
Abstract
Background
There was significant difference in muscle development between fat-type and lean-type pig breeds.
Methods and results
In current study, transcriptome analysis and bioinformatics analysis were used to compare the difference in longissimus dorsi (LD) muscle at three time-points (38 days post coitus (dpc), 58 dpc, and 78 dpc ) between Huainan (HN) and Large white (LW) pig breeds. A total of 24500 transcripts were obtained in 18 samples, and 2319, 2799, and 3713 differently expressed genes (DEGs) were identified between these two breeds at 38 dpc, 58 dpc, and 78 dpc, respectively. And the number and foldchange of DEGs were increased, the alternative splice also increased. The cluster analysis of DEGs indicated the embryonic development progress of LD muscle between these two breeds was different. There were 539 shared DEGs between HN and LW at three stages, and the top-shared DEGs were associated with muscle development and lipid deposition, such as KLF4, NR4A1, HSP70, ZBTB16 and so on.
Conclusions
The results showed DEGs between Huainan (HN) and Large white (LW) pig breeds, and contributed to the understanding the muscle development difference between HN and LW, and provided basic materials for improvement of meat quality.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pork is the main source of animal protein, especially for Chinese consumers, 60% of animal protein comes from pork [1]. Researching the regulation mechanism of muscle development will not only help to increase meat yield, but also help to improve meat quality. Muscle development is a complicated process, including the formation of muscle fibers in embryonic period, the muscle fibers development after birth, and muscle regeneration in adulthood [2]. The embryonic stage is the most important, the number of muscle fibers is generally fixed at this stage. After birth, muscle development is mainly form new muscle fibers (the proliferation, differentiation, and fusion of skeletal muscle satellite cells) and proliferation of muscle fibers [3]. Meat yield is closely related to the number and volume of muscle fibers [4]. Previous researches indicated that there were two growth waves during prenatal muscle development, the first wave was 35–60 days post coitus (dpc) to form the primary muscle fibers, and the second wave was 54–90 dpc to form the secondary fibers [3, 5]. Compared with Western pig breeds (lean-type), Chinese local pig breeds (fat-type) showed thinner muscle fibers, more intramuscular fat deposition content, redder meat color, but slower growth rate, and higher feed-to-meat ratio [6]. The genetic differences between these two pig breeds provided good research materials for meat quality. It had been reported that, during embryonic stage, primary fibers formed earlier in Lantang (LT, Chinese local pig breeds, more obese) pig, but its second fibers were less and thinner than that in Landrace (LR, more lean) [7]. Compared with Yorkshire (YK, lean-type), Tongcheng (TC, fat-type) pigs had more myoblasts at 30 dpc, and the primary muscle fibers could be found in both breeds at 40 dpc. YK pigs showed the secondary muscle fibers at 55 dpc, but TC had secondary muscle fibers much later [7]. These researches indicated that the appearance time and the formation process of myoblasts, the primary and secondary muscle fibers were different between Chinese indigenous pigs and Western pigs, these differences were closely related to the differences in muscle fiber volume and meat yield between these two breeds. Meanwhile, the embryonic muscle development was associated with intramuscular fat content. Wigmore and Stickland found that, the embryonic adipogenesis was synchronized with the formation of secondary muscle fibers [3]. In production, during late pregnancy improving female animals’ nutritional level could increase the intramuscular fat content of the offspring. These indicated that the embryonic stage has an important influence on muscle fiber development and intramuscular fat deposition. The number and volume of muscle fibers, and intramuscular fat content affect meat quality and yield, so embryonic muscle development is a critical period for studying meat quality traits.
Huainan (HN) pigs, an excellent Chinese indigenous breed, were included in “fine livestock and poultry breeds in Henan province” in 1986 [8]. HN pigs were mainly distributed in the upper reaches of the Huai River, and were famous for its roughage- and heat- resistance, large litter size, high intramuscular fat content, thin muscle fibers [9]. Previously we had researched the effect of castration on development of muscle and adipose in HN pigs [10, 11], until now there hasn’t report about the transcriptomic difference between HN and western pig breeds. In this study, we firstly compared the gene expression profiling of longissimus dorsi muscle between HN and Large White (LW) pigs at 38, 58, and 78 dpc. The results were helpful to understand the differences in molecular regulation mechanism of embryonic muscle development between fat-type and lean-type pig breeds, and provided basic materials for improving meat quality traits.
Materials and methods
Materials
Five healthy female HN and LW pigs were obtained from the Henan Xing Rui Agricultural and Animal Husbandry Technology Co., LTD in Henan province, China. All of these sows (in their second or third parity) were raised under the same condition, and they were inseminated with fresh semen from the boars of the same breed. At 38, 58, and 78 dpc, one sow from each breed were randomly selected and killed by sodium pentobarbital (50 mg/kg body weight). Three male and three female fetuses were selected for sample preparation. At 38 dpc, it’s difficult for distinguish the sex of the fetuses, so all of the fetuses were used for sample preparation, and their sex was identified by SRY (sex determining region Y) gene. LD muscle at the same position were quickly dissected and immediately frozen in liquid nitrogen.
Methods
Total RNA isolation, library preparation and Illumina sequencing
According to the manufacturer’s instruction, total RNA was isolated from the LD muscle by using TRIzol reagent (Invitrogen, Carlsbad, USA). Agarose gel electrophoresis, Agilent 2100 Bioanalyzer (Agilent Technologies, Massy, France), and Nano-Drop ND-2000 spectrophotometer (Nano-Drop products, Wilmington, USA) were performed to detect the purity, integrity, and quality of the RNA. Qubit RNA BR was used to assess the concentration of the RNA, only samples with an RNA integrity number (RIN) of greater than eight were used for sequencing.
Ribo-ZeroTM kit (Epicentre, Madison, WI, USA) was used to remove ribosomal RNA (rRNA), and NEBNext Ultra Directional RNA Library Prep Kit for Illumina (New England BioLabs Inc., Beverly, MA, USA) was used to create a sequencing library. For each breed, one female RNA sample and one male RNA sample from the same stage were mixed, so there were three mixed samples for sequencing at each stage. The 18 libraries were sequenced with a HiSeq 2500 platform (125 bp PE, Illumina, San Diego, CA, USA).
Reads mapping and transcript assembly
The clean reads were obtained by removing the low-quality sequences and adapter sequences for the raw reads using FASTQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and the NGS QC TOOLKIT (http://59.163.192.90:8080/ngsqctoolkit/). TopHat2 software was applied to map clean reads to the porcine reference genome (Sscrofa 11.1) with default parameters. Cufflinks was applied to perform the transcript assembly and abundance estimation.
SNP and InDel analysis and variable splices prediction
GATK software was used to identify the single nucleotide polymorphism (SNP) site and insertion-deletion (InDel). rMATS software was used to obtained the variable splicing types and corresponding expressions of each sample. And Cufflink was applied to estimate the differentially expressed isoforms.
Differentially expressed mRNA identification and pathway analysis
Differentially expressed mRNAs (DEMs) were detected by using DESeq software package with Padj < 0.05 and | log2FoldChange | > 0.25. The cluster analysis of DEGs was performed by using “ConensusClusterPlus” software package. DAVID tool was applied to perform Gene Ontology (GO) terms and the Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses of DEMs. Circle plots of GO terms and expression trends of the shared DEGs was performed by using OmicShare tools, a free online platform for data analysis (https://www.omicshare.com/tools). The enriched terms were considered statistically significant when the p value was less than 0.05. The shared DEGs was visualized by using the R package “VennDiagrams”. The heat map of the top-26 shared DEGs was performed by using the heat map package.
Validation of DEGs expression in RNA-sequencing by RT-qPCR (reverse transcription real-time quantitative PCR)
To validate DEMs’ expressional trend, RT-qPCR was performed to analyze the expressional levels of four genes, NR4A1 (nuclear hormone receptor nuclear receptor subfamily 4, group a, member 1), KLF4 (kruppel-like factor 4), HSP70.2 (heat-shock protein 70.2), and ZBTB16 (zinc finger and BTB domain containing 16) were selected from the top-26 shared DEGs in Fig. 3b at 78 dpc between HN and LW pigs. The primers for these genes and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were list in Table 1. The RNA used in RT-qPCR was the same as that used in the RNA-sequencing. PrimeScript® RT reagent Kit (Perfect Real Time) (TaKaRa, Kusatsu, Japan) was used for the qPCR. Each reaction including 2 × SYBR Premix Ex Taq™ 12.5 µL, forward primer (10 µmol/L) 1.0 µL, reverse primer (10 µmol/L) 1.0 µL, cDNA 2 µL and ddH2O 8.5 µL. The amplification including an initial denaturation step, 95 ℃ for 40 s, and 40 cycles (95 °C for 5 s and 60 °C for 30 s). Each amplification was performed in triplicate, and the relative expression of mRNAs were calculated using the 2−△△Ct method.
Statistical analysis
R package was used to analyzed the data, and results were presented as mean ± SE, and it was considered as significant when p < 0.05.
Results
SNP, InDel, and alternative splice detection
A total of 18 LD muscle samples from the HN and LW fetus at 38, 58, and 78 dpc were used for transcriptome sequencing. Against the Sus scrofa reference genome 11.1, the ratio of mapped to “+” and “−” strand of each sample was similar (Fig. 1a). There were more SNPs and InDel in 78 dpc of HN (Fig. 1b and c).
Five alternative splicing events, SE (skippedexon), MXE (mutually exclusive exon), A5SS (alternative 5′ splice site), A3SS (alternative 3′ splice site), and RI (retained intron) were found in all of 18 samples (Fig. 1d), and these five alternative splicing events with the same trends in each sample, SE was the highest, and then was MXE, RI was the lowest. Compared the difference of alternative splicing events between HN and LW at 38 dpc, 58 dpc, and 78 dpc, the difference was highest at 78 dpc.

Features of transcript detected in LD muscle of HN and LW at 38, 58, and 78 dpc. a Mapping statistics of each sample against the Sus scrofa reference genome 11.1. The numbers of SNPs (b) and InDel (c) detected in each sample. d The number of alternative splice at different comparison. SE Skipped exon, MXE Mutually exclusive exon, A5SS Alternative 5′ splice site, A3SS Alternative 3′ splice site, RI Retained intron
The DEGs detection
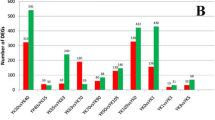
Approximately 24,500 transcripts were detected in all of samples, and the FPKM (fragments per kilobase of exon model per million mapped fragments) distribution of mRNAs in these six groups was similar (Fig. 2a). There were 22459 shared mRNAs between these two pig breeds, and 425 and 864 unique mRNAs in HN and LW, respectively (Fig. 2b).
As the embryo develops, there were 2319, 2799, and 3713 DEGs between these two breeds at 38dpc, 58 dpc, and 78 dpc, respectively. The number of DEGs between these two breeds was increased (Fig. 2c). The cluster analysis indicated that the conservation of three replicates for each stage was good. At each time point, three samples from same breed were clustered together firstly, and then the samples from different breeds clustered together. The samples from HN at 58 dpc were clustered with the samples from these two breeds at 38 dpc, but the samples from LW at 58 dpc were clustered with the samples from these two breeds at 78 dpc (Supplement Fig. 1). For each stage, the lowly altered DEGs (1.2 < FC < 1.4) accouted for the largest proportion of DEG, but at 78 dpc, the proportion of lowly altered was reduced, suggesting that the difference at 78 dpc was more significantly (Fig. 2d). There were 539 shared DEGs between these two breeds at three stages, of which 143 upregulated and 331 downregulated (Fig. 2e, Supplementary Table 1). The shared 539 DEGs were performed cluster analysis at different stages for HN and LW, respectively. The results indicated that there were 8 expression profiles in both breeds (Fig. 3a and b). The heat map of the top-26 shared DEGs (with|log2FoldChange|> 0.8 at each stage ) were shown in Fig. 3c.

The differently expressed genes (DEGs) in LD muscle between HN and LW pigs. a FPKM distribution of genes detected in HN and LW at each time point. b Venn map of genes shared by these two breeds. c The total number of DEGs between HN and LW breeds at 38, 58, and 78 dpc. d Districution of DEGs’ foldchange between these two breeds at 38, 58, and 78 dpc. e Venn maps of the number of DEGs (left panel), upregulated DEGs (right panel, red) and downregulated DEGs (right panel, green) between these two breeds at three time-points. (Color figure online)

The features of the shared DEGs at 38, 58, and 78 dpc between HN and LW pigs. a Expression trends of the shared DEGs in HN. b Expression trends of the shared DEGs in LW. c Heat map of the top-26 shared DEGs. The color bar showed the foldchange of DEGs. (Color figure online)
GO annotation and KEGG pathway mapping
According to GO enrichment of DEGs between these two breeds (Fig. 4), the protein-DNA complex, nucleosome, and protein binding were enriched for all of three stages. KEGG analysis indicated that (Table 2) calcium, HIF-1 (hypoxia inducible factor-1), insulin, rap1 pathways were continuously higher in HN in comparison to LW. At 38 and 58 dpc, ErbB (erythroblastic leukemia viral oncogene homolog), estrogen, lysine, thyroid hormone, Wnt, PI3K-AKT (phosphatidylinositol-3-kinase), focal adhension pathways were continuously lower in HN than in LW, but at 78 dpc, the difference wasn’t significantly. At 78 dpc, the pathway related with glycolipid metabolism was significantly different between these two breeds, such as fatty acid related pathway, biosynthesis of amino acid, TCA (tricarboxylic acid cycle) cycle, insulin, glyoxylate and dicarboxylated metabolism and so on.


Circle plot of enriched GO pathway between HN and LW at 38, 58, and 78 dpc. The outer circle showed the name of enriched GO pathway, and yellow scale showed biological process, blue scale showed molecular function, and green scale showed cellular component. The number in red bar (the second circle) indicated the number of background genes in this pathway, the more genes, the longer the bar, the smaller Q value, the redder the color. The number in purple bars of (the third circle) indicated the DEGs in this pathway. The fourth circle indicated the RichFactor value of each pathway, and each small grid of the background auxiliary line represented 0.1. (Color figure online)
For each breed, KEGG analysis results indicated that adrenergic signaling in cardiomyocytes and HIF-1 signaling were continuously upregulated respectively (Table 3), and metabolic pathway were continuously downregulated in both breeds. For both breeds, PI3K-AKT pathway was downregulated at 58 dpc but upregulated at 78 dpc. In HN pigs, calcium, glycolysis/gluconeogenesis, cGMP-PKG (cyclic guanosine monophosphate-protein kinase G) were continuously upregulated, but only regulated at 78 dpc in LW pigs. TCA cycle, NAFLD (nonalcoholic fatty liver disease), MAPK (mitogen activated protein kinase) were upregulated at 58 dpc in HN, but upregulated at 78 dpc in LW. Some pathways showed opposite trends at 58 vs. 38 dpc and 78 vs. 58 dpc, such as PI3K-AKT, WNT, TGF-beta (transforming growth factor beta). Type II diabetes mellitus, mTOR (mechanistic target of rapamycin), insulin, cAMP (cyclicadenosine monophosphate) only upregulated at 78 vs. 58 in HN, glycerolipid metabolism, ErbB (erythroblastic oncogene B), arginine and proline metabolism only downregulated at 78 vs. 58 dpc in LW.
Validation of the expressional level of DEMs
To validate the DEMs’ expressional trends in RNA-sequencing, the expressional level of four top-26 shared DEGs between HN and LW at 78 dpc were detected by RT-qPCR (Fig. 5). The expressional level of KLF4 and ZBTB16 was upregulated in HN, and the expressional level of NR4A1 and HSP70.2 was downregulated in HN.

Validation of the expression of the genes by RT-qPCR. The data represented the Mean ± SE, ** indicates p < 0.01, * indicates p < 0.05
Discussions
The molecular regulation mechanism of muscle development is complicated and is not clear so far. We firstly used HN and LW pigs as model animals toperforme the transcriptome sequencing of LD muscle at 38, 58, and 78 dpc.
Five kinds of alternative splice had been detected in different corporations, SE and MXE account for the largest proportion (Fig. 1d). A5SS wasn’t detected in H58 vs. HN38, LW58 vs. LW38, and LW78 vs. LW58. A3SS wasn’t detected in LW78 vs. LW58. In HN pigs, the difference of alternative splice between different stages wasn’t significantly, only more A3SS in HN58 vs. HN38 than that in H78 vs. H58. Comparison of alternative splice in each time point between HN and LW pigs, it’s obvious that there were much more splice in 78 dpc than that in 38 dpc and 58 dpc, indicating that the difference was most significantly in 78 dpc. Similarly, the total number of DEGs between these two breeds was gradually increased during embryo development (Fig. 2c). In 78 dpc, the number of DEGs was about two fold than that of in 38 dpc, and the foldchange of DEGs was higher than that in previous two stages (Fig. 2e).
Previous research showed that the primary muscle fiber was found at 35 dpc in LT, 49 dpc in LR. For the formation of primary muscle fibers, LT pigs showed faster develop speed [7]. Another research compared the embryonic muscle development difference between TC and YK, the results showed that there were much more myoblast in TC at 30 dpc [5]. These results indicated that compared with western pig breeds, myogenic differentiation started earlier and primary muscle fibers formed earlier in Chinese local pigs. In current research, the cluster analysis of DEGs indicated that the different stages of two pig breeds were firstly enriched together, 58 dpc in HN were enriched with 38 dpc, but 58 dpc in LW were enriched with 78 dpc (Fig. 2d). The results revealed that the embryonic development progress of LD muscle between these two breeds was different.
There were 539 shared DEGs between these two breeds at three stages, the top differentially expressed genes were shown in Fig. 3b, many of them were associated with muscle development and lipid deposition. For example, KLF4 expressed early during adipogenesis and its expression trend was similar with C/EBP beta. C/EBP beta was activated by KLF4, leading to activation of transcriptional cascade in adipogenesis. Meanwhile C/EBP beta regulated KLF4 through a negative feedback loop [12]. Ubiquitination of KLF4 was necessary for adipogenesis of 3T3-L1 precursor cells [13]. The inhibition of A2b adenosine receptor on murine adipogenesis was relay on KLF4 [14]. Cholecalciferol [15], GSK3 beta (glycogen synthase kinase-3) [16], miR-25 [17] participated in regulation of KLF4.
NR4A1, regulated glycolysis and glucose transport in liver, muscle, and adipose tissue through different signaling pathway [18,19,20]. The expression of NR4A1 was downregulated in diabetic, ob/ob, and db/db mice. Compared with wild mice, NR4A1 knockout mice were more likely obesity [19]. In skeletal muscle, activated by compounds or sport, NR4A1 could promoted glycogen synthesis and participated in insulin resistance [21, 22]. NR4A1 could inhibit adipogenesis and lipid deposition in primary adipocytes, meanwhile NR4A subfamily could regulate lipid metabolism [23]. It was reported that HSP70 was related with tenderness of bovine LD muscle [24], the content of HSP70 in longissimus thoracis and semitendinosus of steer was higher than that in bull [25]. The SNPs in ZBTB16 were associated with body mass index, waist to hip ratio, and LDL cholesterol levels [26]. In rat, losing one copy of Zbtb16 caused reduced body weight and adiposity [27].
HIF-1α enhanced myogenesis by inducing WNT7A, the latter one was essential for muscle differentiation [28]. In current research, HIF-1α was continuously upregulated in both of two breeds. In the comparison between HN and LW, HIF-1α was continuously higher in HN than in LW. Thyroid hormone inhibited muscle satellite cells proliferation [29], promoted myogenic differentiation [30], promoted the expression of fast myosin heavy chain and regulated the conversation of slow-fast muscle fibers [31]. Thyroid hormone also participated in skeletal muscle mitochondrial biosynthesis and enhanced skeletal muscle metabolism, thermogenesis and glucose intake [32, 33]. In this study, thyroid pathway was higher at 38 and 58 dpc in HN than in LW. MAPK participated in promoting myoblast proliferation, and its expression level was highest at 38 dpc and 58 dpc in HN and LW, respectively. These results coincided with the earlier myogenic differentiation in Chinese indigenous pig breed but longer myoblast proliferation in western pig breeds.
Evidences showed that PI3K could promote adipogenesis [34], and its expression was much higher in HN than that in LW at 38 dpc and 58 dpc. This result coincided with higher IMF (intramuscular fat) content in Chinese local pigs.
It was reported that Wnt pathway play a critical role in regulation of muscle fiber type formation, Wnt5a enhanced proliferation of slow muscle fiber, Wnt11 promotes fast muscle fiber formation [35]. Wnt pathway showed higher expressional level in HN than in LW at 38 dpc and 58 dpc. At 38 dpc, Wnt5a showed higher expressional level in HN than in LW. In HN pigs, Wnt pathway was firstly upregulated at 38 dpc and then downregulated at 78 dpc, but in LW pigs, its expression was only upregulated in 58 dpc. Calcium pathway, which could promotes slow muscle fiber formation, was continuously higher in HN than in LW in all stages. These results coincided with the thinner muscle fibers volume in HN.
Conclusion
In general, the transcriptome profiling of LD muscle at different embryonic stages between HN and LW was compared, the results indicated that genes participated in the regulation of differences between HN and LW at embryonic muscle development by regulating proliferation, myogenesis, adipogenesis and muscle fiber transformation. HIF-1α, thyroid、MAPK, PI3K, Wnt pathways might be used as pivotal pathway for research of meat quality.
Data availability
The raw data generated for this study had been submitted to NCBI Sequence Read Archive with accession number SRP243554.
Abbreviations
- LD:
-
Longissimus dorsi
- HN:
-
Huainan
- LW:
-
Large white
- DEGs:
-
Differently expressed genes
- IMF:
-
Intramuscular fat
- dpc:
-
Days post coitus
- LT:
-
Lantang
- LR:
-
Landrace
- YK:
-
Yorkshire
- TC:
-
Tongcheng
- SRY:
-
Sex determining region Y
- RIN:
-
RNA integrity number
- rRNA:
-
Ribosomal RNA
- SNP:
-
Single nucleotide polymorphism
- InDel:
-
Insertion-deletion
- DEMs:
-
Differentially expressed mRNAs
- Go:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- RT-qPCR:
-
Reverse transcription real-time quantitative PCR
- NR4A1:
-
Nuclear hormone receptor nuclear receptor subfamily 4, group a, member 1
- KLF4:
-
Kruppel-like factor 4
- HSP70.2:
-
Heat-shock protein 70.2
- ZBTB16:
-
Zinc finger and BTB domain containing 16
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- SE:
-
Skippedexon
- MXE:
-
Mutually exclusive exon
- A5SS:
-
Alternative 5′ splice site
- A3SS:
-
Alternative 3′ splice site
- RI:
-
Retained intron
- FPKM:
-
Fragments per kilobase of exon model per million mapped fragments
- HIF-1:
-
Hypoxia inducible factor-1
- ErbB:
-
Erythroblastic leukemia viral oncogene homolog
- PI3K-AKT:
-
Phosphatidylinositol-3-kinase
- TCA:
-
Tricarboxylic acid cycle
- cGMP-PKG:
-
Cyclic guanosine monophosphate-protein kinase G
- NAFLD:
-
Nonalcoholic fatty liver disease
- MAPK:
-
Mitogen activated protein kinase
- TGF-beta:
-
Transforming growth factor beta
- mTOR:
-
Mechanistic target of rapamycin
- cAMP:
-
Cyclicadenosine monophosphate
- ErbB:
-
Erythroblastic oncogene B
References
Cai C, Li M, Zhang YW, Meng S, Yang Y, Gao PF, Guo XH, Cao GQ, Li BG (2020) Comparative transcriptome analyses of Longissimus Thoracis between Pig breeds Differing in muscle characteristics. Front Genet 11:526309. https://doi.org/10.3389/fgene.2020.526309
Zeng Q, Du ZQ (2023) Advances in the discovery of genetic elements underlying longissimus dorsi muscle growth and development in the pig. Anim Genet. https://doi.org/10.1111/age.13365
Wigmore PM, Stickland NC (1983) Muscle development in large and small pig fetuses. J Anat 137(Pt 2):235–245. https://doi.org/10.1007/BF00305930
Li X, Lu L, Tong X, Li R, Jin E, Ren M, Gao Y, Gu Y, Li S (2023) Transcriptomic profiling of meat quality traits of skeletal muscles of the Chinese Indigenous Huai Pig and Duroc Pig. Genes 14:1548. https://doi.org/10.3390/genes14081548
Zhao YQ, Li J, Liu HJ, Xi Y, Xue M, Liu WH, Zhuang ZH, Lei MG (2015) Dynamic transcriptome profiles of skeletal muscle tissue across 11 developmental stages for both Tongcheng and Yorkshire pigs. BMC Genomics 16:377. https://doi.org/10.1186/s12864-015-1580-7
Sun YM, Chen XC, Qin J, Liu SG, Zhao R, Yu TY, Chu GY, Yang GS, Pang WJ (2018) Comparative analysis of long noncoding RNAs expressed during intramuscular adipocytes adipogenesis in Fat-Type and lean-type pigs. J Agric Food Chem 66:12122–12130. https://doi.org/10.1021/acs.jafc.8b0424
Zhao X, Mo DL, Li AN, Gong W, Xiao SQ, Zhang Y, Qin LM, Niu YN, Guo YX, Liu XH, Cong P, He Z, Wang C, Li J, Chen Y (2011) Comparative analyses by sequencing of transcriptomes during skeletal muscle development between pig breeds differing in muscle growth rate and fatness. PLoS ONE 6:e19774. https://doi.org/10.1371/journal.pone.0019774
Wang J, Chen MY, Chen JF, Ren QL, Zhang JQ, Cao H, Xing BS, Pan CY (2020) LncRNA IMFlnc1 promotes porcine intramuscular adipocyte adipogenesis by sponging miR-199a-5p to up-regulate CAV-1. BMC Mol Cell Biol 21:77. https://doi.org/10.1186/s12860-020-00324-8
Hu T, Mei SQ, Sun XJ, Peng XW, Zhang XY, Ma CP, Wang L, Hua L, Li FG (2013) Associations of TCF12, CTNNAL1 and WNT10B gene polymorphisms with litter size in pigs. Anim Reprod Sci 140:189–194. https://doi.org/10.1016/j.anireprosci.2013.05.013
Wang J, Hua LS, Chen JF, Zhang JQ, Bai XX, Gao BY, Li C, Shi ZH, Sheng WD, Gao Y, Xing BS (2017) Identification and characterization of long non-coding RNAs in subcutaneous adipose tissue from castrated and intact full-sib pair Huainan male pigs. BMC Genomics 18:542. https://doi.org/10.1186/s12864-017-3907-z
Xing BS, Bai XX, Guo HX, Chen JF, Hua LS, Zhang JQ, Ma Q, Ren QL, Wang HS, Wang J (2017) Long non-coding RNA analysis of muscular responses to testosterone deficiency in Huainan male pigs. Anim Sci J 88:1451–1456. https://doi.org/10.1111/asj.12777
Birsoy K, Chen Z, Friedman J (2008) Transcriptional regulation of adipogenesis by KLF4. Cell Metab 7:339–347. https://doi.org/10.1016/j.cmet.2008.02.001
Tahmasebi S, Ghorbani M, Savage P, Yan K, Gocevski G, Xiao L, You L, Yang XJ (2013) Sumoylation of Krüppel-like factor 4 inhibits pluripotency induction but promotes adipocyte differentiation. J Biol Chem 288:12791–12804. https://doi.org/10.1074/jbc.M113.465443
Eisenstein A, Carroll SH, Johnston-Cox H, Farb M, Gokce N, Ravid K (2014) An adenosine receptor-Krüppel-like factor 4 protein axis inhibits adipogenesis. J Biol Chem 289:21071–21081. https://doi.org/10.1074/jbc.M114.566406
Kim JH, Kang S, Jung YN, Choi HS (2016) Cholecalciferol inhibits lipid accumulation by regulating early adipogenesis in cultured adipocytes and zebrafish. Biochem Biophys Res Commun 469:646–653. https://doi.org/10.1016/j.bbrc.2015.12.049
Cervantes-Camacho C, Beltrán-Langarica A, Ochoa-Uribe AK, Marsch-Moreno M, Ayala-Sumuano JT, Velez-delValle C, Kuri-Harcuch W (2015) The transient expression of Klf4 and Klf5 during adipogenesis depends on GSK3β activity. Adipocyte 4:248–255. https://doi.org/10.1080/21623945.2015.1007823
Liang WC, Wang Y, Liang PP, Pan XQ, Fu WM, Yeung VS, Lu YF, Wan DC, Tsui SK, Tsang SY, Ma WB, Zhang JF, Waye MM (2015) MiR-25 suppresses 3T3-L1 adipogenesis by directly targeting KLF4 and C/EBPα. J Cell Biochem 116:2658–2666. https://doi.org/10.1002/jcb.25214
Men L, Hui W, Guan X, Song T, Wang X, Zhang S, Chen X (2020) Cardiac Transcriptome Analysis reveals Nr4a1 mediated glucose metabolism dysregulation in response to High-Fat Diet. Genes (Basel) 11:720. https://doi.org/10.3390/genes11070720
Chao LC, Wroblewski K, Zhang Z, Pei L, Vergnes L, Ilkayeva OR, Ding SY, Reue K, Watt MJ, Newgard CB, Pilch PF, Hevener AL, Tontonoz P (2009) Insulin resistance and altered systemic glucose metabolism in mice lacking Nur77. Diabetes 58:2788–2796. https://doi.org/10.2337/db09-0763
Zhang L, Wang Q, Liu W, Liu FY, Ji AL, Li YZ (2018) The Orphan Nuclear Receptor 4A1: A Potential New Therapeutic Target for Metabolic Diseases. J Diabetes Res 2018: 9363461. doi: 10.1155/2018/9363461.
Mohankumar K, Lee J, Wu CS, Sun Y, Safe S (2018) Bis-Indole-Derived NR4A1 ligands and metformin exhibit NR4A1-Dependent glucose metabolism and uptake in C2C12 cells. Endocrinology 159:1950–1963. https://doi.org/10.1210/en.2017-03049
Mey JT, Solomon TPJ, Kirwan JP, Haus JM (2019) Skeletal muscle Nur77 and NOR1 insulin responsiveness is blunted in obesity and type 2 Diabetes but improved after exercise training. Physiol Rep 7:e14042. https://doi.org/10.14814/phy2.14042
Fumoto T, Yamaguchi T, Hirose F, Osumi T (2007) Orphan nuclear receptor Nur77 accelerates the initial phase of adipocyte differentiation in 3T3-L1 cells by promoting mitotic clonal expansion. J Biochem 141:181–192. https://doi.org/10.1093/jb/mvm018
Jia X, Veiseth-Kent E, Grove H, Kuziora P, Aass L, Hildrum KI, Hollung K (2009) Peroxiredoxin-6–a potential protein marker for meat tenderness in bovine longissimus thoracis muscle. J Anim Sci 87:2391–2399. https://doi.org/10.2527/jas.2009-1792
Guillemin N, Jurie C, Cassar-Malek I, Hocquette JF, Renand G, Picard B (2011) Variations in the abundance of 24 protein biomarkers of beef tenderness according to muscle and animal type. Animal 5:885–894. https://doi.org/10.1017/S1751731110002612
Bendlová B, Vaňková M, Hill M, Vacínová G, Lukášová P, VejraŽková D, Šedová L, Šeda O, Včelák J (2017) ZBTB16 gene variability influences obesity-related parameters and serum lipid levels in Czech adults. Physiol Res 66(Suppl 3):S425–s431. https://doi.org/10.33549/physiolres.933731
Liška F, Landa V, Zídek V, Mlejnek P, Šilhavý J, Šimáková M, Strnad H, Trnovská J, Škop V, Kazdová L, Starker CG, Voytas DF, Izsvák Z, Mancini M, Šeda O, Křen V, Pravenec M (2017) Downregulation of Plzf Gene ameliorates metabolic and cardiac traits in the spontaneously hypertensive rat. Hypertension 69:1084–1091. https://doi.org/10.1161/HYPERTENSIONAHA.116.08798
Cirillo F, Resmini G, Angelino E, Ferrara M, Tarantino A, Piccoli M, Rota P, Ghiroldi A, Monasky MM, Ciconte G, Pappone C, Graziani A, Anastasia L (2020) HIF-1α directly controls WNT7A expression during myogenesis. Front Cell Dev Biol 8:593508. https://doi.org/10.3389/fcell.2020.593508
Dentice M, Ambrosio R, Damiano V, Sibilio A, Luongo C, Guardiola O, Yennek S, Zordan P, Minchiotti G, Colao A, Marsili A, Brunelli S, Del Vecchio L, Larsen PR, Tajbakhsh S, Salvatore D (2014) Intracellular inactivation of thyroid hormone is a survival mechanism for muscle stem cell proliferation and lineage progression. Cell Metab 20:1038–1048. https://doi.org/10.1016/j.cmet.2014.10.009
Lee EJ, Pokharel S, Jan AT, Huh S, Galope R, Lim JH, Lee DM, Choi SW, Nahm SS, Kim YW, Park SY, Choi I (2017) Transthyretin: a transporter protein essential for proliferation of myoblast in the myogenic program. Int J Mol Sci 18:115. https://doi.org/10.3390/ijms18010115
Carmody C, Ogawa-Wong AN, Martin C, Luongo C, Zuidwijk M, Sager B, Petersen T, Roginski Guetter A, Janssen R, Wu EY, Bogaards S, Neumann NM, Hau K, Marsili A, Boelen A, Silva JE, Dentice M, Salvatore D, Wagers AJ, Larsen PR, Simonides WS, Zavacki AM (2019) A Global loss of Dio2 leads to unexpected changes in function and Fiber types of slow skeletal muscle in male mice. Endocrinology 160:1205–1222. https://doi.org/10.1210/en.2019-00088
Lombardi A, Moreno M, de Lange P, Iossa S, Busiello RA, Goglia F (2015) Regulation of skeletal muscle mitochondrial activity by thyroid hormones: focus on the old triiodothyronine and the emerging 3,5-diiodothyronine. Front Physiol 6:237. https://doi.org/10.3389/fphys.2015.00237
Islam H, Hood DA, Gurd BJ (2020) Looking beyond PGC-1α: emerging regulators of exercise-induced skeletal muscle mitochondrial biogenesis and their activation by dietary compounds. Appl Physiol Nutr Metab 45:11–23. https://doi.org/10.1139/apnm-2019-0069
Wang XJ, Liang F, Fan H, Wang ZJ, Gou XL, Ning Y (2020b) Proteinase-activated receptor 2 promotes 3T3-L1 preadipocyte differentiation through activation of the PI3K/AKT signalling pathway and MAT2A gene expression. Arch Physiol Biochem 126:468–475. https://doi.org/10.1080/13813455.2020.1817094?journalCode=iarp20
Anakwe K, Robson L, Hadley J, Buxton P, Church V, Allen S, Hartmann C, Harfe B, Nohno T, Brown AM, Evans DJ, Francis-West P (2003) Wnt signalling regulates myogenic differentiation in the developing avian wing. Development 130:3503–3514. https://doi.org/10.1242/dev.00538
Funding
This research was supported by National Key R&D Program of China (2021YFD1301200), the Key Program for Science and Technology Development of Henan (212102110010) and Special Fund for Science and Technology Innovation Team in Henan Academy of Agricultural Science (2022TD34, 2023TD22).
Author information
Authors and Affiliations
Contributions
This research was designed by JFC and BSX; experiments were performed by JFC, JW, WJ, and JC; samples were collected by QM and QLR; data collection and analysis were performed by QXL and JJS; manuscript was wrote by JFC; manuscript was corrected by DLM, BSX and JQZ.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
All procedures involving animals described in the present study were performed according to the procedures of Institutional Animal Care and Use Committee of Institute of Animal Science, Henan Academy of Agricultural Sciences.
Additional information
Publisher’s Note
Springer nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, J.F., Wang, J., Chai, J. et al. Transcriptome profiling of longissimus dorsi during different prenatal stages to identify genes involved in intramuscular fat deposition in lean and obese pig breeds. Mol Biol Rep 51, 386 (2024). https://doi.org/10.1007/s11033-023-09088-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11033-023-09088-8