reaction equation for the simulation representing an irreversible 2nd order reaction; B: Differential equations for reaction A; C: Binary rate constant array and molecule arrays corresponding to reaction rate constant and substrates and products, respectively; D: Randomized rate constant array and randomized molecule arrays; E: Algorithm of the applied method within the SMK program
The corresponding differential equations, the reaction constants k and the concentration of the reactants were chosen as input to the program for each set. The kinetic data used for the simulations is based on the experimentally investigated kinetics of the Soai reaction (Soai et al. 1995).
A statistical sampling from one dimensional, binary concentration arrays and binary reaction constant arrays was performed during the simulation. The reaction constant k is defined for 1 mol of molecules. Computationally, this leads to a limit, because it would require defining a large array of addressable single molecules with 6.023 ⋅ 1023 elements. Because of the size of the Avogadro number NA, we decided to define molecule arrays with 602 214 076 elements. The intervals were set at \(1.00\cdot {10}^{15}\) and each set was calculated n = 602 214 076 times, which corresponds to the probability of a reaction kinetic in the bulk material. Before the simulation the concentrations of the reactants and products, and the reaction rates of the individual reactions are split into two different types of arrays—one fixed at NA /1⋅ 1015 = 6.02214⋅ 108, the molecule arrays, and the other type, the rate constant array, with a flexible number of elements (see arrows from B to C). Both types of the described arrays are then randomized, and the algorithm performs a random selection of an array element of each array k, A and B. If the combination of the randomly selected elements is k = 1, A = 1 and B = 1, one product molecule is formed, and one randomly chosen element of the product array(s) will be set from 0 to 1. Every other combination (elements of \(k,\mathrm{ A} and/or \mathrm{B}\ne 1\)) signifies no reaction and no product molecule formation. The resulting simulated product concentration courses are then saved into an output file.
For reaction constant values smaller than one (k < 1), the number of elements in the reaction constant array is truncated at\({10}^{\left(\frac{1}{\mathrm{log}k}\right)}\). If the reaction constant k is greater than one (k > 1), the program performs appropriate repetitions, e.g., for k = 600, 600 repetitions of the inner loop (n = 602 214 076 times) to calculate the kinetics are performed. The program solves the ordinary differential equations (ODEs), and the obtained simulated concentration courses are then saved and the enantiomeric excess ee is determined by subtracting the number of molecules of the minor product from the major product and subsequently dividing by the sum of the major and minor products (\(ee= \frac{{N}_{major}-{N}_{minor}}{{N}_{major}+{N}_{minor}}\)).
To investigate the effect of randomness on symmetry breaking, simulations were repeated several times to obtain statistical data for a given parameter set. This is important for the initial symmetry breaking processes, which randomly produce molecules of R or S configuration as a seed for the consecutive reactions. To verify the reproducibility of the here described approach and algorithm, we implemented in the program code the feature to store and reassign the seed of the random number generator. We performed simulations by storing the seed of the randomisation generator and reassigning this seed to a set of simulation with the same parameters. These calculations (10 repetitions) provided the identical results for the reaction progress and ee values.
We investigated scenarios with a defined substrate feedstock (1 mol substrate) and a single enantioselective catalyst molecule ((R)-Cat or (S)-Cat) to simulate kinetic effects for catalysis on a molecular basis.
Results and Discussion
We explored four scenarios, that could lead to symmetry breaking (see Schemes 1, 2, 3, 4 (scenarios A-D)) depending on the reaction mechanism and the reaction rate constant k values. Different kinetics were examined for each scenario, and it was determined if and at what point an ee would occur in the system.
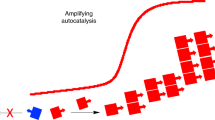
The first scenario of interest is classic autocatalysis (see Scheme 1). The reactants A and B first react in a preceding reaction with a small reaction rate constant k1 to form the enantiomers (R)-C or (S)-C in very low concentrations. This is followed by autocatalytic amplification with a significantly higher reaction rate k2.
The ODEs, which were used for the simulation of scenario A are shown in Eqs. (1)–(4):

Scenario A: classical autocatalysis. In a first stochastic reaction, A and B react to the reaction product enantiomers (R)-C or (S)-C with the same probability, determined by the reaction rate constant k1. In the consecutive autocatalytic process, the reaction products (R)-C or (S)-C catalyse the conversion of A and B to (R)-C or (S)-C and control at the same time the selectivity. (R)-C catalyses only the formation of the product (R)-C and (S)-C the formation of (S)-C, respectively. Both processes, the stochastic and the autocatalytic process, are competitive for the same reactants
The simplified reaction network shown in Scheme 1 was simulated by permutation of the reaction rate constants, whose exact magnitudes are shown in Fig. 2, while keeping the initial concentration of the reactants constant to obtain the reaction progress on a molecular level. The ee value of each system was then determined and the enantiomer that was predominantly formed after 10 s was indicated for each set. The screening of the selected reaction parameters resulted in 16 sets of total ee value courses which can be plotted versus the simulated reaction time (see Fig. 2).

Scenario A: simulation with SMK: ee versus reaction time was plotted for each parameter set; the respective stereo descriptor shows per representation which stereoisomer was formed predominantly after 10 s [the following reaction rate constants kx were permuted: k1: \(1.00\cdot {10}^{-9}\) (A-D), \(1.00\cdot {10}^{-8}\) (E-H), \(1.00\cdot {10}^{-7}\) (I-L), \(1.00\cdot {10}^{-6}\) (M-P), and k2: \(1.00\cdot {10}^{-3}\) (A, E, I, M), \(1.00\cdot {10}^{-2}\) (B, F, J, N), \(1.00\cdot {10}^{-1 }\) (C, G, K, O), \(1.00\cdot {10}^{0}\) (D, H, L, P)]
These simulations demonstrate for scenarios, where the stochastic process is very slow (Fig. 2A-D) and the consecutive selective process is fast, an efficient amplification of an initial very small imbalance in the ratio of the enantiomers takes place. In contrast, if the reaction rates are getting closer for the stochastic and the selective process, the resulting amplification and ee value are converging to zero. It is important to point out that the initial stochastic process is a bimolecular reaction of A and B, while the consecutive selective reaction is a trimolecular reaction, where the formed product molecules participate in the reaction as a catalyst. The consequence is, that the later process is rather unlikely to occur, but the product molecule acts as catalyst and therefore accelerates the reaction and transferred to a molecular level the probability for a successful reaction is increased, because of the lowered Gibbs activation energy ΔGǂ. In general, this is an important finding for an experimental design and proper selection of reactions that an initially inefficient process, which can trigger a selective and efficient process, leads to amplification of an enantiomeric imbalance and symmetry breaking.
Such a behaviour in a chemical system is not limited to symmetry breaking but can be observed also in ligation and replication of RNA and DNA, where short oligomers can serve as templates, catalysing the extension of oligomers to longer strains.
Especially in set D of scenario A (see Fig. 2, top right), a rapid occurrence of symmetry breaking could be observed, with reaction rate constants k1 = \(1.00\cdot {10}^{-9}\) and k2 = \(1.00\cdot {10}^{0}\). For this parameter set D, the total number of formed molecules was plotted versus the reaction time (see Fig. 3D2). Figure 3D2 shows also the expected kinetic trac, here obtained by the statistical sampling from the molecule arrays and rate constant array to reflect the reaction of single molecules.

Scenario A, reaction D: D1: ee versus reaction time; ee after 10 s: 53.5% ee with predominantly (R)-enantiomer; D2: number of molecules versus time: major product ((R)-enantiomer, red) and minor product ((S)-enantiomer, blue)
An ee of 53.5% is obtained in favour of the (R)-enantiomer (R)-C with a total formation of 18,202 (R)-enantiomers (R)-C and 5517 (S)-C. A more detailed analysis reveals, that the initial stochastically induced formation of (R)-C occurs after 0.017 s (100% ee for a single enantiomer molecule), and is already compensated after 0.019 s by the stochastic formation of (R)-C. After 0.122 s another (R)-C enantiomer is formed, and then rapid amplification starts after 0.212 s. After 1 s an amplification of the initial imbalance can be observed, which leads to a rapid increase in the formation of (R)-C enantiomers. Interestingly, caused by stochastic formation of (S)-C enantiomers and their amplification, two major setbacks with respect to symmetry breaking can be observed, since the number of molecules is also still relatively small (after 2 s: 34 (R)-C and 16 (S)-C; after 5 s: 446 (R)-C and 128 (S)-C), and thus any fluctuation causes a big change in the ee. This gets more robust with increasing number of molecules.
It can be envisioned, that a process, which already forces an imbalance in favor of one of the enantiomers in the initial step, e.g., by polarized light or enantiotopic faces of a crystal, can be efficiently amplified by a fast consecutive reaction or reaction cascade.
In the second reaction scenario (scenario B), substrates A and B react in an initial stochastic reaction to either [(R)-C] or [(S)-C], which each subsequently catalyse the reaction to form more (R)-C or (S)-C, respectively (see Scheme 2). Heterochiral dimerization of the formed compounds was additionally considered exemplarily, since heterochiral dimers from an enantiomerically enriched mixture may have a positive effect on the ee of the main monomeric enantiomer (Puchot et al. 1986; Kitamura et al. 1989; Noble‐Terán et al. 2016). Scenarios B and C are comparable to the Soai reaction.
The ODEs, that were used for the simulation are shown in Eqs. (5)–(9):

Scenario B: autocatalytic reaction scheme with heterochiral dimer formation. This scenario is similar to scenario A with the addition of an equilibrium to form a heterochiral dimer, which efficiently removes the minor enantiomer from a mixture of enantiomers
For comparison we choose the same reaction rates for the two consecutive reactions and a rate of formation of the heterochiral dimers of k3 = \(1.00\cdot {10}^{-2}\), to efficiently remove the racemate. The first four parameter sets (sets A-D) (see Fig. 4), look differently in terms of step size in comparison to sets E-P, since only a small number of total molecules were formed in these reactions. In parameter sets I-L, considerably more molecules were formed in the first 10 s of the simulated reactions, but the ee values are significantly lower.

Scenario B; simulation with SMK: ee versus time; the respective stereo descriptor shows per representation which stereoisomer was formed predominantly after 10 s [the following reaction rate constants kx were permuted: k1: \(1.00\cdot {10}^{-9}\) (A-D), \(1.00\cdot {10}^{-8}\) (E-H), \({10}^{-7}\) (I-L), \(1.00\cdot {10}^{-6}\) (M-P), and k2: \(1.00\cdot {10}^{-3}\) (A, E, I, M), \(1.00\cdot {10}^{-2}\) (B, F, J, N), \(1.00\cdot {10}^{-1 }\) (C, G, K, O), \(1.00\cdot {10}^{0}\) (D, H, L, P) and k3: \(1.00\cdot {10}^{-2}\)]; parameter sets A-D yield a significantly lower total number of molecules, than parameter sets E-P
The dimerization of [(R)-C] and [(S)-C] to the heterochiral dimer can be considered as a correction mechanism to remove the minor enantiomer from a reaction mixture and ideally to relatively enrich the major enantiomer over the minor enantiomer. However, this mechanism leads also to a significant decrease in catalytic activity, which slows down the autocatalytic amplification.
In the third reaction scenario (scenario C), substrates A and B react to either (R)-C or (S)-C, that subsequently gives the catalyst (R)-Dcat or (S)-Dcat upon reaction with substrate A, which are able to catalyse the formation of more (R)-C, or (S)-C respectively. The hemiacetal formation from the product alcohol and the aldehyde to form the accelerating ligand for the zinc reagent in the Soai reaction, is a prominent example for such a scenario.
The ODEs, that were used for the simulation are shown in Eqs. (10)–(15):
This set of simulations exhibits parameter sets with enormously high ee values (see Fig. 5, e.g., reaction D: 61.1% ee) while simultaneously yielding large numbers of molecules. The first two parameter sets (A, B) (see Fig. 5), on the other hand, show large stepwise variations in the ee values, which is because only very few molecules are formed in total. In the simulated reactions J-L and N-P, the stochastic seeding process proceeds too rapidly and no significant ee occurs.

Scenario C, simulation with SMK: ee versus time; the respective stereo descriptor shows per representation which stereoisomer was formed predominantly after 10 s [the following reaction rate constants kx were permuted: [k1: \(1.00\cdot {10}^{-9}\) (A-D), \(1.00\cdot {10}^{-8}\) (E-H), \(1.00\cdot {10}^{-7}\) (I-L) \(1.00\cdot {10}^{-6}\) (M-P), and k2: \(1.00\cdot {10}^{-3}\) (A, E, I, M), \(1.00\cdot {10}^{-2}\) (B, F, J, N), \(1.00\cdot {10}^{-1 }\) (C, G, K, O), \(1.00\cdot {10}^{0}\) (D, H, L, P) and k3: \(1.00\cdot {10}^{-1}\)]
Overall, scenario C seems to be very efficient when again the initial stochastic seeding reaction is slow, and the consecutive amplification is fast. The additional step to form a transient catalyst improves the selectivity, because for the formation the reactant A is consumed (but also later released), which decreases the probability for the initial stochastic process. On the other hand, the high concentration of substrate A at the beginning of the reaction guarantees the breakthrough of one of the pre-catalysts, while in the course of the reaction this process is slowly reduced to the background. For data set D (Fig. 5) an ee of 61.1% is observed, corroborating the excellent selectivity obtained by this reaction cascade.
It is interesting to mention, that this is related to the case where homochiral product inhibition is observed. If the active catalyst is a more complex entity, i.e. an oligomer or can even act as an allosteric modulator corresponding to the substrate-product adduct, such effects can be overwritten (Issac and Chmielewski 2002; Li and Chmielewski 2003).
The last scenario examined (scenario D, Scheme 4) was a reaction cascade as broadly occurring in nature. The reactants A and B react in a preceding reaction to form the respective catalyst (R)-Ccat and (S)-Ccat. These catalysts enable the reaction of molecules D and E to give either (R)-F or (S)-F, respectively.
The ODEs, that were used for the simulation are shown in Eqs. (16)–(23):

Scenario C: A and B react to from the enantiomers (R)-C or (S)-C with the same probability, which is determined by the reaction rate constant k1. The initially formed molecules (R)-C or (S)-C react with A to form the catalyst (R)-Dcat or (S)-Dcat, respectively. These catalyse the conversion of A and B to (R)-C or (S)-C and control the selectivity. (R)-Dcat catalyses only the formation of the product (R)-C and (S)-Dcat the formation of (S)-C

Scenario D: a reaction cascade; A and B react to the enantiomers (R)-Ccat or (S)-Ccat with the same probability, determined by the reaction rate constant k1. (R)-Ccat or (S)-Ccat subsequently each catalyse the conversion of D and E and control the selectivity. (R)-Ccat only catalyses the formation of (R)-F and (S)-Ccat the formation of (S)-F
The results of the simulations (see Fig. 6) show a drastically different result compared to scenarios A-C (see Schemes 1, 2, 3; Figs. 2, 4, 5 and 6). In this scenario only a low concentration in A and B (1 mM) was used as starting conditions to simulate a process, which is triggered by a few molecules and then enables another process by the product molecules, which act as a catalyst. This however leads to the formation of only a few molecules in total within the simulated reaction time (10 s).

Scenario D: simulation with SMK: ee versus time; the respective stereo descriptor shows per representation which stereoisomer was formed predominantly after 10 s, where applicable [the following reaction rate constants kx were permuted: k1: \(1.00\cdot {10}^{-9}\) (A-D), \(1.00\cdot {10}^{-8}\) (E-H), \(1.00\cdot {10}^{-7}\) (I-L), \(1.00\cdot {10}^{-6}\) (M-P), and k2: \(1.00\cdot {10}^{-3}\) (A, E, I, M), \(1.00\cdot {10}^{-2}\) (B, F, J, N), \(1.00\cdot {10}^{-1 }\) (C, G, K, O), \(1.00\cdot {10}^{0}\) (D, H, L, P)]
For example, parameter set D in scenario D, which can be described as the "most inefficient" process with very few product molecules, exhibits a high ee. This is highly interesting, because with the results obtained from scenarios A, B and C in mind, this could be a starting point to achieve very high enantioselectivities in a cascade of consecutive reactions. It can be envisioned, that such a process with building up enantioselectivities is highly robust. The question is, why a process which is highly inefficient leads to very high enantioselectivities and finally to a largely homochiral system? This might be explained by the fact that such a process is able to thin out the initially random formation of product enantiomers. This process, which continues to proceed in the background, leads in a long-term with a high probability to the formation of a racemic seed. With decreasing probability of a random process by inducing the amplification with a single molecule seed, the production of molecules with the same sense of chirality is guaranteed. This is very similar to fertilization of an egg cell and subsequent cell division, which also occurs at a single molecule level and is well proven in nature.
In parameter sets A, B, E, F, H-J, M and N, no symmetry breaking occurred. In parameter set P (see Fig. 6), an already built-up ee with a majorly formed (R)-enantiomer decreases again to 0, which is caused by the overtaking nonselective background reaction. As can be seen again, a faster amplifying downstream process is efficiently able to amplify the ee and lead to symmetry breaking.
Subsequently, a set of a specific scenario, here the parameter set D of scenario A (Scheme 1), was chosen exemplarily, repeated and a statistical analysis was performed to investigate the randomness of the seeding process and the variation from set to set. For scenario A (Scheme 1) a set consisting of 50 repetitions with the reaction rate constants \({k}_{1}=1.00\cdot {10}^{-9}\), \({k}_{2}=1.00\cdot {10}^{0}\) was calculated and evaluated. The statistical analysis of the obtained ee values for scenario A, see Fig. 7, corroborates as that the resulting formation of a preferred enantiomer is random. From this we can conclude that there is no bias in the simulation process.

Statistical analysis for scenario A: 50 repetitions of the simulation with the parameter set D, \({k}_{1}=1.00\cdot {10}^{-9}\), \({k}_{2}=1.00\cdot {10}^{0}\)
The average ee in the set of repeated simulations for 50 repetitions was 35% ee with a standard deviation σ of 26% ee. The ratio of the total number of (R)-enantiomers (243919) and (S)-enantiomers (230542) formed after a long simulation run (500 s), indicate that an overall ratio of (R)- versus (S)-enantiomers is approximately 1:1.
Remarkably, simulation no. 42 exhibits an ee of 94.1% ee for the (S)-enantiomer, which shows, that even very high ee values can occur. Despite the deviations and considerable scatter, it is remarkable that such high ee values occur randomly under these conditions.
Since only a few molecules are formed, especially in the simulations of the latter scenarios, we have extended the simulation times to 80 s for each of the examples presented here (Fig. 8). These reaction profiles confirm the trend already observed in the first 10 s and it can be shown that a bias in the enantiomeric ratio manifests itself over a longer period of time. It is also noteworthy that the scenarios presented here lead to a symmetry break within a few seconds, which in some cases intensifies or converges.

Extended simulated reaction profiles of 80 s for A scenario A (k1: \(1.00\cdot {10}^{-9}\), and k2: \(1.00,\;1000\;mM\;A,1000\;mM\;B\)), B scenario B (k1: \(1.00\cdot {10}^{-9}\), k2: \(1.00\), and k3: \(0.1,\;1000\;mM\;A,\;1000\;mM\;B\)), C scenario C (k1: \(1.00\cdot {10}^{-8}\), k2: \(1.00\), and k3: \(0.1,\;1000\;mM\;A,\;1000\;mM\;B\)), and D scenario D (k1: \(1.00\cdot {10}^{-7}\), k2: \(1.00,\;1\;mM\;A,\;1\;mM\;B,\;1000\;mM\;D,\;1000\;mM\;E\))
Summary and Conclusions
We have developed an efficient simulation software tool to predict reactions occurring on a molecular level based on a stochastic algorithm. With this tool we investigated four different reaction scenarios. All of them are initiated by a stochastically occurring reaction, which yields product enantiomers. These enantiomers serve in scenario A as catalyst in an autocatalytic process, in scenario B a dimerization of the enantiomers to a homochiral dimer is implemented, scenario C considers the formation of a transient catalyst, and scenario D simulates a reaction cascade. The results demonstrate that interlocking processes, which e.g., form catalysts, transient catalysts, autocatalytic systems, or reaction cascades that build on each other and lead to a kinetic acceleration, can very well amplify a statistically occurring symmetry breaking. In general, it was observed that a process in which downstream reactions are faster is efficient to amplify and achieve symmetry breaking. The scenarios we studied clearly show this relationship: If rates of the competing processes converge, a racemic mixture of products is obtained. An interesting observation is, that the "most inefficient" initial processes lead to high ee values and represent the most efficient scenarios in terms of achieving symmetry breaking. This occurs, when single enantiomers are formed slowly, and the consecutive process of amplification is fast and selective. The highest ee values and most efficient symmetry breaking processes were found for the scenarios forming a transient catalyst and the reaction cascade of coupled processes.
These results represent an important guidance for the experimental identification and elucidation of symmetry breaking processes leading to homochiral systems, which were essential for the emergence of life.
References
Athavale SV, Simon A, Houk KN, Denmark SE (2020) Demystifying the asymmetry-amplifying, autocatalytic behaviour of the Soai reaction through structural, mechanistic and computational studies. Nat Chem 12(4):412–423. https://doi.org/10.1038/s41557-020-0421-8
Bailey J (2001) Astronomical sources of circularly polarized light and the origin of homochirality. Orig Life Evol Biosph 31:167–183. https://doi.org/10.1023/A:1006751425919
Bailey J, Chrysostomou A, Hough JH, Gledhill TM, McCall A, Clark S, Ménard F, Tamura M (1998) Circular polarization in star-formation regions: Implications for biomolecular homochirality. Science 281.5377:672–674. https://www.science.org/doi/10.1126/science.281.5377.672. Accessed 1 July 2017
Bissette AJ, Fletcher SP (2013) Mechanisms of autocatalysis. Angew Chem Int Ed 52:12800–12826. https://doi.org/10.1002/anie.201303822
Blackmond DG (2019) Autocatalytic models for the origin of biological homochirality. Chem Rev 120(11):4831–4847. https://doi.org/10.1021/acs.chemrev.9b00557
Blackmond DG (2010) Kinetic aspects of non-linear effects in asymmetric synthesis, catalysis, and autocatalysis. Tetrahedr Asymmetr 21.11–12:1630–1634. https://doi.org/10.1016/j.tetasy.2010.03.034
Buhse T (2003) A tentative kinetic model for chiral amplification in autocatalytic alkylzinc additions. Tetrahedr Asymmetr 14(8):1055–1061. https://doi.org/10.1016/S0957-4166(03)00128-9
Buhse T, Cruz JM, Noble-Teran ME, Hochberg D, Ribó JM, Crusats J, Micheau JC (2021) Spontaneous deracemizations. Chem Rev 121(4):2147–2229. https://doi.org/10.1021/acs.chemrev.0c00819
Frank FC (1953) On spontaneous asymmetric synthesis. Biochem Biophys Acta 11:459–463. https://doi.org/10.1016/0006-3002(53)90082-1
Gehring T, Quaranta M, Odell B, Blackmond DG, Brown JM (2012) Observation of a transient intermediate in soai’s asymmetric autocatalysis: insights from 1h nmr turnover in real time. Angew Chem Int Ed 51:9539–9542. https://doi.org/10.1002/anie.201203398
Geiger Y (2021) One Soai reaction, two mechanisms? Chem Soc Rev 51(4):1206–1211. https://doi.org/10.1039/D1CS01038G
Geiger Y, Achard T, Maisse-François A, Bellemin-Laponnaz S (2020) Hyperpositive nonlinear effects in asymmetric catalysis. Nat Catal 3(5):422–426. https://doi.org/10.1038/s41929-020-0441-1
Gillespie DT (2007) Stochastic simulation of chemical kinetics. Annu Rev Phys Chem 58:35–55. https://doi.org/10.1146/annurev.physchem.58.032806.104637
Girard C, Kagan HB (1998) Nonlinear effects in asymmetric synthesis and stereoselective reactions: ten years of investigation. Angew Chem Int Ed 37(21):2922–2959. https://doi.org/10.1002/(SICI)1521-3773(19981116)37:21%3c2922::AID-ANIE2922%3e3.0.CO;2-1
Guillaneux D, Zhao SH, Samuel O, Rainford D, Kagan HB (1994) Nonlinear effects in asymmetric catalysis. J Am Chem Soc 116(21):9430–9439. https://doi.org/10.1021/ja00100a004
Haq S, Liu N, Humblot V, Jansen APJ, Raval R (2009) Drastic symmetry breaking in supramolecular organization of enantiomerically unbalanced monolayers at surfaces. Nat Chem 1(5):409–414. https://doi.org/10.1038/nchem.295
Issac R, Chmielewski J (2002) Approaching Exponential Growth with a Self-Replicating Peptide. J Am Chem Soc 124:6808–6809. https://doi.org/10.1021/ja026024i
Islas JR, Lavabre D, Grevy JM, Lamoneda RH, Cabrera HR, Micheau JC, Buhse T (2005) Mirror-symmetry breaking in the Soai reaction: A kinetic understanding. Proc Natl Acad Sci 102(39):13743–13748. https://doi.org/10.1073/pnas.0503171102
Kitamura M, Okada S, Suga S, Noyori R (1989) Enantioselective addition of dialkylzincs to aldehydes promoted by chiral amino alcohols. Mechanism and nonlinear effect. J Am Chem Soc 111.11:4028–4036. https://doi.org/10.1021/ja00193a040
Kurata M, Yoshizawa A (2020) The formation of a chiral supramolecular structure acting as a template for chirality transfer. Chem Commun 56(59):8289–8292. https://doi.org/10.1039/D0CC02413A
Lente G (2004) Homogeneous chiral autocatalysis: a simple, purely stochastic kinetic model. J Phys Chem A 108(44):9475–9478. https://doi.org/10.1021/jp046413u
Lente G (2005) Stochastic kinetic models of chiral autocatalysis: a general tool for the quantitative interpretation of total asymmetric synthesis. J Phys Chem A 109(48):11058–11063. https://doi.org/10.1021/jp054613f
Li X, Chmielewski J (2003) Peptide Self-Replication Enhanced by a Proline Kink. J Am Chem Soc 125:11820–11821. https://doi.org/10.1021/ja036569s
Meinert C, Hoffmann SV, Cassam-Chenaï P, Evans AC, Giri C, Nahon L, Meierhenrich UJ (2014) Photonenergy-controlled symmetry breaking with circularly polarized light. Angew Chem 126(1):214–218. https://doi.org/10.1002/ange.201307855
Noble-Terán ME, Buhse T, Cruz JM, Coudret C, Micheau JC (2016) Nonlinear effects in asymmetric synthesis: a practical tool for the discrimination between monomer and dimer catalysis. ChemCatChem 8(10):1836–1845. https://doi.org/10.1002/cctc.201600216
Puchot C, Samuel O, Dunach E, Zhao S, Agami C, Kagan HB (1986) Nonlinear effects in asymmetric synthesis. Examples in asymmetric oxidations and aldolization reactions. J Am Chem Soc 108(9):2353–2357. https://doi.org/10.1021/ja00269a036
Ribó JM, Hochberg D, Crusats J, El-Hachemi Z, Moyano A (2017) Spontaneous mirror symmetry breaking and origin of biological homochirality. J R Soc Interface 14(137):20170699. https://doi.org/10.1098/rsif.2017.0699
Sallembien Q, Bouteiller L, Crassous J, Raynal M (2022) Possible chemical and physical scenarios towards biological homochirality. Chem Soc Rev 51:3436–3476. https://doi.org/10.1039/D1CS01179K
Satyanarayana T, Abraham S, Kagan HB (2009) Nonlinear effects in asymmetric catalysis. Angew Chem Int Ed 48(3):456–494. https://doi.org/10.1002/anie.200705241
Scholtes JF, Trapp O (2021) Asymmetric induction and amplification in stereodynamic catalytic systems by noncovalent interactions. Synlett 32:971–980. https://doi.org/10.1055/a-1274-2777
Soai K, Shibata T, Morioka H, Choji K (1995) Asymmetric autocatalysis and amplification of enantiomeric excess of a chiral molecule. Nature 378(6559):767–768. https://doi.org/10.1038/378767a0
Storch G, Trapp O (2017) By-design enantioselective self-amplification based on non-covalent product-catalyst interactions. Nat Chem 9:179–187. https://doi.org/10.1038/nchem.2638
Storch G, Trapp O (2018) Supramolecular chirality transfer in a stereodynamic catalysts. Chirality 30:1150–1160. https://doi.org/10.1002/chir.23007
Trapp O (2020) Efficient Amplification in Soai’s Asymmetric Autocatalysis by a Transient Stereodynamic Catalyst. Front Chem 8:1173. https://doi.org/10.3389/fchem.2020.615800
Trapp O (2022) Self-amplification of Enantioselectivity in Asymmetric Catalysis by Supramolecular Recognition and Stereodynamics. Supramole Catal New Direct Dev 55–67. https://doi.org/10.1002/9783527832033.ch4
Trapp O, Lamour S, Maier F, Siegle AF, Zawatzky K, Straub BF (2020) In situ mass spectrometric and kinetic investigations of Soai's asymmetric autocatalysis. Chem A Eur J 26.68:15871–15880. https://doi.org/10.1002/chem.202003260
Funding
Open Access funding enabled and organized by Projekt DEAL. We acknowledge financial support from the Max‐Planck‐Society (Max‐Planck‐Fellow Research Group Origins of Life), the Volkswagen Stiftung (Initiating Molecular Life), the Deutsche Forschungsgemeinschaft DFG/German Research Foundation (Project-ID 364653263 – TRR 235, Emergence of Life) and Germany's Excellence Strategy (ORIGINS, EXC-2094 – 390783311).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huber, L., Trapp, O. Symmetry Breaking by Consecutive Amplification: Efficient Paths to Homochirality. Orig Life Evol Biosph 52, 75–91 (2022). https://doi.org/10.1007/s11084-022-09627-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11084-022-09627-6