
Abstract
Pyrite and organic matter closely coexist in some hydrothermally-altered gabbroic xenoliths from the Hyblean Plateau, Sicily. The representative sample consists of plagioclase, Fe-oxides, clinopyroxene, pyrite and minor amounts of many other minerals. Plagioclase displays incipient albitization, clinopyroxene is deeply corroded. Pyrite grains are widely replaced by spongy-textured magnetite, which locally hosts Ca-(and Fe-)sulfate micrograins and blebs of condensed organic matter. Whole-rock trace element distribution evidences that incompatible elements, particularly the fluid-mobile Ba, U and Pb, are significantly enriched with respect to N-MORB values. The mineralogical and geochemical characteristics of the sample, and its U-Pb zircon age of 216.9 ± 6.7 MA, conform to the xenolith-based viewpoint that the unexposed Hyblean basement is a relict of the Ionian Tethys lithospheric domain, mostly consisting of abyssal-type serpentinized peridotites with small gabbroic intrusions. Circulating hydrothermal fluids there favored the formation of hydrocarbons trough Fischer-Tropsch-type organic synthesis, giving also rise to sulfidization episodes. Subsequent variations in temperature and redox conditions of the system induced partial de-sulfidization, Fe-oxides precipitation and sulfate-forming reactions, also promoting poly-condensation and aromatization of the already-formed hydrocarbons. Here we show organic matter adhering to a crystal face of a microscopic pyrite grain. Pyrite surfaces, as abiotic analogues of enzymes, can adsorb and concentrate organic molecules, also acting as catalysts for a broad range of proto-biochemical reactions. The present data therefore may support established abiogenesis models suggesting that pyrite surfaces carried out primitive metabolic cycles in suitable environments of the early Earth, such as endolithic recesses in mafic rocks permeated by hydrothermal fluids.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Scientific reports on abiogenesis traditionally focus more on biological and biochemical aspects, even though the main topic actually extends over the entire realm of the natural sciences (e.g. Ferris et al. 1996; Hazen and Sverjensky 2010), including mineralogical, petrologic and geochemical studies. These can shed light on the inorganic background on which the biochemical processes began. For example, Lindgren et al. (2011) reported that in some rocks from Ireland, Carboniferous in age, pyrite acts as a substrate for the concentration of organic matter. Such an intriguing occurrence conjures up, and even supports, those models suggesting, albeit with different modulation, that pyrite (and other metal sulfides) played a key role in abiogenesis (e.g. Wächtershäuser 1988; Blöchl et al. 1992; Heinen and Lauwers 1996; Holm and Anderson 1998; Tessis et al. 1999; Schoonen et al. 1999; Martin 2012).
The present paper documents a different case of pyrite associated with organic matter, occurring in a xenolith of hydrothermally altered gabbro from the Hyblean Plateau, Sicily. In particular, evidence is provided of organic matter adhering to a pyrite crystal face, a few micrometers in size, that is a pre-requisite for processes such as condensation of biomolecular precursors, prebiotic catalysis and autocatalytic metabolism. In addition, a comprehensive set of petrologic data is here reported and discussed, in order to outline the complex igneous and hydrothermal history of the sample.
Pyrite as a Key Mineral for Abiogenesis
As known, the isometric polymorph of the iron disulfide, pyrite, is a semiconductor, partially ionic crystal. It displays NaCl-type cubic structure (lattice constant a = 5.419 Å, space group Pa3) with Fe2+ at the cation site and S22− dimers at the anion site aligned along the cube diagonal <111>. Pyrite surfaces can selectively adsorb and concentrate different organic molecules, hence promoting a broad range of chemical reactions (Holm et al. 1993; Bebié and Schoonen 2000; Cody et al. 2000, 2004; Zhang et al. 2003; Schulte and Rogers 2004; Pollet et al. 2007; Murphy and Strongin 2009; Wang et al. 2010; Kundell 2011; Liu et al. 2012; Suzuki et al. 2018).
Although the iron and sulfur atoms in the crystal lattice of the pyrite are arranged according to an isotropic 3D geometry, the occurrence of dumbbell shaped S–S pairs can give rise to anisotropic patterns at atomic scale (“atomic terrace edges”: Herbert et al. 2014), on the crystal surfaces. For example, on the (001) crystal face, one of the two sulfur atoms of the S22− dimer is situated above, and the other below, the terminal plane of iron atoms. The apparent tilting of such S–S pairs along particular crystallographic directions produces on the surface a regular array of ridges and grooves, two atoms in thickness (Philpott et al. 2004). These are possible catalytic sites for a variety of chemical reactions, even those displaying (proto)biochemical relevance, also favoring unidirectional arrangement of newly formed polymers (Philpott et al. 2004).
In addition, the pyrite surfaces often displays sulfur vacancy (VS) and iron vacancy (VFe) defect sites. A defective pyrite {100} surface can be due, for example, to more reduced sulfur S2− originating from the breaking of the surface S22− dimers, coupled with the oxidation of surface Fe2+ ions to Fe3+. Therefore in a VS site there are two 4-coordinated (defective) surface iron sites (e.g. Nesbitt et al. 1998; Stirling et al. 2007; Murphy and Strongin 2009; Xian et al. 2019). In addition, vacancy sites can coalesce as vacancy clusters, which are considered more stable than dispersed individual ones (Herbert et al. 2014).
Different defect sites often coexist on the same pyrite surface, which can therefore bind chemically different species (Bebié and Schoonen 2000). Some organic compounds are capable of migrating laterally over the pyrite surface without losing their overall cooperative surface bonding (Wächtershäuser 1988). Such a surface migration process can give rise to the casual contact of two different compounds, which can polymerize producing more complex compounds. Some organic units can be indirectly surface bonded by being attached to directly surface-bonded constituents, therefore increasing the complexity degree of the system. Wächtershäuser (1988) highlighted that organic compounds bound to pyrite surface can persist or spread only if the rate of production equals or exceeds the rate of depletion by detachment. In this respect, weakly bonding constituents give way to stronger bonding ones, giving therefore rise to a process of “natural selection”. Such surface processes may eventually give rise to organic compounds displaying biochemical relevance, such as nucleic acids and proteins.
Both molecular and dissociative adsorption can occur in the pyrite surface (Stirling et al. 2015; Zhao et al. 2014; Li et al. 2018), the latter being generally more energetically favorable on defect sites (e.g. Xian et al. 2019). Adsorption and heterogeneous catalysis on the pyrite surface also depend on different physicochemical parameters of the system. For example, relatively high temperature can decrease activation free energy barriers, high pressure may favor polymerization reactions (e.g. Pollet et al. 2007). Dry or wet conditions can also modify the reactivity of the pyrite surfaces. On dry pyrite surfaces the lowest empty atomic orbitals are on the Fe2+ sites whereas, in wet conditions, water molecules bind on surface iron atoms, hence exposing the surface sulfur atoms to potential nucleophilic attacks (Guevremont et al. 1998; Stirling et al. 2015).
The interfacial free energy of a given organic compound and pyrite surface has been described as a balance between attractive Lifshitz–van der Waals force, repulsive or attractive electrostatic force and acid/base interaction force (Sharma and Rao 2003). In the presence of a liquid phase, as it occurs in hydrothermal conditions, the van der Waals interaction energy is negligible. The electrostatic forces can be depicted by the zeta-potential (ζ), which is the potential at plane of shear between the interacting materials. Electrophoresis experiments by Sharma and Rao (2003) indicated that, in presence of molecular oxygen, the pyrite zeta-potential has an inversed relationship with pH of the hydrothermal solution (ζ > 0 if pH < 7; ζ > 0 if pH > 7). On the other hand, experiments by Bebié and Schoonen (2000) showed that the pyrite zeta-potential, under anoxic conditions, is negative at any pH values. The presence of a bridge of divalent cations, such as Ca2+ and Mg2+, is therefore necessary to allow adhesion of (poly)anionic organic molecules on a pyrite surface in the early Earth environments (Bebié and Schoonen 2000). It is worth to notice that Ca2+ and Mg2+ (and Fe2+) are the most common divalent cations in mafic silicate rocks.
Aforementioned auto-metabolic processes imply the occurrence of abiotic organic compounds already bound on the surface of pyrite, as potential reactants to produce (bio)polymers. Some authors (e.g. Wächtershäuser 1988) hypothesized that the abiotic production of simple organic compounds, such as low molecular weight hydrocarbons and formic acid, may occur on the same surface of pyrite, as the exothermic behavior of reaction of iron monosulfide and hydrogen sulfide under anaerobic conditions:
can even drive the endothermic reduction of carbon dioxide, according, for example, to the following reaction:
Nevertheless, it has been demonstrated on thermodynamic basis that under hydrothermal conditions such a process is precluded by high activation barriers (Schoonen et al. 1999; Michalkova et al. 2011). The organic molecules capable of adhering to the surface of pyrite to initiate a “surface metabolism” reaction pathway should therefore have formed elsewhere, through a different (abiotic) synthesis.
Reactions on pyrite surface may be chirally selective, since pyrite crystals often exhibit striations, usually parallel to [001] on their {210} faces, as a result of selective adsorbing of impurities (e.g. Endo and Sunagawa 1973). In this respect Hazen and Sholl (2003) suggested that symmetry breaking on a chirally-selective mineral surface is a plausible geochemical mechanism for the chiral selection, and subsequent homochiral polymerization, of amino acids in the prebiotic Earth.
More in general, a pyrite-mediated abiogenesis is quite consistent with the ubiquitous role of iron and sulfur in a number of key biologic compounds, such as some proteins (e.g. ferredoxins) and related enzymes, (Schulte and Rogers 2004; Suzuki et al. 2018). Correspondingly, Weiss et al. (2016), through the study of a number of protein-coding genes and protein clusters from sequenced prokaryotic genomes, inferred that the biochemical pathway of the “last universal common ancestor” (LUCA: e.g. Lane et al. 2010) included iron–sulfur clusters.
Submarine Hydrothermal Systems as Primary Loci for Abiotic Organic Synthesis and Metal Sulfides Formation: The Early Earth Perspective
Scientific results of modern marine geology enterprises revealed a number of seafloor hydrothermal vents along slow- and ultra-slow oceanic ridges, where serpentinized mantle ultramafics and mafic igneous intrusives were tectonically exhumed at sea-floor (e.g. Natland and Dick 2001; Mével 2003; Snow and Edmond 2007; Silantyev et al. 2011; Andreani et al. 2014). Serpentinite-gabbro hosted hydrothermal systems can be divided in two main types on the basis of the physicochemical characteristics of their vent fluids (e.g. Rona et al. 2010). In one of two types of hydrothermal systems, fluids exhibit a relatively low temperature (T = 40–90 °C), being also strongly basic (pH = 9–11.5) and very poor in transition metals, silica and Mg. Minerals precipitated at the vent sites give rise to complex systems of overlapping and branching columnar chimneys, known as “white smokers”, mostly consisting of carbonate and sulfate minerals. In this case, the subsurface hydrothermal circulation is though to be driven by the heat released by the exothermic serpentinization reactions (e.g. Evans 2004). The “Lost City” hydrothermal system located 15 km west of the spreading axis of the MAR at 30°N, can be considered the archetypal of white smokers (e.g. Kelley 2015).
The other type of hydrothermal fluids circulating in serpentinites may resemble those emitted by basalt-hosted systems, hence displaying high temperature (up to 407 °C: German et al. 2004), acidic (pH = 2–5) and anoxic character, with high contents in sulfur and transition metals, especially Fe (Seyfried et al. 2011). Abundant metal-sulfides precipitate when the hydrothermal fluids mixes with ambient cold seawater, giving rise to seafloor chimneys dark in color (“black smokers”: e.g. Kelley 2015; Firstova et al. 2016). The serpentinization reactions occurred prior to, and independently of, the sulfide mineralization event, and the hydrothermal circulation is thought to be driven by the conductive cooling of deep-seated igneous bodies (German et al. 2004). The Rainbow is a typical black smoker structure sited on a serpentinite protrusion (“oceanic core complex”: OCC) in the MAR (36°14’ N) (Marques et al. 2007; Seyfried et al. 2011).
Di-hydrogen, methane and other light organic compounds are common components of fluids issuing from hydrothermal seafloor vents, both white- and black-smokers, sited upon serpentinized mantle rocks (e.g. Fu et al. 2007; Konn et al. 2009; Cannat et al. 2010). Serpentinite-hosted hydrothermal systems are thought to be ideal places for the FTT abiotic organic synthesis, which consists of the catalytic hydrogenation of dissolved inorganic carbon species (e.g. Szatmari 1989; Holm and Charlou 2001; Charlou et al. 2002; McCollom and Seewald 2007; McCollom 2013; Etiope and Schoell 2014; Sforna et al. 2018). Di-hydrogen necessary for the FTT synthesis in hydrothermal conditions is mostly released by different reactions implying the anaerobic oxidation of ferrous iron by the protons of water (e.g. Schikorr-type reaction: Schikorr 1933). Typical cases of H2 production are related to the hydration of anhydrous mafic silicate minerals. Considering, for instance, the fayalite end-member of the olivine (Palandri and Reed 2004; Bach et al. 2006; Frost and Beard 2007; McCollom and Seewald 2007):
Magnetite, being one of the products of (3), is one of the most effective catalyst for the FTT organic synthesis, even in hydrothermal systems, together with Ni-Fe and Cr-Fe oxides, Ni-Fe and PGE-bearing alloys (Foustoukos and Seyfried Jr 2004; Fu et al. 2007; Lazar et al. 2012; McCollom 2013). The role of metal sulfides as FTT catalysts is still debated, since more than 2 wt% of dissolved sulfur in the hydrothermal fluid de-activates most metal-oxide and native-metal catalysts (e.g. McCollom and Seewald 2007).
The molecular hydrogen, abiotic organic compounds and sulfur produced in the abyssal serpentinite systems provide energy sources for metabolic reactions of microbial ecosystems dominated by anaerobic methanotrophs and cryptoendolithic chemolithoautothrophic, sulfur-oxidizing, microorganisms (e.g. Brazelton et al. 2011; Ménez et al. 2018). This fact inevitably gives rise to the “contamination” of abiotic hydrocarbons by a variety of biogenic organic compounds (e.g. Gold 1999).
It is generally considered very likely that submarine hydrothermal systems hosted in mafic–ultramafic rocks were present, even very abundant, on the ultramafic oceanic crust of the pre-biotic Earth (e.g. Russel et al. 2005). This would result, as in modern cases, in chemically reactive environments rich in H2, CO2, S, P, N, Fe and other transition metals, far from equilibrium conditions, displaying temperature, redox and pH gradients (e.g. Kelley 2015). The early ocean was particularly rich in dissolved CO2 from the primitive atmosphere and also rich in hydrogen from serpentinization reactions affecting the widespread ultramafic substratum, including komatiite lava flows. The production of abiogenic organic compounds was therefore much more favored than it can be in modern times (Russel et al. 2005). The coexistence of pyrite (and other sulfides) and organic compounds was certainly quite common in Early Earth submarine environments, especially in the fractured mafic and ultramafic rocks hosting the ascending limbs of high-T hydrothermal cells, and in the related vents at the seafloor. Accordingly, some authors (e.g. Lane et al. 2010; Dodd et al. 2017) suggested that LUCA was a thermophile organism, which inhabited a submarine, anaerobic, hydrothermal vent setting.
Ancient Submarine Hydrothermal Events Inferred from the Study of Hyblean Ultramafic and Gabbroic Xenoliths
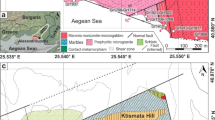
Diatremes from the Hyblean Plateau (Sicily: Fig. 1), Late Miocene in age (Carbone and Lentini 1981), carried out, as a rule for such unusual eruptive systems, a number of rock fragments (xenoliths), generally having the size of a fist, deriving from the underlying pre-Triassic basement, which is not exposed anywhere in this region nor in the surrounding areas. A 30 years study of the Hyblean deep-seated xenoliths has led researchers to conclude that the Mesozoic to Cenozoic sedimentary and volcanic succession of this Central Mediterranean area (Fig. 1) is set on a “fossil” oceanic lithosphere likely belonging to the Oman-Levantine-Sicily branch of the Permian Tethys Ocean (e.g. the review provided by Manuella et al. 2015; Giampiccolo et al. 2017). In this respect, gabbroic xenoliths have played a crucial role to put forward the aforementioned hypothesis. Scribano et al. (2006a) highlighted that the oxide-rich variety, with sheared (“mortar”) texture, closely resembles gabbro from modern oceanic fracture zones (FZs) along slow-spreading ridges (e.g. Natland and Dick 2001). Moreover, secondary mineral assemblages occurring in various Hyblean xenoliths record a complex hydrothermal history likely developed in an abyssal serpentinite system (Scribano et al. 2006b, 2009; Manuella et al. 2015, 2018). Organic matter, often associated with reduced carbon species (including nanodiamonds: Simakov et al. 2015), occurs in some serpentinized ultramafic and spilitized gabbroic xenoliths. An abiotic origin of the aforementioned organic matter via FTT synthesis associated to serpentinization reactions has been tentatively put forward by Scribano and Ioppolo (2006) and hence experimentally substantiated by Ciliberto et al. (2009) and Scirè et al. (2011). Nevertheless, none of aforementioned samples, bearing abiotic organic matter, exhibit significant amounts of sulfide minerals.

Location (a) and geological sketch map (b) of the sample area. Arrow indicates the site (Valle Guffari) of the xenolith-bearing tuff-breccia deposit where the sample has been collected. Legend: 1) Major fault lines; 2) Rudists-bearing limestones (Upper Cretaceous); 3) Submarine mafic volcanic rocks (Cretaceous); 4) Cenozoic limestones; 5) Marly–limestones and marls (Oligocene – Miocene); 6) Xenolith-bearing diatremes and/or related tuffeccia deposits (Tortonian); 7) Plio-Pleistocenic OIB-type and E-MORB-type volcanic rocks; 8) Neogene–Quaternary open–shelf clastics; 9) Folded and thrusted marine sedimentary rocks (Mesozoic to Cenozoic) and Messinian salt deposits (Simplified after Lentini and Carbone 2014) (c) “GoogleEarth” satellite view of Valle Guffari area. The arrow indicates the tuff-breccia outcrop where the sample was taken
Here we consider a xenolith of hydrothermally altered gabbro, displaying an assemblage of redox-sensitive sulfur-bearing minerals which coexist with a sizable amount of organic matter. Although we collected in the same area several xenoliths exhibiting the same petrologic and geochemical characteristics as the sample, the latter has been selected as representative of the entire group.
Material and Methods
The sample (SM2) is a gabbroic xenolith from the tuffsite-breccia deposits outcropping in Valle Guffari, Hyblean area (Sicily, Southern Italy: Fig. 1). The aforementioned tuff-breccia is related to a shallow-submarine diatreme structure, Upper Miocene in age (Carbone and Lentini 1981; Suiting and Schmincke 2009; Suiting and Schmincke 2010). The sample exhibited rough oval shape, ~18 cm in length, ~10 cm in width. It caught our attention due to the intriguing odor of bitumen released in the course of cutting to make a thin section. Heating on a methane flame of a hand specimen of the same confirmed the above-mentioned circumstance, with the additional information due to a transient scent of sulfur, issuing just before the more persistent odor of bitumen. No visible impregnation of hydrocarbons has been detected in the outcropping zone of the sample, in Valle Guffari (Fig. 1), ruling therefore out a late impregnation of hydrocarbons in the outcrop area. Moreover, this is located in a steep wall along a narrow mountain-path, being quite outside any form of pollution due to human activities.
Routine petrography study was performed by the means of optical microscope and scanning electron microscope (SEM). Mineral chemistry was obtained by SEM fitted with Energy dispersion system (EDS), microprobe fitted with wavelength dispersion system (WDS) and Laser Ablation inductively coupled plasma mass spectrometry (LA-ICPMS). Whole rock chemistry was done via ICPMS and ICPAES. U-Pb zircon age has been obtained by LA-ICPMS. Micro-Raman spectra were useful to assess some mineralogical and organic geochemistry issues.
Going into more details, we used a TESCAN-VEGA\\LMU SEM at the Department of Geological Sciences, Catania University (Italy). Observations were made in backscattered electron mode under high vacuum conditions at accelerating voltage 20 kV and beam current 0.2 nA. SEM was equipped with an EDAX Neptune XM4 60 microanalysis working in energy dispersive spectrometry (EDS). Qualitative analyses on organic compounds were performed on uncoated thin sections and on rough fragments of the sample. Wavelength-dispersive analyses of major elements of diverse minerals were performed using the JEOL JXA 8230 electron probe micro-analyzer (EPMA) at the Department of Biological, Ecological and Earth Sciences of the Università della Calabria (Cosenza, Italy). Calibrations were carried out using native metals and silicates as standard materials. Analyses were obtained under the following instrumental conditions: an accelerating voltage of 15 kV, a beam current of 10 nA, a beam spot of 5 μm, and an analysis time of 100 s. A detection limit of 0.01 wt.% is for all the detected elements.
Selected portions of the whole-rock were crushed and powdered with an agate mortar. Weighted aliquots were sent to the laboratory of SGS (Canada). The powdered samples were there analyzed by ICP-AES and ICP-MS technical procedures, with different digestion methods. In particular, digestion by lithium metaborate fusion was used to analyze major elements and most trace elements. Sodium peroxide fusion was used to analyze REE. Two-acids digestion, which is based on a combination of 2:1 HNO3: HCl, was used to analyze major and different trace elements in order to get information on the composition of non-silicate minerals, since most silicate minerals remain undigested by such weak solute. The accuracy of the methods was determined by analyzing certified reference materials, while their precision was determined with replicate analyses (and found to be generally better than 10%). More information is given in the SGS analytical services guide (com/−/media/global/documents/brochures/sgs-analytical-guide.pdf).
In situ trace-element analyses were performed in key minerals by laser ablation ICP-MS technique at Istituto Nazionale di Geofisica e Vulcanologia (INGV) in Palermo. Selected fragment of the sample, of about 80 μm in thickness, were polished before analysis. The analytical system consisted of an Agilent-7500 CX quadrupole mass spectrometer coupled with an ArF excimer laser ablation system (GeoLas Pro). During analysis, samples were maintained in a helium atmosphere, with a laser output energy of 10 J/cm2, a repetition rate of 10 Hz, and a 130-μm-diameter circular spot. We used Ca, Si, and Fe as internal standards and NIST 612 as an external standard. The NIST 612 analyses were carried out at the start, middle, and end of each analytical session. The precision was determined during each analysis session from the variance of ~15 NIST 612 measurements, which gave a relative standard deviation of b5%. The accuracy, calculated using the BCR-2 international standard, was b10% for most of the elements.
Zircon U-Pb age was obtained at University of Pavia by Laser ablation model “Geolas 200 Q-Microlas”, type “Excimer ARF” and ICP-MS model “Element I, ThermoFinningan Mat”, type “Single-collector double-focusing magnetic sector field”. Spot diameter was 20 and 10 μm. Determined isotopes were: 202Hg, 204(Pb + Hg), 206Pb, 207Pb, 208Pb, 232Th, 238U. Data reducing software was GLITTER (van Achterbergh et al. 2001). Seven spot analyses were made in a single zircon grain to ensure a statistically valid result.
Phase Analyses and Whole-Rock Chemistry of the Sample
Plagioclase
About 75% by volume of the sample consists of a porphyroclastic aggregate of plagioclase grains, 0.5–2.5 mm in size (Fig. 2a,c). Porphyroclasts often display finely polysynthetic mechanical twinning according to the albite and pericline laws, whereas subgrains are generally untwinned. Both porphyroclasts and subgrains display undulose extinction, being also crosscut by an unusual checkerboard-like fracture system (Fig. 2d), and hence clouded by a number of secondary fluid inclusions whose nature is not yet clarified. Plagioclase feldspar displays patchy zoning (An mol% = 28–50: Table 1) containing 750–1200 ppm of Sr and 60–150 ppm of Ba (Table 2).

Petrographic details of the sample seen at different magnification. a Magnified picture of the cut surface of a small fragment of the sample. Pale colored areas consist of plagioclase feldspar, the differently colored areas include Fe–oxide(s), sulfides, relicts of pyroxene and other minerals as reported in the text. b A more magnified area of the previous picture, highlighting the textural relationships between plagioclase (Pl), Fe–oxide (Fx) and pyrite (Pt) grains. c Microphotograph of part of a thin section of the sample (plane pol. light) displaying an aggregate of corroded pyroxene grains (Cpx) surrounded by an opaque rim. Pale colored areas consist of inequigranular plagioclase (Pl) grains. d Microphotograph (plane pol. light) showing checkerboard-type fracture system in a plagioclase grain of the same thin-section as in (c), and lots of secondary fluid inclusions. e SEM image of a portion of clinopyroxene grain evidencing corrosion features and breakdown products (X = amorphous or cryptocrystalline silicate matter). f SEM image displaying a particular of one of the numerous Fe-oxide(Fx) areas in the sample, with associated Ca-Fe-sulfate micrograins (Sf)
Fe-Oxides
About 12% by volume of the sample consists of irregular patches, dull gray in color, largely formed by Fe-oxide minerals (Fig. 2a,b). Detailed electron microscope observations highlighted that these Fe-oxides consist of an irregularly packed assemblage of rosette-like aggregates of Fe-oxide platelets, <1 mμ in size, which give rise to a finely porous texture (Fig. 2f). Even though no investigation on magnetic susceptibility of the sample has been performed, it has been empirically observed that a hand-specimen of the whole-rock, about 2.5 × 2.5 × 0.5 cm in size, placed on the cover glass of a standard field–geology compass (Bézard), induces a ~25° deflection to the compass needle. Moreover, the largest Fe-oxide patches of the sample can attract and hold a stapler pin from a distance ≤0.5 cm. Such a magnetic behavior of the sample indicates that the Fe-oxide domains largely consist of magnetite.
Microprobe spots in the spongy-textured Fe-oxide(s) areas detected almost only Fe, but low quantity of Ti (0.06 < TiO2wt% < 0.60) and Al (0.47 < Al2O3 wt% < 1.76: Table 1). As previously mentioned, the magnetic behavior of the sample Fe-oxides strongly points to a magnetite-rich composition, although the occurrence of some hematite and/or a crystalline or amorphous hydroxide phase cannot be ruled outs. LA-ICPMS analyses revealed trace amounts of Cr (0.63 < Cr ppm <77) and Ni (171 < Ni ppm <2357), with Ni/Cr > 30.
The Fe-oxide(s) micro-Raman spectrum (Fig. 3a), exhibits a strong background noise, probably due to fluorescence related to pore-filling organic matter. Two bands are clearly above the background, the one with a peak at about 643 cm−1, the other displaying a broad, asymmetric shape (Fig. 3a), in the spectral range 750–775 cm−1. Hanesch (2009) highlighted that poor crystallinity leads to a broadening of the Fe-oxyhydroxides bands. The main peak of magnetite is reported at 667 cm−1, with minor bands at 540 cm−1 and 310 cm−1 (de Faria et al. 1996; RRUFF ID: R060191). Nevertheless, Hanesch (2009) highlighted that magnetite is at risk of being damaged by the laser-beam, e.g. by heating induced oxidation (Shebanova and Lazor 2003; Gasparov et al. 2000). The morphology of the spectrum and position of peaks displayed in Fig. 3a can be therefore an artifact due to the transformation of magnetite into a mixture of poorly crystalline hematite (α-Fe2O3) and maghemite (γ-Fe2O3) nanoparticles (e.g. de Faria et al. 1996; Hanesch 2009; Chernyshova et al. 2007). The occurrence in the sample of some original hematite replacing pyrite together with magnetite, cannot be however ruled out. Sparse, strongly corroded, relicts of ilmenite, of likely igneous origin (Scribano et al. 2006a) also occur in the sample, consisting of TiO2 = 46 wt%; FeO (total) = 42.82 wt%; MgO = 8.97 wt%.

Micro-Raman spectra of different components of the sample (a = Fe-oxide; b = pyrite; c,d = two different blebs of organic matter)
Iron Sulfide
Pyrite occurs as aggregates of inequigranular (1–250 μm in size), grains generally anhedral (Fig. 4a,c). A few euhedral pyrite grains, as small cubes displaying either smooth or pitted (Fig. 4e) faces, were also observed between the dominant anhedral pyrite micrograins. The relatively coarse subgrains are irregularly arranged, giving rise to wide interstitial areas filled by selvages of the smallest (≤3 μm in size) grains (Fig. 3c) which locally display framboidal texture (e.g. Butler and Rickard 2000). Both coarse and fine pyrite grains are more or less replaced by spongy-textured magnetite (Fig. 4a, d) hosting clusters of Ca- and Fe-sulfate micrograins, organic matter (Fig. 2f), and fibrous, Mg-rich, silicate minerals tentatively ascribed to the fibrous-clay (sepiolite – palygorskite) group. Pyrite massive grains closely approaches the theoretical FeS2 composition (e.g. Fe = 46.21 wt%; S = 53.79 wt%: S/F = 2.000), whereas some spots in the alteration rim are out of pyrite stoichiometry because of some sulfur deficiency. LA-ICPMS spot analyses (Table 2) revealed very low contents in Cu (224–771 ppm), Zn (66–131) and negligible Pb (2–9 ppm). Micro-Raman spectrum, with main bands at 343 cm−1 and 376 cm−1 (Fig. 3b), confirms that the considered sulfide is the FeS2 cubic polymorph (pyrite: Vogt et al. 1983).

SEM images (BSE mode) of fragments and thin section of the sample, displaying pyrite and pyrite–Fe-oxide(s) assemblages. a SEM image (secondary electrons) of pyrite (Pt) micrograins surrounded by their breakdown products largely consisting of Fe-oxide(s) (Fx), immersed in a plagioclase (Pl) matrix. b EDS elemental map of sulfur, iron and calcium distribution in the same as in (a). c Typical aspect of an aggregate of pyrite grains occurring in a fragment of the sample. d Detail of the boundary surface between a pyrite relict and the surrounding spongy Fe-oxide selvage. Arrow (bottom left corner) indicates exolution blades of galena. e Framboidal and ehuedral pyrite coexisting in one of the numerous pyrite clusters f Detail of the previous image evidencing truncated pyrite microcrystals at a framboid surface (Butler and Rickard 2000)
Pyroxene
Clinopyroxene forms about 6% by volume of the sample mineral assemblage, consisting of aggregates of a few highly corroded grains, 1–3 mm in size, immersed in a turbid matter, or as fragments of individual grains (Figs. 2c,e and 5a,b). The aforementioned turbid matter, often hosting amygdalae of hyaline calcite, consists of clay minerals, probable limonite and blebs of an amorphous or cryptocrystalline silicate matter. The pyroxene grains display very fine exsolution lamellae of Ca-poor pyroxene and/or ilmenite, often coexisting in parallel arrays. Such ex-solution lamellae are impossible to avoid during the microprobe analyses and certainly influence the analytical result (Table 1). Pyroxene mineral chemistry can be summarized as the following: (42 ≤ Wo mol% ≤43.5; 43.6 ≤ En mol% ≤45.5; 12.2 ≤ Fs mol% ≤ 12.8), with Na2O~ 1 wt%, Al2O3 = 3.8 wt%, TiO2 = 0.5 wt%. Representative trace element analyses obtained by LA-ICPMS spot analyses are reported in Table 2. The unusually high content in Ba (=48 ppm: Table 2) in some spot analyses may indicate the occurrence of secondary mineral (e.g. amphibole or its retrograde products), less than one micrometer in size, replacing the pyroxene.

Microphotographs documenting the petrographic aspects of the only zircon grain in the sample suitable for U-Pb dating. a Magnified picture of a cut surface of an hand-specimen of the sample (Pl = plagioclase; Zrn = zircon; Cpx = clinopyroxene). b SEM image (BSE mode) of the central zone of the same area as in (a). c Concordia diagram (238U/206 Pb vs. 207Pb/206Pb) based on eight LA–ICPMS spot analyses in the same zircon grain as in (a,b). More information is given in the text
Zircon
Only one euhedral zircon grain (ZrO2 = 66.13; HfO2 = 0.80; SiO2 = 32.7) was observed in the sample, and hence selected for U-Pb dating (Fig. 5). Seven spot LA-ICMS analyses were performed in the single zircon grain for uranium - lead dating. Methodology details are report in the text. The spots were distributed evenly in the grain and no zones with different ages were detected. Concordia diagram indicates a 238U–206Pb age of 216 ± 6.7 Ma age (Fig. 5).
Other Minor Minerals
Scarce amounts of apatite and many other minerals also occur in the sample. Apatite bears some thorium (Th = 51 ppm), uranium (U = 19 ppm) yttrium (Y = 664 pm) and REE (in particular Ce = 593 ppm and Nd = 626 ppm). There are different Cu-bearing minerals including a Cu-oxide, whose chemical composition (CuO = 91.9 wt%; Zn = 5.2 wt%; FeO = 1.24 wt%; NiO = 1.66 wt%) and acicular texture indicate the tenorite polytype. Tenorite is often associated with irregular patches, turquoise-blue in color at the hand-specimen scale, of a copper-bearing silicate phase, tentatively ascribed to chrysocolla accordingly to EDS analyses. Moreover, some amygdalae filled by single calcite grains are rimmed by a film of a Cu-rich phase, likely a copper carbonate, impossible to analyze precisely because it is smaller than the spot beam. Covellite (Cu = 59.64 wt%; S = 40.36 wt%) micrograins are also dispersed in several areas of the sample. Different metal-sulfides micrograins, less than 25 mμ in size, also rarely occur in the sample, including cadmian sphalerite, realgar, calcopyrite and galena. Monazite-(Ce), and a LREE-rich oxide mineral tentatively ascribed to the Aeschynite-group minerals were also detected by a carefully SEM investigation.
Whole-Rock Chemistry
Due to the relatively high modal abundance of non-silicate minerals in the sample, two portions of the powdered sample were processed by two different digestion methods prior to obtain ICP-MS chemical analyses. The one method gives rise to the digestion of the entire mineral assemblage, the other does not digest most silicate minerals, but some clay minerals and silicate glass. The comparison between the different analytical results, in the light of the modal and normative mineral assemblages and mineral-chemistry data, provided us valuable, though not definitive, information on the distribution of major elements between silicate and non-silicate minerals in the sample.
Whole rock major element analysis (Table 1) of the “fully-digested” portion of the sample displays relatively low contents in magnesium (MgO = 1.99 wt%), calcium (CaO = 7.88 wt%) and silica (SiO2 = 45 wt%), and relatively high contents in iron (Fe2O3 as total iron = 11.72 wt%) and in sodium (Na2O = 4.9 wt%) according to the high modal abundances of plagioclase feldspar and Fe-rich, non silicate minerals. On the other hand, major element analysis on the “partially-digested” fraction (Table 1) yielded very low totals (sum of oxides = 17.55 wt%), iron being the most abundant element (11.18 wt%, as total Fe2O3). This fact indicates that most of the iron of the rock is allocated in the oxide and sulfide minerals. Large part of the MgO in the sample occurs in the partially digested portion as well, probably occurring in clay minerals. On the other hand, the MgO obtained by subtracting the amount of the partially digested sample to that of the fully digested one, is 0.82 wt%, being therefore compatible with the estimated modal occurrence of clinopyroxene in the sample (about 5% by volume). Most of CaO (=6.41 wt%) is allocated in the “fully digested”, silicate rich fraction, accordingly to the modal abundance of plagioclases. CaO in the mostly non-silicate fraction is 1.47 wt%. (Table 1). Looking into more details, 0.15 wt% of CaO may be allocated to phosphates (apatite in most part), as calculated on normative (CIPW) basis. About 0.38 wt% of CaO occurs as carbonate, consistently with the 0.25 wt% CO2 detected in the whole rock by coulometry. The rest (=0.98 wt%) of the non-silicate CaO may be allocated to sulfate (gypsum and/or anhydrite) which has been detected by EDS spot-analyses (Figs. 2f) between the pyrite breakdown products.
A problematic aspect of the major element distribution in the whole-rock is related to the relatively low value of the sum of the analyzed oxides (Total = 95.1 wt%: Table 1), which includes the mass fraction lost on ignition (LOI = 2.29 wt%), as recorded in the analysis of the “fully digested” fraction. In this respect, we put forward the hypothesis that about 5 wt% of the sample mass, possibly in form of hardly reactive organic compounds, such as PAH-rich polycondensates, escaped even the “strong” digestion process (by Li-methaborate fusion), hence persisting unanalyzed.
Considering results on portions of the sample digested by Li-metaborate and Na- peroxide fusion, respectively, trace element distribution (Table 2) evidences a significant REE fractionation (CeN/YbN = 14.6) with positive Eu anomaly (Fig. 6). This fact agrees with the high modal abundance of plagioclase (~70 vol%) in this rock. Some fluid-mobile elements are more or less enriched with respect to MORB values (e.g. Rb = 6 x MORB; Ba = 38 x MORB; Sr = 8.6 x MORB; Th = 5.5 x MORB; Pb = 14 x MORB), whereas the amounts of less mobile elements (e.g. MREE, HREE, Y, Nb) are MORB-like or depleted at different extend with respect to MORB values (Fig. 6).

Multielement variation diagrams of whole-rock and some mineral phases of the sample. a Classification of the sample (circle) through the Nb/Y vs. SiO2 of Whinchester and Floyd (1977). b Morb-normalized compatibility diagram of two fractions of the whole-rock, processed by Li-methaborate fusion (WR1) and two-acids digestion (WR2), respectively. c REE patterns (CI normalized) of the sample whole-rock (WR1) and one of its pyroxene grains. Patterns indicated as cpx1 and cpx2 represent results of spot analyses by La-ICPMS performed in two different zones of the same pyroxene grain. More information is given in the text
Comparisons between trace element abundances between fully-digested and partially-digested portions of the sample indicate that Sr is almost totally allocated in the silicate fraction (e.g. plagioclase) whereas V, Co, Zn and Cu, mostly occur, as expected, in non-silicate (e.g. sulfide and oxide) minerals.
Organic Matter
Looking at a cut surface of the sample by the means of a (10x) magnifying glass, organic matter appears as irregular patches, 0.3 to 1.5 mm in width, black in color, with a glassy luster. In a standard thin section, under the optical microscope, the organic matter looks quite opaque. Blebs of organic matter, 20 ― 200 μm in size, often associated with Ca-, and minor Fe-sulfate, and Mg-rich clay minerals, often occur in the oxide-hydroxide areas (Fig. 7a). More interestingly, SEM observations revealed blebs of organic matter adhering to crystal-faces of euhedral pyrite grains (Fig. 7b). Organic matter also fills pores, less than one micron in size, in the spongy-textured Fe-oxides.

SEM images (BSE mode) illustrating textural relationships between organic matter (OM) and coexisting minerals in the sample. a View of a typical assemblage of spongy-textured Fe-oxide (Fx) micrograins, organic matter (OM) and Ca- Fe-sulfate micrograins (Sf). b Organic matter (OM) attached in one of the faces of a small, ehuedral pyrite grain belonging to a pyrite micrograins aggregate as in Fig. 2a. c A relatively large bleb of organic matter filling a void in a plagioclase rich area of the sample. More explanation is reported in the text.
Micro-Raman analyses were performed in two blebs of organic matter in the 500–2500 cm−1 Raman shift region. The samples presented an intense fluorescence signal. Fluorescence profiles exhibit a broad bulge, with a “crown” of several peaks, in the spectral range 1250–1650 cm−1 (Fig. 3c) and 1250–1500 cm−1 (Fig. 3d), respectively, where chemically heterogeneous carbonaceous materials display their characteristics Raman bands (e.g. Ferrari and Robertson 2000; Cloutis et al. 2016). Unfortunately, such low quality spectra do not allow sorting out closely juxtaposed peaks, and hence multiple interpretations exist for the various peaks. For example, peak at 1280 cm−1 may indicate CH2 wagging (Dollish et al. 1974; Sforna et al. 2018 and references there reported), peaks at 1315 and 1353 cm−1 CH3 bending (Cloutis et al. 2016), peak at 1380 cm−1 symmetric CH3 bending, peaks at 1405 and 1464 cm−1 C―H bending (Cloutis et al. 2016). Bands at 1300 and 1350 cm−1 may be related to deformation of phenyl rings and C-C stretching (Cloutis et al. 2016). In addition, Jelicka et al. (2006) and Gremlich and Yang (2000) assigned the bands at 1386 and 1468 cm−1 and a weak band at around 1400 cm−1 to symmetric stretching vibrations of COO― functional groups. On the other hand, one of peaks in the range about 1370 - 1380 cm−1 the “disorder” (D) band of the reduced carbon (e.g. Ferrari 2001; Reich and Thomsen 2004). Moreover, peaks at 1424, 1573 and 1630 cm−1 can be associated to C=C stretching (Cloutis et al. 2016). The first order Raman band of the “ordered “carbon materials (“G” band: e.g. Ferrari 2001), in the spectral range between 1560 and 1630 cm−1, here displays very low intensity in one of the two spectra (Fig. 7c), lacking at all in the other one (Fig. 3d).
Obvious physical characteristics of the sample organic matter (e.g. its capability of being cut in thin section and polished, the bitumen odor released on heating) conjure up macromolecular organics rich in PAHs, such as petroleum asphaltenes (e.g. Rodrigues Coelho et al. 2006). Accordingly, the high fluorescence noise here recorded (Fig. 3c,d) may be related to organic compounds displaying conjugated bonds (Matrajt et al. 2004 and references reported there), hence consisting of a number of PAHs with large ring systems and/or functional groups, which exert mesomeric effects (e.g. Busemann et al. 2007; Cloutis et al. 2016). On the other hand, the Raman bands of the sample organics are not consistent with compounds rich in PAHs, which display strong “G” band, since the electronic and vibrational properties of PAHs tend towards those of graphite, as the system becomes large enough to contain six aromatic rings in conjugation (e.g. Loppnow et al. 2004). On the above grounds, the present set of data best corresponds to some macromolecular compounds, tentatively indicated as “natural hydrocarbon (petroleum) resins” (e.g. Andersen and Speight 2001) characterized by relatively large number of saturated bonds within their fused ring systems, hence displaying much lower aromaticity than asphaltenes (e.g. Hashmi and Firoozabadi 2013).
Discussion
The porphyroclastic texture of the sample and its modal composition correspond to those observed in the “sheared oxide-gabbros”, which are relatively common among the Hyblean xenolith suite (Scribano et al. 2006a). On the other hand, several petrologic and mineralogical features (such as evidence of Na-metasomatism in the plagioclase, relatively high abundance of pyrite and, more in general, of ores, some of which being quite unusual in mafic igneous rocks; the incipient-to-advanced breakdown of pyroxene and ilmenite) strongly indicate that the putative gabbro protolith underwent a complex alteration history. In consideration of the alteration effects on chemical composition, the sample can be classified by using the SiO2 vs. Nb/Y diagram (Winchester and Floyd 1977), where it plots in the subalkaline basalt (gabbro) field, close to the alkaline basalt field (Fig. 6a).
The Late Triassic zircon age implies an original sub-seafloor setting, in accordance with the geological history of the Hyblean region as inferred from field and borehole investigations (e.g. Vai 1994, 2003; Bianchi et al. 1987), geophysical data (Catalano et al. 2000; Giampiccolo et al. 2017) and studies on deep-seated xenoliths (Scribano et al. 2006a, b; Manuella et al. 2015).
Previous studies on Hyblean xenoliths (Scribano et al. 2006b, 2009; Ciliberto et al. 2009; Scirè et al. 2011; Manuella 2011, 2015, 2016, 2018) indicated that an “abyssal” serpentinite system was active in this area since the early Mesozoic time. Pyrite occurring in the sample is most probably related to S- and metal-rich hydrothermal fluids circulating in the shallow sub-seafloor, as inferred from the study of present (Marques et al. 2007; Rona et al. 2010; Kawasumi and Chiba 2017) and “fossil” (e.g. Barriga 1990) oceanic settings. A temperatures ≤160 °C is suggested for the hydrothermal fluids related to the pyrite formation according to experimental data by Taylor et al. (1979) and Rickard (1997).
The Pyrite Perspective
Some of the pyrite–forming reactions in abyssal-type hydrothermal systems, such as that suggested by Holm and Anderson (1998), imply an iron monosulfide precursor
Products of reaction (4) entail some molar volume increasing with respect to reactants, that may explain the unusual “checkerboard type” fracture system in the plagioclase porphyroclasts (Fig. 2d). Butler and Rickard (2000) suggested that, since the FeS precursor dissolves as cluster complexes of quantum-sized particles displaying an electroactive behavior, the newly formed pyrite grains assume anhedral, even “framboidal”, texture. According to experiments by Qian et al. (2010), the morphology of the pyrite crystals could be also related to the pH value of the circulating hydrothermal solution. A pH value >4 favors formation of anhedral grains, whereas ehuedral grains (Figs. 4e and 7b) can be formed at pH < 4.
Although part of the magnetite in the sample may be a co-product of the pyrite–forming reaction (4), textural relationships between the two mineral phases indicate that large part of the magnetite is a product of the pyrite breakdown (Figs. 4a–d). In this respect, it is reasonable to hypothesize that dramatic variation of physicochemical parameters of the aforementioned hydrothermal system (related, for example, to an increasing of the water/rock ratio) induced desulfidization and hence pyrite grains were extensively replaced by Fe-oxides and some sulfates. On these grounds, the Fe-oxide formation may be regarded as the reverse of the magnetite sulfidization pathway suggested by Qian et al. (2010):
Or:
Some energy required for pyrite desulfidization reactions (5, 6) may have been likely provided by an hypothetical shallow-sited igneous intrusion, which is a common occurrence in oceanic core-complexes (OCCs) hosting hydrothermal systems (e.g. Grimes et al. 2008).
Desulfidization of pyrite may also occur through the hydroxylation on defect sites with no need of external energy inputs. Experimental results by Xian et al. (2019) validated the hypothesis by Borda et al. (2003) that, under hydrothermal anaerobic conditions, the oxidation of H2O by the defective Fe sites on the pyrite surface produces OH• radicals:
Borda et al. (2003) also suggested that the combination of two (OH•) can produce H2O2 and subsequently O2. Molecular oxygen can therefore induce desulfidization in defect-free pyrite surfaces:
From the data-set reported by Qian et al. (2010), it is possible to infer that pyrite desulfidization and magnetite production produces an equivalent molar volume decrease up to ~60%, which may explain the porous texture of the Fe-oxide in the sample (Figs. 2 and 4).
Pyrite dissolution kinetics can be also described in term of an electrochemical mechanism (Rimstidt 2003). The anodic reaction produces ferrous and sulfate ions (Caldeira et al. 2009):
Hence the related cathodic reaction (Caldeira et al. 2009):
The sulfate anion (SO42−) released by the pyrite breakdown (7, 9), can react with dissolved metal cations to produce sulfate minerals. In the study case, Ca2+ ions were produced by the process of albitization of calcic plagioclase, Ca-sulfate (Fig. 2f) being therefore the main reaction product:
The formation of an unknown, presumably scarce, amount of Fe-sulfate, as indicated by EDS spot-analyses, suggests that part of Fe2+ deriving from the breakdown of pyrite escaped the Fe-oxide formation.
Abiotic Organic Compounds
Condensed organic compounds, such as aliphatic hydrocarbons and asphaltenes, were previously detected in some Hyblean gabbroic and ultramafic xenoliths by the means of impact-direct pyrolysis mass spectrometry (EI-DPMS), micro- Fourier transform infrared spectra (μ-FTIR), thermogravimetric and termodifferential analyses, X-ray photoelectron spectroscopy (XPS) (Ciliberto et al. 2009; Scirè et al. 2011). The same authors concluded that the FTT synthesis in an abyssal hydrothermal system is the most likely process to originate the aforementioned organic matter. The same FTT synthesis may be therefore invoked for the origin of the organic matter in the sample. In this respect, molecular hydrogen has likely derived from a Schikorr-type reaction involving divalent iron released during pyroxene breakdown (Fig. 2c), and hence inorganic carbon, as CO2 and carbonate, and adequate catalysts, such as magnetite, were certainly present in the same system. The molecular hydrogen may have been present in the circulating hydrothermal fluid, being therefore due to the serpentinization of the peridotite wall rocks.
On the other hand, sulfides were in negligible amount in the xenoliths studied by Ciliberto et al. (2009) and Scirè et al. (2011), while they are relatively abundant in the present case. The role of sulfides as FTT catalyst has been discussed by several authors (Cody et al. 2004; McCollom and Seewald 2007; Bromfield and Coville 1999; McCue and Anderson 2014; Holm and Anderson 1998). Cody et al. (2004) concluded that pyrite is not an effective FTT catalyst. On these grounds we put forward the hypothesis that the organic matter attached on the pyrite surface (Fig. 3b) was not produced in the same surface, but it has been already formed in different places of the same sample via FTT synthesis not involving sulfides, as previously mentioned. FTT synthesis is thought to produce relatively light, hence very mobile, hydrocarbon species (e.g. Chaumette et al. 1995; McCollom 2013). This fact may explain the occurrence of organic matter in any capillary fractures, fluid inclusions and micrometric pores in the sample, even though the organic matter presently displays a dense, highly viscous hence hardly mobile, behavior. On these premises, early formed light (and mobile) hydrocarbons, except the fraction likely entrapped in fluid inclusions, got severe degradation (e.g. aromatization and poly-condensation) through the entire history of the hydrothermal system, giving eventually rise to the extant blebs of solid-like matter sparse in the rock. In this respect, Scirè et al. (2011) typified as asphaltenes (Calemma et al. 1995) the organic matter occurring in some hydrothermally altered, ultramafic xenoliths from the same area, locally hosting clusters of nanodiamonds (Simakov et al. 2015).
Implications on Abiogenesis
Natural hydrocarbon resins, as those abiotically formed in the sample, consist of complex organic molecules bearing highly aromatic and acidic moieties, with sulfur, oxygen, and nitrogen heteroatoms, being therefore highly polar species with high surface activity (Andersen and Speight 2001). This fact likely favors the adhesion of resins to adequate defective pyrite surfaces. In this respect, it is noteworthy the probable occurrence of COOH− groups in the sample organics, as indicated by micro-Raman spectra (Fig. 3c,d). The synthesis of complex prebiotic molecules on the pyrite surface here is facilitated, since such “hydrocarbon resins” display an intrinsic structural and compositional complexity being therefore apt to be transformed into more complex organic molecules, with possible biological relevance. On the other hand, the pyrite dissolution reactions (7, 8) suggested by Caldeira et al. (2009), as protons and electrons producers, may also give rise to chemiosmoisis and hence fundamental proto-biochemical compounds, according to Lane et al. (2010).
Some cubic pyrite micrograins occurring in the sample display pitted surface (Fig. 4d). A typical “pit” appears as an irregular half-bubble much less than one micrometer in size. There are different ways for pits formation on the pyrite surfaces. Experimental results under reducing conditions by Herbert et al. (2014) suggested that at a temperature of ∼240 °C, vacant cation and anion sites coalesce into pits of <100 nm in lateral dimension and of either one-half or one lattice parameter in depth. Repeatedly clustering of point defects can eventually give rise to pits as large as those observed in the sample. Such pits look like ideal isolated niches to synthesize prebiotic compounds, and hence to host and “protect” finite molecular entities functional to biological processes (e.g. Koonin and Martin 2005). Moreover, the “coarse” pyrite aggregates (Fig. 4c) also display a lot of open cavities, much less than one micrometer in size, locally giving rise to a sort of “honeycomb” structure (e.g. Figure 4e), which may serve as anchors for primitive cellular membranes (Russell and Hall 1997; Martin and Russell 2003; Koonin and Martin 2005). Accordingly, The FTT synthesis can give rise to chains of hydrophobic hydrocarbons, possible precursors of biological membranes (e.g. Lane et al. 2010).
The possible formation of reactive oxygen species (ROS), even molecular oxygen, on pyrite defect sites (e.g. Xian et al. 2019) has been considered by Borda et al. (2003) as a potential way for the oxygen generation and local accumulation on early Earth prior to the “great oxidation event” (GOE). In this respect, endolithic conditions should be ideal sites for local O2 accumulation. One may speculate that the selective evolution of endolithic chemolithotrophs occurring by change in the aforementioned oxygenated recesses, would therefore have eventually give rise to facultative anaerobes, as the high electronegativity of oxygen makes it ideal as terminal electron acceptor.
The Mesozoic age of the sample suggests that organic compounds with biological relevance can spontaneously arise every time on Earth, provided that adequate physicochemical conditions occur, such as in endolithic niches in abyssal hydrothermal systems. The newly formed biomolecules are however unlikely to give rise to new evolutionary lines, as it happened in the Early Earth, since they will be easily involved in metabolic reactions of nearby living organisms.
It is also opportune to mention that the Fe-oxides and Ca- Fe-sulfate mineral assemblage, produced by anaerobic sulfide oxidation, as observed in the sample, may have implication even in extraterrestrial contexts, such as in the widespread occurrence of sulfates associated with Fe-oxides in the Martian soils (e.g. Bibring et al. 2007; Weitz et al. 2014; Schmidt et al. 2018).
Conclusions
The set of petrologic data here discussed suggest that the sample was initially part of a small tholeiite-gabbro igneous intrusion in a ultramafic-mafic oceanic core complex (OCC), belonging to the Ionian section of the Tethys Ocean. The intrusive body was later permeated by H2-rich hydrothermal fluids which promoted the formation of organic compounds, such as low-molecular-weight aliphatic hydrocarbons, by FTT synthesis. A subsequent temperature increasing of the circulating fluids, probably due to the emplacement of an igneous sill at greater depth, favored diffuse sulfidization episodes in the country rocks, including the sample. In a later stage of the activity of the hydrothermal system, pyrite replaced previous iron monosulfides as a consequence of a temperature decreasing of the circulating fluids. A further declining of the temperature and an increasing of the water/rock ratio, gave rise to more important changes in the redox conditions of the system, such as extensive de-sulfidization, Fe-oxides precipitation, sulfate formation, also promoting poly-condensation and aromatization of the already formed, hydrocarbons.
The occurrence of both pyrite and abiotic organic matter in the same rock recalls an established school of thought which considers pyrite as a key mineral for abiogenesis. The present data also cautiously suggest that rocks likewise the sample, permeated by hydrothermal fluids, were favorable sites for the concentration of adequate reactants and development of pyrite-mediated prebiotic reactions in the shallow subsurface of the early Earth. In addition, polycondensed organic compounds resembling petroleum resins, ultimately deriving from FTT abiotic synthesis, may have been effective starting reactants for proto-biochemical reactions in the surface of pyrite crystals.
References
Andersen SI, Speight JG (2001) Petroleum resins: Separation character and role in petroleum. Petrol Sci Technol 19:1–34. https://doi.org/10.1081/LFT-100001223
Andreani M, Escartin J, Delacour A, Ildefonse B, Godard M, Dyment J, Fallick AE, Fouquet Y (2014) Tectonic structure lithology and hydrothermal signature of the rainbow massif (mid-Atlantic ridge 36 140N). Geochem Geophys 15:3543–3571. https://doi.org/10.1002/2014GC005269
Bach W, Paulick H, Garrido CJ, Ildefonse B, Meurer WP, Humphris SE (2006) Unraveling the sequence of serpentinization reactions: petrography mineral chemistry and petrophysics of serpentinites from MAR 15°N ( ODP Leg 209 Site 1274 ). Geophys Res Lett 33:L1330633:4–7. https://doi.org/10.1029/2006GL025681
Barriga FJAS (1990) Metallogenesis in the Iberian Pyrite Belt. In: Eds DRD, Garcia M (eds) Pre- Mesozoic Geology of Iberia. Springer-Verlag, Berlin, pp 369–379
Bebié J, Schoonen MAA (2000) Pyrite surface interaction with selected organic aqueous species under anoxic conditions. Geochem Trans 8:1–7. https://doi.org/10.1039/b005581f
Bianchi F, Carbone S, Grasso M, Invernizzi G, Lentini F, Longaretti G, Merlini S, Mostardini F (1987) Sicilia orientale: pro lo geo- logico Nebrodi-Iblei. Mem Soc Geolol It 38:429–458
Bibring J-P, Arvidson RE, Gendrin A, Gondet B, Langevin Y, Le Mouelic S, Mangold N, Morris RV, Mustard JF, Poulet F, Quantin C, Sotin C (2007) Coupled ferric oxides and sulfates on the Martian surface. Science 317:1206–1210
Blöchl E, Keller M, Wächtershäuser G, Stetter KO (1992) Reactions depending on iron sulfide and linking geochemistry with biochemistry. Proc Natl Acad Sci U S A 89:8117–8120
Borda MJ, Elsetinow AR, Strongin DR, Schoonen MA (2003) A mechanism for the production of hydroxyl radical at surface defect sites on pyrite. Geochim Cosmochim Acta 67:935–939
Brazelton WJ, Mehta MP, Kelley DS, Baross JA (2011) Physiological differentiation within a single-species biofilm fueled by serpentinization. mBio 2:127–111. https://doi.org/10.1128/mBio00127-11
Bromfield TC, Coville NJ (1999) The effect of sulfide ions on a precipitated iron Fischer–Tropsch catalyst. Appl Catal A 186:297–307
Busemann H, CMO’D A, Nittler LR (2007) Characterization of insoluble organic matter in primitive meteorites by microRaman spectroscopy. Meteorit Planet Sci 42:1387–1416
Butler I, Rickard D (2000) Framboidal pyrite: Framboidal pyrite formation via the oxidation of iron (II) monosulfide by hydrogen sulphide. Geochim Cosmochim Acta 64:2665–2672
Caldeira CL, VST C, Osseo-Asare K (2009) The role of carbonate ions in pyrite oxidation in aqueous systems. Geochim Cosmochim Acta 74:1777–1789. https://doi.org/10.1016/jgca200912014
Calemma V, Iwanski P, Nali M, Scotti R, Montanari L (1995) Structural characterization of asphaltenes of different origins. Energ Fuel 9:225–230. https://doi.org/10.1021/ef00050a004
Cannat M, Fontaine F, Escartín J (2010) Serpentinization and associated hydrogen and methane fluxes at slow spreading ridges. In: Murton BJ RPADCWDJ (ed) Diversity of Hydrothermal Systems on Slow Spreading Ocean Ridges. Am Geophys Union Washington DC. https://doi.org/10.1029/2008GM000760
Carbone S, Lentini F (1981) Caratteri deposizionali delle vulcaniti del Miocene superiore negli Iblei (Sicilia Sud-Orientale). Geol Romana 20:79–101
Catalano R, Franchino A, Merlini S, Sulli A (2000) A crustal section from the eastern Algeria basin to the Ionian Ocean (Central Mediterranean). Mem Soc Geol It 55:71–85
Charlou J-L, Donval JP, Fouquet Y, Jean-Baptiste F, Holm N (2002) Geochemistry of high H2 and CH4 vent fluids issuing from ultramafic rocks at the rainbow hy- drothermal field (36°14′N MAR). Chem Geol 191:345–359
Chaumette P, Verdon C, Boucot P (1995) Influence of the hydrocarbons distribution on the heat produced during Fischer–Tropsch synthesis. Top Catal 2:301–311. https://doi.org/10.1007/BF01491974
Chernyshova IV, Hochella MF, Madden A S (2007) Size-dependent structural transformations of hematite nanoparticles 1 phase transition. Phys Chem Chem Phys 9(14):91736–1750. https://doi.org/10.1039/b618790k
Ciliberto E, Crisafulli C, Manuella FC, Samperi F, Scirè S, Scribano V, Viccaro M, Viscuso E (2009) Aliphatic hydrocarbons in metasomatized gabbroic xenoliths from Hyblean diatremes Sicily: genesis in a serpentinite hydrothermal system. Chem Geol 258:258–268. https://doi.org/10.1016/jchemgeo200810029
Cloutis E, Szymanski P, Applin D, Goltz D (2016) Identification and discrimination of polycyclic aromatic hydrocarbons using Raman spectroscopy. Icarus 274:211–230. https://doi.org/10.1016/jicarus201603023
Cody GD, Boctor NZ, Filley TR, Hazen RM, Scott JH, Sharma AY, Yoder HS Jr (2000) Primordial carbonylated iron-sulfur compounds and the synthesis of pyruvate. Science 289:1337–1340
Cody GD, Boctor NZ, Brandes JA, Filley TR, Hazen RM (2004) Assaying the catalytic potential of transition metal sulfides for abiotic carbon fixation. Geochim Cosmochim Acta 68:2185–2196. https://doi.org/10.1016/jgca200311020
de Faria DLA, Venâncio Silva S, de Oliveira MT (1996) Raman Microspectroscopy of Some Iron Oxides and Oxyhydroxides. J Raman Spectrosc 28:873–878
Dodd MS, Papineau D, Grenne T, Slack JF, Rittner M, Pirajno F, O'Neil J, Little CTS (2017) Evidence for early life in Earth's oldest hydrothermal vent precipitates. Nature 543:60–64. https://doi.org/10.1038/nature21377
Dollish FR, Fateley WG, Bentley FF (1974) Characteristic Raman frequencies of organic–compounds. John Wiley and Sons, New York
Endo Y, Sunagawa I (1973) Positive and negative striations in pyrite. Am Mineral 58:930–935
Etiope G, Schoell M (2014) Abiotic gas: atypical but not rare. Elements 10:291–296
Evans BW (2004) The serpentinite multisystem revisited: chrysotile is metastable. Int Geol Rev 46:479–506
Ferrari AC (2001) A model to interpret the Raman spectra of disordered amorphous and nanostructured carbons materials. Res Soc Symp Proc 675:1–12
Ferrari AC, Robertson J (2000) Interpretation of Raman spectra of disordered and amorphous carbon. Phys Rev 61:14095–14107. https://doi.org/10.1103/physrevb6114095
Ferris JP, Hill AR Jr, Liu R, Orgel LE (1996) Synthesis of long prebiotic oligomers on mineral surfaces. Nature 381(6577):59–61
Firstova A, Stepanova T, Cherkashov G, Goncharov A, Babaeva S (2016) Composition and formation of gabbro-peridotite hosted seafloor massive sulfide deposits from the Ashadze-1 hydrothermal field, mid-Atlantic ridge. Minerals 6(19). https://doi.org/10.3390/min6010019
Foustoukos DI, Seyfried WE Jr (2004) Hydrocarbons in hydrothermal vent fluids: the role of chromium-bearing catalysts. Science 304:1002–1005
Frost RB, Beard JS (2007) On silica activity and serpentinization. J Petrol 48:1351–1368
Fu Q, Sherwood Lollar B, Horita J, Lacrampe-Couloume G, Seyfried WE Jr (2007) Abiotic formation of hydrocarbons under hydrothermal conditions: constraints from chemical and isotope data. Geochim Cosmochimi Acta 71:1982–1998
Gasparov LV, Tanner DB, Romero DB, Berger H, Margaritondo G, Forro L (2000) Infrared and Raman studies of the Verwey transition in magnetite. Phys Rev B Condens Matter Mater Phys 62:7939–7944
German CR, Lin J, Parsons L (2004) Mid-Ocean ridges hydrothermal interactions between the lithosphere and oceans. Geophys Monogr 148. Am Geophys union, Washington DC, 318 pp.
Giampiccolo E, Brancato A, Manuella FC, Carbone S, Gresta S, Scribano V (2017) New evidence for the serpentinization of the Palaeozoic basement of southeastern Sicily from joint 3-D seismic velocity and attenuation tomography. Geophys J Int 211:1375–1395
Gold T (1999) The deep hot biosphere. Springer, New York, p 243. https://doi.org/10.1007/978-1-4612-1400-7
Gremlich H-U, Yang B (2000) Infrared and Raman spectroscopy of biological materials. Practical spectroscopy series 24, CRC press 600 pp
Grimes CB, John BE, Cheadle MJ, Wooden JL (2008) Protracted construction of gabbroic crust at a slow spreading ridge: Constraints from 206Pb/238U zircon ages from Atlantis Massif and IODP Hole U1309D (30° N MAR). Geochem Geophys Geosyst 9:Q08012. https://doi.org/10.1029/2008GC002063
Guevremont JM, Strongin DR, Schoonen MAA (1998) Thermal chemistry of H2S and H2O on the (100) plane of pyrite: unique reactivity of defect sites. Am Mineral 3:1246–1255
Hanesch M (2009) Raman spectroscopy of iron oxides and (oxy)hydroxides at low laser power and possible applications in environmental magnetic studies. Geophys J Int 177(3):941–948. https://doi.org/10.1111/j1365-246X200904122x
Hashmi SM, Firoozabadi A (2013) Self-assembly of resins and asphaltenes facilitates asphaltene dissolution by an organic acid journal of colloid and Interface. Science 394:115–123. https://doi.org/10.1016/jjcis201211069
Hazen RM, Sholl DS (2003) Chiral selection on inorganic crystalline surfaces. Nat Mater 2:367–374
Hazen RM, Sverjensky DA (2010) Mineral Surfaces Geochemical Complexities and the Origins of Life. CSH Perspect Biol 2(5). https://doi.org/10.1101/cshperspecta002162
Heinen W, Lauwers A-M (1996) Organic sulfur compounds resulting from the interaction of iron sulfide hydrogen sulfide and carbon dioxide in an anaerobic aqueous environment. Orig Life Evol Biosph 26:131–150
Herbert FW, Krishnamoorthy A, Ma W, Van Vliet KJ, Yildiz B (2014) Dynamics of point defect formation, clustering and pit initiation on the pyrite surface. Elettrochim Acta 127:416–425. https://doi.org/10.1016/j.electacta.2014.02.048
Holm NG, Anderson EM (1998) Hydrothermal systems. In: Brack A (ed) The molecular origins of life assembling pieces of the puzzle. Cambridge University press, pp 86–99
Holm NG, Charlou J-L (2001) Initial indications of abiotic formation of hydrocarbons in the rainbow ultramafic hydrothermal system mid-Atlantic ridge. Earth Planet Sci Lett 191:1–8
Holm NG, Ertem G, Ferris JP (1993) The binding and reactions of nucleotides and polynucleotides on iron oxide hydroxide polymorphs. Orig Life Evol Biosph 23:195–215
Jelicka J, Edwards HGM, Villar SEJ (2006) Raman spectroscopic study of mellite- a naturally ocuuring aluminium benzenehexacarboxylate from lignite-claystone series of the tertiary age. Spectrochim Acta A Mol Biomol Spectrosc 65:229–234
Kawasumi S, Chiba H (2017) Redox state of seafloor hydrothermal fluids and its effect on sulfide mineralization. Chem Geol 451:1–188. https://doi.org/10.1016/jchemgeo201701001
Kelley DS (2015) Black smokers: incubators on the seafloor. https://www.researchgate.net/publication/268358305_Black_Smokers_Incubators_on_the_Seafloor (Access March, 2017)
Konn C, Charlou JL, Donval JP, Holm NG, Dehairs F, Bouillon S (2009) Hydrocarbons and oxidized organic compounds in hydrothermal fluids from rainbow and lost City ultramafic hosted vents. Chem Geol 258:299–314
Koonin EV, Martin W (2005) On the origin of genomes and cells within inorganic compartments. Trends Genet 21:647–654. https://doi.org/10.1016/jtig200509006
Kundell FA (2011) A suggested Pioneer organism for the Wächtershäuser. Orig life Evol Biosph 41:175–198. https://doi.org/10.1007/s11084010-9217-y
Lane N, Allen JF, Martin W (2010) How did LUCA make a living? Chemiosmosis in the origin of life. Bioessays 32:271–280. https://doi.org/10.1002/bies200900131
Lazar C, McCollom TM, Manning CE (2012) Abiogenic methanogenesis during experimental komatiite serpentinization: implications for the evolution of the early Precambrian atmosphere. Chem Geol 326:102–112
Lentini F, Carbone S (2014) Geologia della Sicilia con i contributi di Branca S (vulcanico) e Messina A (basamenti cristallini). ISPRA Mem Descrit C Geol It 95:7–414
Li Y, Chen J, Chen Y, Zhao C, Zhang Y, Ke B (2018) Interactions of oxygen and water molecules with pyrite surface: a new insight. Langmuir 34(5):1941–1952. https://doi.org/10.1021/acs.langmuir.7b04112
Lindgren P, Parnell J, Holm NG, Broman C (2011) A Demonstration Of An Affinity Between Pyrite And Organic Matter. In: A Hydrothermal Setting. Geochem Trans, vol 12, pp 1–13. https://doi.org/10.1186/1467-4866-12-3
Liu T, Temprano I, Jenkins SJ, King DA, Driver SM (2012) Nitrogen adsorption and desorption at iron pyrite FeS2 {100} surfaces. Phys Chem Chem Phy 14:11491–11499
Loppnow GR, Shoute L, Schmidt KJ, Savage A, Hall RH, Bulmer JT (2004) UV Raman spectroscopy of hydrocarbons. Philos Trans Royal Soc A 362:2461–2476. https://doi.org/10.1098/rsta20041449
Manuella FC (2011) Vein mineral assemblage in partially serpentinized peridotite xenoliths from Hyblean plateau (southeastern Sicily Italy). Per Miner 80:247–266
Manuella FC, Scribano V, Carbone S, Brancato A (2015) The Hyblean xenolith suite (Sicily): an unexpected legacy of the Ionian–Tethys realm. Int J Earth Sci 104:1317–1336
Manuella FC, Ottolini L, Carbone S, Scavo L (2016) Metasomatizing effects of ser- pentinization-related hydrothermal fluids in abyssal peridotites: new contributions from Hyblean peridotite xenoliths (southeastern Sicily). Lithos 264:405–421
Manuella FC, Della Ventura G, Galdenzi F, Carbone S (2018) Sr-rich aragonite veins in Hyblean serpentinized peridotite xenoliths (Sicily Italy): evidence for abyssal-type carbonate metasomatism. Lithos (accepted manuscript). https://doi.org/10.1016/jlithos201812024
Marques AFA, Barriga FJAS, SD DS (2007) Sulfide mineralization in an ultramafic-rock hosted seafloor hydrothermal system: from serpentinization to the formation of cu–Zn–(co)-rich massive sulfides. Mar Geol 245:20–39
Martin W (2012) Hydrogen, metals, bifurcating electrons, and proton gradients: the early evolution of biological energy conservation. FEBS Lett 586:485–493
Martin W, Russell MJ (2003) On the origins of cells: a hypothesis for the evolutionary transitions from abiotic geochemistry to chemoautotrophic prokaryotes and from prokaryotes to nucleated cells. Philos Trans R Soc Lond Ser B Biol Sci 358(1429):59–85
Matrajt G, Borg J, Raynal PI, Djouadi Z, d’Hendecourt L, Flynn G, Deboffle D (2004) FTIR and Raman analyses of the Tagish Lake meteorite: relationship with the aliphatic hydrocarbons observed in the diffuse interstellar medium. Astron Astrophys 416:983–990. https://doi.org/10.1051/0004-6361:20034526
McCollom TM (2013) Laboratory simulations of abiotic hydrocarbon formation in Earth's deep subsurface. Rev Mineral Geochem 75:467–494
McCollom TM, Seewald JS (2007) Abiotic synthesis of organic compounds in deep-sea hydrothermal environments. Chem Rev 107:382–401
McCue AJ, Anderson JA (2014) Sulfur as a catalyst promoter or selectivity modifier in heterogeneous catalysis. Catal Sci Technol 4:272–294. https://doi.org/10.1039/c3cy00754e
Ménez B, Pisapia C, Andreani M, Jamme F, Vanbellingen QP, Brunelle A, Richard L, Dumas P, Réfrégiers M (2018) Abiotic synthesis of amino acids in the recesses of the oceanic lithosphere. Nature 564:59–63. https://doi.org/10.1038/s41586-018-0684-z
Mével C (2003) Serpentinization of abyssal peridotites at mid-ocean ridges. C R Geosci 335:825–852. https://doi.org/10.1016/jcrte200308006
Michalkova A, Kholod Y, Kosenkov D, Gorb L, Leszczynski J (2011) Viability of pyrite pulled metabolism in the ‘iron-sulfur world’ theory: quantum chemical assessment. Geochim Cosmochim Acta 75:1933–1941
Murphy R, Strongin DR (2009) Surface reactivity of pyrite and related sulfides. Surf Sci Rep 64:1–45. https://doi.org/10.1016/j.surfrep.2008.09.002
Natland JH, Dick HJB (2001) Formation of the lower ocean crust and the crystallization of gabbroic cumulates at a very slowly spreading ridge. J Volcanol Geotherm Res 110:191–233
Nesbitt HW, Bancroft GM, Pratt AR, Scaini MJ (1998) Sulfur and iron surface states on fractured pyrite surfaces. Am Mineral 83:1067–1076
Palandri J, Reed M (2004) Geochemical models of metasomatism in ultramafic systems: serpentinization rodingitization and sea floor carbonate chimney precipitation. Geochim Cosmochim Acta 68:1115–1133
Philpott MR, Goliney IY, Lin TT (2004) Molecular dynamics simulation of water in a contact with an iron pyrite FeS2 surface. J Chem Phys 120:1943–1950
Pollet R, Boehme C, Marx D (2007) Ab initio simulations of desorption and reactivity of glycine at a water-pyrite interface at “iron sulfur world” prebiotic conditions. Orig Life Evol Biospheres 36:363–379. https://doi.org/10.1007/s11084-006-9010-0
Qian G, Brugger F, Skinner WM, Chen G, Pring A (2010) An experimental study of the mechanism of the replacement of magnetite by pyrite up to 300 °C. Geochim Cosmochim Acta 74:5610–5630. https://doi.org/10.1016/jgca201006035
Reich S, Thomsen C (2004) Raman spectroscopy of graphite. Philos Trans R Soc Lond 362:2271–2288. https://doi.org/10.1098/rsta20041454
Rickard D (1997) Kinetics of pyrite formation by the H2S oxidation of iron (II) monosulphide in aqueous solutions between 25°C and 125°C: the rate equation. Geochim Cosmochim Acta 61:115–134
Rimstidt JD, Vaughan DJ (2003) Pyrite Oxidation: A State-Of-The-Art Assessment of the reaction mechanism. Geochimic Cosmochim Acta 67:873–880
Rodrigues Coelho R, Hovell I, de Mello Monte MB, Middea A, Lopes de Souza A (2006) Characterisation of aliphatic chains in vacuum residues VRs of asphaltenes and resins using molecular modelling and FTIR techniques. Fuel Process Technol 87:325–333
Rona PA, Devey CW, Dyment J, Murton BJ (Eds) (2010) Diversity of Hydrothermal Systems on Slow Spreading Ocean Ridges. Am Geophys Union Washington DC. https://doi.org/10.1029/2008GM000760
Russel MJ, Hall AJ, Boyce AJ, Fallick AE (2005) On Hydrothermal Convection Systems and the Emergence of Life. Econ Geol 100:419–438. 0361–0128/01/3503/419–20 $600
Russell MJ, Hall AJ (1997) The emergence of life from iron monosulphide bubbles at a submarine hydrothermal redox and pH front. J Geol Soc Lond 154:377–402
Schikorr G (1933) Über eisen(II)-hydroxid und ein ferromagnetisches eisen(III)-hydroxyd. Z Anorg Allg Chem 212:33–39
Schmidt G, Fueten F, Stesky R, Flahaut J, Hauber E (2018) Geology of Hebes Chasma Mars: 1 Structure Stratigraphy and Mineralogy of the Interior Layered Deposits. JGR Planets 123:2893–2919. https://doi.org/10.1029/2018JE005658
Schoonen MAA, Xu Y, Bebie J (1999) Energetics and kinetics of the prebiotic synthesis of simple organic acids and amino acids with the FeS-H2S/FeS2 redox couple as reductant. Orig Life Evol Biosph 29:5–32
Schulte MD, Rogers KL (2004) Thiols in hydrothermal solution: standard partial molal properties and their role in the organic geochemistry of hydrothermal environments. Geochim Cosmochim Acta 68:1087–1097
Scirè S, Ciliberto E, Crisafulli C, Scribano V, Bellatreccia F, Della Ventura G (2011) Asphaltene-bearing mantle xenoliths from Hyblean diatremes Sicily. Lithos 125:956–968. https://doi.org/10.1016/jlithos201105011
Scribano V, Ioppolo S (2006) Possibile produzione di idrocarburi abiogenici in seguito alla serpentinizzazione di rocce ultrafemiche/femiche nel basamento ibleo (Sicilia): evidenze dagli xenoliti. Atti 85° Congresso Soci It Min Petr (In Italian)
Scribano V, Sapienza GT, Braga R, Morten L (2006a) Gabbroic xenoliths in tuff-breccia pipes from the Hyblean plateau: insights into the nature and composition of the lower crust underneath southeastern Sicily Italy. Mineral Petrol 86:63–88. https://doi.org/10.1007/s00710-005-0092-6
Scribano V, Ioppolo S, Censi P (2006b) Chlorite/smectite-alkali feld- spar metasomatic xenoliths from Hyblean Miocenic diatremes (Sicily Italy): evidence for early interaction between hydrothermal brines and ultramafic/mafic rocks at crustal levels. Ofioliti 31:161–171. https://doi.org/10.4454/ofiolitiv31i2338
Scribano V, Viccaro M, Cristofolini R, Ottolini L (2009) Metasomatic events recorded in ultramaficxenoliths from the Hyblean area (southeastern Sicily Italy). Mineral Petrol 95:232–250
Seyfried WE, Pester NJ, Ding K, Rough M (2011) Vent fluid chemistry of the rainbow hydrothermal system (36°N, MAR): phase equilibria and in situ pH controls on subseafloor alteration processes. Geochim Cosmochimic Acta 75:1574–1593
Sforna MC, Brunelli D, Pisapia C, Pasini V, Malferrari D, Ménez B (2018) Abiotic formation of condensed carbonaceous matter in the hydrating oceanic crust Nat Com 9:5049 https://doi.org/10.1038/s41467-018-07385-6
Sharma PK, Rao KH (2003) Adhesion of Paenibacillus polymyxa on chalcopyrite and pyrite: surface thermodynamics and extended DLVO theory. Colloids Surf B 29:21–38
Shebanova ON, Lazor P (2003) Raman study of magnetite (Fe3O4): laser- induced thermal effects and oxidation. J Raman Spectrosc 34:845–852. https://doi.org/10.1002/jrs1056
Silantyev SA, Novoselov AA, Mironenkom MV (2011) Hydrothermal systems in peridotites at slow-spreading ridges modeling phase transformations and material balance: role of gabbroids. Petrology 19:227–248
Simakov S, Kouchi A, Mel’nik NN, Scribano V, Kimura Y, Hama T, Suzuki N, Saito H, Yoshizawa T (2015) Nanodiamond finding in the Hyblean shallow mantle xenoliths. Sci Rep 5. https://doi.org/10.1038/srep10765
Snow JE, Edmond HN (2007) Ultraslow-spreading ridges rapid paradigm changes. Oceanography 20:90–101
Stirling A, Bernasconi M, Parrinello M (2007) Defective pyrite (100) surface: an ab initio study. Physi Rev B 75:165406. https://doi.org/10.1103/PhysRevB.75.165406
Stirling A, Rozgonyi T, Matthias M, Bernasconi M (2015) Pyrite in contact with supercritical water: the desolation of steam. Phys Chem Chem Phys 17:1735–17379. https://doi.org/10.1039/c5cp01146a
Suiting I, Schmincke H-U (2009) Internal vs external forcing in shallow marine diatreme formation: a case study from the Iblean Mountains (SE-Sicily Central Mediterranean). J Volcanol Geotherm Res 186:361–378
Suiting I, Schmincke HU (2010) Iblean diatremes 2: shallow marine vol- canism in the Central Mediterranean at the onset of the Messinian salinity crisis (Iblean Mountains SE-Sicily)-a multidisciplinary approach nt. J Earth Sci 99:1917–1940
Suzuki T, Yano T, Hara M, Ebisuzaki T (2018) Cysteine and cystine adsorption on FeS2 (100). Surf Sci 674:6–12. https://doi.org/10.1016/j.susc.2018.03.011
Szatmari P (1989) Petroleum formation by Fischer–Tropsch synthesis in plate tectonics. AAPG Bull 73:989–998
Taylor P, Rummery TE, Owen DG (1979) Reactions of iron monosulfide solids with aqueous hydrogen sulfide up to 160°C. J Inorg Nucl Chem 41:1683–1687
Tessis AC, Penteado-Fava A, Pontes-Buarque M, Amorim HS, Bonapace JAP, Souza-Barros F, Vieyra A (1999) Pyrite suspended in artificial sea water catalyzes hydrolysis of adsorbed ATP: enhancing effect of acetate. Orig Life Evol Biosph 29:361–374
Vai GB (1994) Crustal evolution and basement elements in the Italian area: palaeogeography and characterization. B Geofis Teor Appl 36:141–144
Vai GB (2003) Development of the palaeogeography of Pangaea from late carboniferous to early Permian. Palaeogeogr Palaeoclimatol Palaeoecol 196:125–155
Van Achterbergh E, Ryan CG, Jackson SE, Griffin WL (2001) Data reduction software for LA-ICP-MS: appendix; In: Sylvester PJ (ed) Laser Ablation –ICP-Mass Spectrometry in the Earth Sciences: Principles and Applications Mineralogical Association of Canada Short Course Series Ottawa Ontario Canada 29 pp 239–243
Vogt H, Chattopadhyay T, Stolz HJ (1983) Complete first-order Raman spectra of the pyrite structure compounds FeS2 MnS2 and SiP2. J Phys Chem Solids 44:869–873
Wächtershäuser G (1988) Before enzymes and templates: theory of surface metabolism. Microbiol Mol Biol Rev 52:452–484
Wang Z, Xie X, Xiao S, Liu J (2010) Adsorption behavior of glucose on pyrite surface investigated by TG, FTIR and XRD analyses. Hydrometallurgy 102:87–90
Weiss MC, Sousa FL, Mrnjavac N, Neukirchen S, Roettger M, Nelson-Sathi S. Martin WF (2016) The physiology and habitat of the last universal common ancestor. Nat Microb 1(9):16116. https://doi.org/10.1038/NMICROBIOL2016116
Weitz CM, Dobrea EN, Wray JJ (2014) Mixtures of clays and sulfates within deposits in western Melas. Chasma Mars Icarus 251:291–314. https://doi.org/10.1016/jicarus201404009
Winchester JA, Floyd PA (1977) Geochemical discrimination of different magma series and their differentiation products using immobile elements. Chem Geol 20:325–343
Xian H, Zhu J, Tan W, Tang H, Liu P, Zhu R, Liang X, Wei J, He H, Teng HH (2019) The mechanism of defect induced hydroxylation on pyrite surfaces and implications for hydroxyl radical generation in prebiotic chemistry. Geochim Cosmochim Acta 244:163–172
Zhang X, Borda MJ, Schoonen MAA, Strongin D (2003) Adsorption of phospholipids on pyrite and their effect on surface oxidation. Langmuir 19:8787–8792
Zhao C, Chen J, Long X, Guo J (2014) Study of H2O adsorption on sulfides surfaces and thermokinetic analysis. J Ind Eng Chem 20:605–609
Acknowledgements
We would like to thank Massimo Tiepolo for having provided in-situ zircon U-Pb dating. Paolo Mazzoleni and Germana Barone kindly let us to use the micro-Raman equipment at Catania University.
Research Funding
Università di Catania, Piano per la ricerca 2016–2018, II annualità (to VS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Scribano, V., Simakov, S.K., Finocchiaro, C. et al. Pyrite and Organic Compounds Coexisting in Intrusive Mafic Xenoliths (Hyblean Plateau, Sicily): Implications for Subsurface Abiogenesis. Orig Life Evol Biosph 49, 19–47 (2019). https://doi.org/10.1007/s11084-019-09581-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11084-019-09581-w