
Abstract
This study, using the MIN6 cell line, examines the effect of glucocorticoids (GCs) on the expression and protein levels of endoplasmic reticulum stress (ERS) related genes. Furthermore, we evaluated the protective role of 4-phenylbutyric acid (4-PBA) on the aforesaid GCs induced changes. Pancreatic islet MIN6 cells were treated with dexamethasone (DEX) at distinct concentrations (0.1 μmol/L and 0.5 μmol/L) for different periods (1 h, 4 h, 12 h, and 24 h). The mRNA and protein levels of ERS related genes were measured using real-time qPCR (qRT-PCR) and western blotting. Similar evaluations were also carried out for the cells treated with 4-PBA combined with DEX. Upon DEX intervention which induces the unfolded protein response (UPR), the expression levels of BIP, ATF6, IRE1, and PERK increased in the MIN6 cells, both in concentration and time-dependent manner. Similarly, ERS associated gene CHOP, which is involved in the apoptotic pathway, also showed increased levels both in concentration and time-dependent manner. However, treatment with 4-PBA decreased the expression levels of ERS related proteins. Quantitative analysis found that all these results were statistically significant (P < 0.05). GCs markedly activates the ERS in the MIN6 cell line in vitro, however, this effect can be significantly alleviated upon treatment with 4-PBA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus, a pressing and growing health problem that affects over 463 million adults (20–79 years old) worldwide has become a global epidemic [1]. The pathogenesis of diabetes is a very complex phenomenon. In addition to genetic and environmental factors, abnormal secretion of insulin counter-regulatory hormones such as glucocorticoids (GCs) is also one of the mechanisms of diabetes pathogenesis.
GCs have a wide range of physiological effects, including regulation of glucose and lipid metabolism, lipolysis, liver gluconeogenesis, amino acid mobilization, and reduced skeletal muscle glucose uptake [2]. GCs function by multiple regulatory mechanisms for glucose metabolism. Mainly, they induce insulin resistance by inhibiting the insulin signal transduction pathway [3]. Besides, GCs may also affect insulin secretion from islet β-cells by regulating the concentration and distribution of cellular calcium ions [4, 5].
Pancreatic β-cells, the sites of insulin synthesis, have abundant endoplasmic reticulum (ER) structures. For maintaining cell viability and function, steady-state maintenance of ER is vital. But, a variety of reasons can lead to disorders of endoplasmic reticulum balance, known as "ER stress" (ERS) [6]. The ERS is an important link in the development of diabetes in the islet β cells [7]. Usually, when the accumulation of misfolded proteins in the ER reaches beyond a critical threshold, β-cells enable various adaptation mechanisms to match the function of the ER according to current cellular needs. This response is known as the UPR [8]. The UPR is mainly mediated by three signaling pathways, namely inositol-requiring enzyme 1 (IRE1); RNA-dependent protein kinase R-like ER kinase (PERK); and activating transcription factor 6 (ATF6) signal network [9]. Factors, such as increased unfolded/misfolded protein, Ca2+ depletion, or enhanced insulin biosynthesis affect ER homeostasis of islet β cells. This triggers the release of BIP, a major ER chaperone protein to activate UPR [10]. Simultaneously, long-term ERS may also induce apoptosis. The C/EBP homologous protein (CHOP), also known as DNA damage-inducible transcript 3 (DDIT3), plays an important pro-apoptotic role in the ERS-mediated apoptosis [11].
4-Phenylbutyric acid (4-PBA) is a non-selective chemical small molecule with a chaperone-like effect in increasing protein stability. It helps unfolded protein remodeling and has been approved by the U.S. Food and Drug Administration (FDA) for the prevention and treatment of diseases caused by the accumulation of misfolded proteins such as in motor neuron disease. Moreover, researches have also shown that 4-PBA can attenuate the GCs induced apoptosis of MC3T3-E1 osteoblast-like cells and inhibit ER stress and mitochondrial dysfunction [12].
In this study, using mouse insulinoma cell line MIN6, the effects of DEX which is a GC on ER-related proteins and respective insulin secretion-related genes (GLU2 and PDX1) were investigated in vitro. Also, we explored whether 4-PBA would affect the aforesaid regulation and if so, can it provide new targets for the prevention and treatment of diabetes, especially in those with abnormal GCs secretion.
Materials and methods
Cells and culture
The mouse insulinoma cell line MIN6 (a kind gift from Professor Zhuoxian Meng of Zhejiang University) was grown in 85% DMEM medium (Gibico) containing 25 mM glucose supplemented with 15% fetal calf serum (Gibico), 0.5% β-Mercaptoethanol (Gibico) and 0.2% antibiotics (Penicillin and Streptomycin, Gibico) at 37 °C in a humidified atmosphere containing 5% CO2. For preparation for DEX and 4-PBA, see supplementary material.
Pharmacological intervention
The experimental groupings were as follows: the control group MIN6 was left untreated; the DEX group MIN6 cell line was treated with 0.1 µmol/L and 0.5 µmol/L DEX (Sigma, USA), and they all intervened for 1 h, 4 h, 12 h, and 24 h. In the 4-PBA + DEX group, DEX was co-incubated with 2.5 mmol/L 4-PBA (Sigma, USA). The 4-PBA group was only treated with 2.5 mmol/L 4-PBA.
Real-time quantitative PCR (qRT-PCR)
The total cell RNA was extracted using the Trizol reagent kit. The concentration and purity of the total RNA were measured and it was reverse-transcribed into cDNA for analysis using the qRT-PCR. The comparative Ct (2 − ΔΔCt) method was used to calculate the relative mRNA amounts.
The primer sequences used in the experiments were as follows: β-actin upstream:5′-CGGGGACCTGACTGACTACC-3′ and downstream: 5′-AGGAAGGCTGGAAGAGTGC-3′; BIP upstream: 5′-AGGACAAGAAGGAGGATGTGGG-3′ and downstream: 5′-ACCGAAGGGTCATTCCAAGTG-3′; ATF6 upstream: 5′-TGGGCAGGACTATGAAGTAATG-3′ and downstream: 5′-CAACGACTCAGGGATGGTGCTG-3′; PERK upstream: 5′-CGATCAAATGGAAGCCCTTA-3′, and downstream: 5′-TGCGGATGTTCTTGCTGTAG-3′; IRE-1 upstream: 5′-AGTATTCCACCAGCCTCTATGC-3′, and downstream: 5′-CACACACTCTCCTTTGTCTCCA-3′; CHOP upstream: 5′- TTCACTACTCTTGACCCTGCGTC -3′ and downstream: 5′- CACTGACCACTCTGTTTC-3′; PDX1 upstream: 5′-CGGACATCTCCCCATACG-3′ and downstream: 5′-AAAGGGAGCTGGACGCGG-3′; GLUT2 upstream: 5′-TTCCAGTTCGGCTATGACATCG-3′ and downstream: 5′-CTGGTGTGACTGTAAGTGGGG-3′.
Western blotting
MIN6 cells, in the logarithmic growth phase, were seeded into 6-well plates at a cell density of 1 × 106 cells per well. After an overnight adherent growth, the solvent control group, 0.5 μM DEX + solvent control group (referred to as the DEX group), 0.5 μM DEX + 2.5 mM 4-PBA group, and 2.5 mM 4-PBA group were set. All interventions were at 4 h, 12 h, and 24 h. The cells were washed twice with pre-chilled PBS and then added to cold RIPA lysis buffer containing 1% protease inhibitor for 10 min. The collected cell lysate was centrifuged at 4 °C to extract the total protein. From this, 30 μg of protein, mixed with the loading buffer (4:1), and heated to denaturation, were separated using a 10% acrylamide sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE). Next, the protein was transferred onto polyvinylidene difluoride (PVDF) membranes which were blocked in 5% skim milk for 1 h at room temperature. Then, the primary antibodies BIP, ATF6, IRE1, PERK, CHOP Rabbit mAb (all 1:1000, CST) were incubated with the PVDF membranes at 4 °C overnight, respectively. After this incubation, the membrane was washed with TBS-T (Tris-buffered saline with Tween-20) and incubation with secondary antibody (1:5000, Boster) was carried out for 1 h. The membrane was washed again with the TBS-T for another 30 min. Finally, the protein band was visualized using the chemiluminescent reaction.
Statistics
Data are presented as mean ± SD. Comparisons were performed using Student’s t-test or one-way ANOVA with Dunnett post-hoc test using the GraphPad Prism 6 program. A P-value < 0.05 was deemed statistically significant.
Results
DEX upregulates ERS-related genes in MIN6 cells
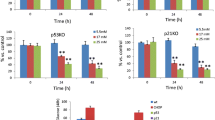
The qPCR results show that compared to the control group, both low and high concentrations (0.1 μmol/L and 0.5 μmol/L, respectively) of DEX intervention up-regulated the mRNA levels of unfolded protein response-related genes and CHOP in MIN6 cells (Fig. 1). The increased levels of CHOP can be directly correlated with increased cell apoptosis. As per time points analysis, DEX intervention at 4, 12, and 24-h groups were significantly higher (P < 0.01) compared to the control group (Fig. 1). Among the concentrations, mRNA levels were higher for 0.5 μmol/L DEX intervention than 0.1 μmol/L DEX intervention for 1 h, 4 h, 12 h, and 24 h in MIN6 cells. There was no statistical difference in control groups at different time. (Supplementary Fig. 1).

The mRNA levels of ERS-related genes in MIN6 cells after treatment with different concentration of DEX or DEX + 4-PBA. Data are expressed as a mean value ± SD of three independent experiments. a Treated with 0.1 μmol/L DEX in the presence or absence of 2.5 mmol/L 4-PBA. Vs the control group *(P < 0.01); Vs 0.1 μmol/L DEX group #(P < 0.01). b Treated with 0.5 μmol/L DEX in the presence or absence of 2.5 mmol/L 4-PBA. Vs the control group *(P < 0.01); Vs 0.5 μmol/L DEX group #(P < 0.01)
4-PBA attenuated DEX-induced ERS and apoptosis in MIN6 cells
Since we find out that the high concentration (0.5 μmol/L) of DEX was more effective in upregulating the level of ERS-related genes (Fig. 1), we selected 0.5 μmol/L DEX for the subsequent experiments and increased the 4-PBA group. The results of western blot analysis showed that the protein levels of BIP, ATF6, IRE1, PERK, and CHOP were significantly reduced in the cells than those in the DEX group (P < 0.01). This effect was comparable to the control group without the DEX intervention and the 4-PBA group alone (Fig. 2). These results clearly indicate that DEX indeed induced ERS in MIN6 cells. However, 4-PBA treatment could reduce the DEX-induced ERS.

Effect of 0.5 μmol/L DEX and/or 4-PBA on the protein levels of BIP, IRE1, PERK, ATF6, and CHOP. a Western blotting analysis of BIP, IRE1, PERK, ATF6, and CHOP protein levels in the control and treated groups in MIN6 Cells. b Quantification of the relative protein levels of BIP, IRE1, PERK, ATF6, and CHOP, normalized with the internal marker β-actin (P < 0.01). Data are expressed as a mean value ± SD of three independent experiments. Vs control group *(P < 0.01); Vs 0.5 μmol/L DEX group #(P < 0.01)
Effect of DEX on GLUT2 and PDX1 genes in MIN6 cells
Compared with the control group, low-concentration (0.1 μmol/L) of DEX down-regulated the mRNA level of GLUT2 for 12 h and 24 h. Interestingly, a high-concentration (0.5 μmol/L) of DEX could reduce the GLUT2 mRNA level even after a comparatively short 4 h intervention (P < 0.01) (Fig. 3). Similarly, low concentration (0.1 μmol/L) of DEX had little effect on PDX1 levels in MIN6 cells. Again, here too, high concentration (0.5 μmol/L) of DEX showed marked reduction in the mRNA level of PDX1 (Fig. 3). However, 4-PBA did not affect PDX1 and GLUT2 levels in DEX-induced MIN6 cells (Fig. 3).

mRNA levels of PDX1 and GLUT2 in MIN6 Cells. a Treated with 0.1 μmol/L DEX and/or 4-PBA. b Treated with 0.5 μmol/L DEX and/or 4-PBA. Data are expressed as a mean value ± SD of three independent experiments. Vs control group *(P < 0.01)
Discussion
In this study, firstly, we found that long-term exposure to DEX induces ERS and ERS-mediated apoptosis in MIN6 cells, both in a time and concentration-dependent manner. Interestingly, 4-PBA could alleviate these DEX-induced UPR changes in the MIN6 cells in vitro. This is evident by the simultaneous reduction of BIP and the three pathway-related genes, downstream of the ER stress response, ATF6, IRE1, and PERK to the normal control level. Moreover, 4-PBA also inhibited the DEX-induced ERS-mediated apoptosis. Concerning insulin synthesis and release, low concentrations of DEX did not affect the expression levels of GLUT2 and PDX1 genes in MIN6 cells. However, a high concentration of DEX markedly reduced the expression levels of these. It appears that the distinct DEX concentrations may have different degrees of effect on insulin synthesis and secretion in the β-cells. Moreover, unlike the protective effect of the up-regulation of ER related genes upon DEX treatment, 4-PBA has no significant protective effect on DEX-induced down-regulation of insulin biosynthesis-related genes. This could be since GLUT2 and PDX1 genes are not directly associated with ER stress, or due to the limitation of only assessing the gene expression levels rather than the secretion function of the islet cells.
GCs-induced hyperglycemia is mostly associated with progressive insulin resistance in the surrounding tissues, but recent studies also suggest the involvement of the islet β-cell dysfunction [13]. GCs mediated apoptosis leads to a reduced number of pancreatic β-cells that in turn affect insulin biosynthesis [14]. GCs also cause β-cell dysfunction by disrupting the cell redox balance leading to ER stress. In a study using INS-1E cells, it was found that prednisolone activates ER-stress ATF6 and IRE1 pathways, and simultaneously up-regulates the expression of apoptosis-related factors CHOP and caspase-3 for cell apoptosis [15]. There is no way to mimic the in vivo GC concentration precisely in vitro. Because of the different distribution of drugs in vivo, the concentration around islet cells in vivo is still unknown even though the blood concentration of GC can be measured. The effective treatment concentration in vitro study was selected based on documents and pre-tests. In this study, we found that both high and low concentrations of DEX could activate the three classical UPR pathways, including ATF6, IRE1, and PERK, and also up-regulated CHOP. However, 4-PBA showed a good inhibitory effect on DEX-induced ER stress-related proteins by downregulating CHOP both at mRNA and protein levels, which is a pro-apoptotic factor in ERS [12, 16].
If UPR activation fails to restore ER homeostasis under decompensated ERS, then UPR shifts from an adaptive program to a pro-apoptotic program. Chronic PERK activation increases the expression of CHOP, by activation of the transcription factor ATF4 (activating transcription factor 4). CHOP also plays an important role in the ER stress-induced β-cell apoptosis [17, 18]. For instance, targeted disruption in the CHOP gene delays the disease onset in the Akita mouse, a diabetic model related to ER stress [17]. In summary, activation of UPR reduces the synthesis and transport of newly synthesized proteins to the ER. It also simultaneously increases the folding capacity of organelles by increasing the synthesis of ER molecular chaperones along with increased processing of the irreversibly misfolded proteins.
ERS is an important factor in the pathogenesis of diabetes. Therefore, prevention or reduction of ERS in β-cells could be a novel strategy for the prevention and/or treatment of diabetes. Moreover, 4-PBA has shown to improve the inhibition of palmitate-induced glucose-stimulated insulin secretion (GSIS) in primary rat islets. It does so by reducing the ER stress in obese mice to restore normal insulin sensitivity and blood glucose [19]. Interestingly, in humans too, PBA has shown to improve insulin resistance and β-cell dysfunction that resulted from the prolonged elevation of free fatty acids. These effects were due to PBA dependent reduction in ERS [20]. As an inhibitor of ER stress, 4-PBA eliminates intracellular UPR, and inhibits ER stress by regulation of several vital proteins, including BIP, PERK, ATF6, IRE1 and CHOP. Upregulation of some ER stress markers, especially BIP and CHOP, was observed in MIN6 cells after DEX intervention, suggesting that ER stress was induced by DEX. In summary, our study demonstrated that suppression of ER stress with 4-PBA inhibits GC-induced apoptosis by attenuating ER stress. We also observed that 4-PBA alone inhibit expression of CHOP, PERK, ATF6, IRE1 and BIP at 24 h, some even at 12 h (CHOP). 4-PBA has non-specific effects only in the presence of DEX. But DEX mediated ER stress is more obvious, and 4-PBA can alleviate this effect. We hope to provide a novel clue for molecular intervention against DEX-induced islet cell damage by inhibiting ER stress. Therefore, 4-PBA can be considered as a new therapy for diabetes. In our study too, we show that PBA relieves GCs-induced β-cell damage by alleviating the ER stress. However, the effect of 4-PBA on β-cell function remains to be studied further.
References
Saeedi P, Petersohn I, Salpea P et al (2019) Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the international diabetes federation diabetes atlas, 9th edition. Diabetes Res Clin Pract 157:107843
Fichna M, Fichna P (2017) Glucocorticoids and beta-cell function. Endokrynol Pol 68(5):568–573
Ramshanker N, Jessen N, Voss TS et al (2019) Effects of short-term prednisolone treatment on indices of lipolysis and lipase signaling in abdominal adipose tissue in healthy humans. Metabolism 99:1–10
Ullrich S, Berchtold S, Ranta F et al (2005) Serum- and glucocorticoid-inducible kinase 1 (SGK1) mediates glucocorticoid-induced inhibition of insulin secretion. Diabetes 54(4):1090–1099
Fine N, Doig CL, Elhassan YS et al (2018) Glucocorticoids reprogram β-Cell signaling to preserve insulin secretion. Diabetes 67(2):278–290
Walter P, Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334(6059):1081–1086
Oakes SA, Papa FR (2015) The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol 10:173–194
Hetz C, Papa FR (2018) The unfolded protein response and cell fate control. Mol Cell 69(2):169–181
Hsu SK, Chiu CC, Dahms HU et al (2019) Unfolded Protein Response (UPR) in survival, dormancy, immunosuppression, metastasis, and treatments of cancer cells. Int J Mol Sci 20(10):2518
Hotamisligil GS (2010) Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140(6):900–917
Li Y, Guo Y, Tang J, Jiang J, Chen Z (2014) New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim Biophys Sin (Shanghai) 46(8):629–640
Yang J, Wu Q, Lv J, Nie H (2017) 4-Phenyl butyric acid prevents glucocorticoid-induced osteoblast apoptosis by attenuating endoplasmic reticulum stress. J Bone Miner Metab 35(4):366–374
van Raalte DH, Nofrate V, Bunck MC et al (2010) Acute and 2-week exposure to prednisolone impair different aspects of beta-cell function in healthy men. Eur J Endocrinol 162(4):729–735
Guo B, Zhang W, Xu S, Lou J, Wang S, Men X (2016) GSK-3β mediates dexamethasone-induced pancreatic β cell apoptosis. Life Sci 144:1–7
Linssen MM, van Raalte DH, Toonen EJ et al (2011) Prednisolone-induced beta cell dysfunction is associated with impaired endoplasmic reticulum homeostasis in INS-1E cells. Cell Signal 23(11):1708–1715
Mueller M, Castro RE, Thorell A et al (2018) Ursodeoxycholic acid: effects on hepatic unfolded protein response, apoptosis and oxidative stress in morbidly obese patients. Liver Int 38(3):523–531
Oyadomari S, Koizumi A, Takeda K et al (2002) Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest 109(4):525–532
Cunha DA, Hekerman P, Ladrière L et al (2008) Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J Cell Sci 121(Pt 14):2308–2318
Ozcan U, Yilmaz E, Ozcan L et al (2006) Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313(5790):1137–1140
Xiao C, Giacca A, Lewis GF (2011) Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and beta-cell dysfunction in humans. Diabetes 60(3):918–924
Acknowledgements
We thank MogoEdit, a language editing company for their help in improvising this manuscript. This work was supported by the National Key R&D Program of China (2018YFC1314800 and 2018YFC1314805), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (Grant No. JX10231803).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, X., Xu, Y., Gu, Y. et al. 4-Phenylbutyric acid protects islet β cell against cellular damage induced by glucocorticoids. Mol Biol Rep 48, 1659–1665 (2021). https://doi.org/10.1007/s11033-021-06211-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-021-06211-5