
Abstract
Common bean (Phaseolus vulgaris L.), one of the most important grain legume crops for direct human consumption, faces many challenges as a crop. Domesticated from wild relatives that inhabit a relatively narrow ecological niche, common bean faces a wide range of biotic and abiotic constraints within its diverse agroecological settings. Biotic stresses impacting common bean include numerous bacterial, fungal, and viral diseases and various insect and nematode pests, and abiotic stresses include drought, heat, cold, and soil nutrient deficiencies or toxicities. Breeding is often local, focusing on improvements in responses to biotic and abiotic stresses that are particular challenges in certain locations and needing to respond to conditions such as day-length regimes. This review describes the major breeding objectives for common bean, followed by a description of major genetic and genomic resources, and an overview of current and prospective marker-assisted methods in common bean breeding. Improvements over traditional breeding methods in CB can result from the use of different approaches. Several important germplasm collections have been densely genotyped, and relatively inexpensive SNP genotyping platforms enable implementation of genomic selection and related marker-assisted breeding approaches. Also important are sociological insights related to demand-led breeding, which considers local value chains, from farmers to traders to retailers and consumers.
Similar content being viewed by others

Introduction
Common bean (Phaseolus vulgaris L.) is the arguably the most important edible grain legume worldwide, with global production estimated to be 26.8 million metric tons in 2016 (http://faostat.fao.org/). Common bean (CB) accounts for a high proportion of daily protein intake in many countries, particularly in Latin America, Africa, and parts of Asia. Beans are also an economically significant food legume and vegetable crop in Canada, USA, and Europe. Bean consumption is particularly high in African countries—for example, per capita consumption of bean ranges from 50 to 60 kg per year in Rwanda, Kenya, and Uganda (Broughton et al. 2003; Buruchara et al. 2011). CB is very nutrient rich, with both protein and complex carbohydrates, vitamins (e.g., A, C, folate), dietary fiber, and biologically important minerals such as Ca, Mg, K, Cu, Fe, Mg, and Zn (Broughton et al. 2003; Blair 2013). CB also helps improve soil and environmental health through symbiotic nitrogen fixation (SNF).
The challenges faced by CB as a crop are strongly shaped both by its evolutionary history in particular environments of Central and South America and by the set of new agroecosystems where it is now grown. Several lines of evidence indicate that CB was domesticated at least twice—from northern Andean and from Mesoamerican populations (Bitocchi et al. 2013; Schmutz et al. 2014; Ariani et al. 2016; Cortés and Blair 2017). Ecological niches for both wild populations are relatively specialized and narrow—with the Mesoamerican population, for example, being adapted to a bimodal rainfall regime and a mid-season dry period, typically on relatively fertile volcanic soils, in disturbed areas or transitional forest clearings, in a near-equatorial geographical range, while the Andean wild population, growing on the Andean slopes, is more cold-adapted (Bitocchi et al. 2013; Ariani et al. 2016). Domestication bottlenecks have likely further reduced the capacity for responses to some stresses such as drought conditions and particular pathogens (Bitocchi et al. 2013; Schmutz et al. 2014). These evolutionary and domestication histories arguably leave CB with vulnerabilities to a wide range of biotic and abiotic stresses, particularly as the crop has moved into new agroecological niches worldwide. These constraints, in turn, help set the breeding objectives for CB.
Both traditional and molecular breeding methods are in use in CB (Miklas et al. 2006). New genomic tools promise more rapid progress and the ability to solve some previously intractable breeding challenges. Marker-assisted breeding (MAB) should, in principle, increase the efficiency of selection for both major and minor qualitative and quantitative trait loci (QTL) (Xu and Crouch 2008). There are not only technical challenges; however; in developing countries, breeding programs face additional challenges of correctly identifying the highest value breeding targets and of getting uptake of new varieties where they are needed (Rubyogo et al. 2007). Some approaches, under the name of “demand-led breeding” and “wider impact,” have shown successes (Persley and Anthony 2017; Rubyogo et al. 2010).
The review concludes with the following set of recommendations: integration of robust, high-value markers into breeding programs; better characterization of strengths and weaknesses of genomic selection and related methods; better characterization of germplasm resources; solutions for physiological weak points in CB; and combining Marker-Assisted Breeding Methods with Demand-Led Breeding.
Breeding objectives for common bean
Overview of target traits and market classes for breeding
Breeding in CB is guided primarily by improvement with respect to biotic and abiotic stresses, combined with a need to maintain particular quality traits and market class characteristics, which are essential for meeting consumer preference in various markets. For example, most farmers in Africa and Latin America are continuing to plant specific Andean type varieties with lower yield to meet particular quality traits required by consumers, even though the Mesoamerican types commonly yield higher than the Andean types (Beebe 2012). Objectives relative to biotic and abiotic stresses tend to vary by location—varying, for example, by temperature and humidity or by soil fertility and water availability.
Over 80% of bean production in developing countries is from subsistence farming of semi-arid regions and sub-humid to humid growing environments. In these areas, most producers are small-scale farmers who use unimproved bean cultivars. CB tends to face a high incidence of biotic and abiotic stresses, including diseases, insects, drought, and low soil fertility (Singh 1992). Hence, breeding for resistance/tolerance to these stresses has been a major research objective (Singh 1992; Beebe 2012). Table 1 provides an overview of some of the breeding objectives, derived from breeding priorities of the International Center for Tropical Agriculture (CIAT) and Pan-African Bean Research Alliance (PABRA, http://www.pabra-africa.org; described in the “Linkage and association mapping resources” section below), and includes some other traits such as higher content of minerals (iron and zinc), fast cooking time, canning quality, harvest index, and market class/seed color (Beebe et al. 2013; Assefa et al. 2015, 2017).
In the USA, Canada, and European countries, most bean production is by commercial farmers, with much of that production being for export (e.g., small white Navy beans for UK processing and small black beans for Cuba and Mexico) or for specialized markets (e.g. “alubias” white beans for export largely to Argentina and Spain). In these areas, improvement efforts have particularly focused on resistance to major diseases, including white mold, bacterial blight, rust, halo blight, anthracnose, and bean common mosaic virus, and to insect pests such as bean leaf beetle, stinkbugs, and aphids (Table 1).
Major bean classifications are based on market classes and agronomic features (Voysest and Dessert 1991; Santalla et al. 2002). Great variation is found among dry bean market classes—with differences in pod shape, size, and color as well as seed shape (kidney, elongate, and round), seed size (varies from small-medium to large size), seed color (classified into nine groups being white, cream, yellow, brown, pink, red, purple, black, and other = gray/green/etc.), seed pattern or striation (striped, mottled, and bi-color), growth habit, and phenological traits (Singh 2001). Seed size in CB cultivars varies from small (< 25 g/100 seeds) to large (> 40 g/100 seeds). Seed shape varies from round to oblong to kidney-shaped, with different combinations of color patterns (Voysest and Dessert 1991). Seed also varies in terms of surface texture from shiny (brilliant) to opaque to intermediate.
CB genotypes can also be grouped into determinate bush types to indeterminate climbing growth habit. This growth habit classification divides beans into four groups: Type I (determinate bush), Type II (indeterminate bush), Type III (indeterminate semi climber), and Type IV (indeterminate climber) (Singh 1991). Besides growth habit classification, beans are sometimes also classified by origin—specifically, by the major Andean and Mesoamerican gene pools and races within those pools (Singh et al. 1991b; Beebe et al. 2013).
For example, in many developing countries, reduction of cooking time and improvement of mineral composition are of relatively higher importance than in developing countries, and biotic stress challenges such as web blight and nematode are of greater concern in particular locations, while various root rot diseases are a problem worldwide. See references in “Breeding objectives for common bean” section for discussion of relative priorities.
Grain yield and yield-related traits
Grain yield is a product of both plant growth rate and capacity to partition photosynthates to seeds. Significant yield differences are likely due, in part, to differing growth habits and seed sizes for beans from different market classes and gene pools (Beebe 2012). However, yield differences among cultivars within the same gene pool are often small, especially for those in the same maturity group.
Large-seeded cultivars with growth habits I, II, and III belonging to the Nueva Granada and Chile races, respectively, are physiologically less efficient and exhibit narrow adaptation compared with the small-seeded market class genotypes (Beaver et al. 1996; Beebe 2012). Direct selection for seed yield was used to improve CB productivity for Andean bush beans and is thus considered an important selection criterion. However, the progress in increasing CB yield has been modest compared to self-pollinated cereals (Singh 1991). This is due to lower dry matter partitioning efficiency toward grain yield of CB compared to cereals, low response to inputs (nitrogen fertilizer), moderate to low narrow-sense heritability of yield, high intensity of diseases, and large genotype by environment interactions (Singh 1991; Beebe 2012).
Seed yield and yield components are quantitatively inherited and are highly influenced by environments (Singh 1991), so understanding the relation between yield and its components is important to set effective selection criteria and breeding strategies. In several CB studies, high correlations have been found between yield and 100 seed weight, yield and pods/plant, and yield and seeds/plant (Beebe et al. 2013; Assefa et al. 2015; Rao et al. 2017). Hence, yield component traits have been used as selection criteria to improve grain yield and cultivar development.
The majority of efforts toward increasing seed yield under favorable environments has come from improvement in pods/plant, seed/plant, and seed weight (Beebe et al. 2013). However, under unfavorable conditions (e.g., drought), other traits including biomass partitioning indices (pod partitioning index, harvest index, and pod harvest index) have been used as key traits to improve yield (Beebe et al. 2013; Assefa et al. 2013, 2017; Rao et al. 2017).
Improvements in grain yield and related traits in CB have been associated with the improvement of the number of seeds per plant and grain yield per day (Bezaweletaw et al. 2006; Ribeiro et al. 2008). Several studies showed that hybridization of interracial bean varieties had higher yield, particularly in crosses between Mesoamerican with Durango or Jalisco races (Beebe 2012). Increasing yield potential has also been achieved through breeding for abiotic stress tolerance. Beebe et al. (2008) reported that yield could increase under drought conditions through photosynthate remobilization and biomass translocation, implying that yield improvements can be made under drought conditions. Further, CIAT and PABRA have designed new breeding strategies to breed for grain yield and resistance to single biotic and abiotic stresses, based both on grain type and market class. This has led CIAT and PABRA to release numerous new bean varieties in Africa and Latin America (Buruchara et al. 2011).
Biotic stresses
With over 200 different bean diseases reported, the pathogens causing significant yield losses to beans include bacteria, virus, fungi, and plant parasitic nematodes (Table 2). Many of these diseases and insect pests have co-evolved with CB (Beebe 2012; Beebe et al. 2013). Some of the most significant bean diseases in the tropics include bean angular leaf spot (ALS, Phaeoisariopsis griseola), anthracnose (ANT, Colletotrichum lindemuthianum), common bacterial blight (CBB), and viral diseases bean golden mosaic virus (BGMV) and bean common mosaic virus (BCMV) (Beebe and Corrales 1991; Duc et al. 2015; Miklas et al. 2017). In temperate regions, the most common diseases are CBB, halo blight, rust, and white mold (Duc et al. 2015).
Significant progress has been made in developing cultivars with resistance to various diseases using conventional breeding. Some important resistance-mapping studies are summarized in Table 2. Markers associated with established resistance loci can be used for more efficient breeding to develop resistant cultivars. Some early examples of marker-assisted selection for bean diseases include 23 RAPD markers and 5 SCAR markers associated to 15 different resistance genes, described by Kelly and Miklas (1998). Molecular markers and linkage mapping of rust resistance genes have been reviewed by Miklas et al. (2002). Kelly and Vallejo (2004) provided a summary of markers, MAS, map location, and breeding value for anthracnose resistance. Similarly, Miklas et al. (2006) reviewed MAS in breeding for resistance to anthracnose, angular leaf spot, common bacterial blight, halo bacterial blight, bean golden yellow mosaic virus, root rots, rust, and white mold (Table 2).
Abiotic stresses
Drought stress
Abiotic (climatic and edaphic) stress factors are major constraints for bean productivity in most tropical and subtropical countries (Rao 2014). In Central America and in eastern and southern Africa, as much as 60% of the bean growing areas in these regions suffer from periodic drought stress (Assefa et al. 2013; Ambachew et al. 2015; Darkwa et al. 2016). Key traits linked to drought resistance include phenology, root size and depth, root hydraulic conductivity, carbohydrate reserve storage and mobilization, and water absorption efficiency (Beebe et al. 2013). Breeders and physiologists are particularly focused on improving the traits related to photosynthate mobilization from vegetative parts of the plant to the pod walls and seeds under drought conditions (Rao et al. 2017). These photosynthate-mobilizing traits include pod harvest index (PHI), pod partitioning index (PPI), and harvest index (HI) (Beebe et al. 2013) which may be used to select drought-adapted beans (Beebe et al. 2008; Assefa et al. 2013; Rao et al. 2013, 2017; Polania et al. 2016a, 2017). Sources of drought resistance have been found in the Durango race and in tepary bean (Beebe 2012; Rao et al. 2013; Asfaw and Blair 2014; Mukeshimana et al. 2014). Several drought-resistant lines have also been identified in Africa (Asfaw et al. 2012; Mukeshimana et al. 2014).
Breeding for improved adaptation to drought is complex because several traits are involved in resistance mechanisms, and the traits are quantitatively inherited and highly affected by environments (Mir et al. 2012). Use of MAS for improving drought resistance was explored by Schneider et al. (1997), who identified QTLs for drought using Random Amplified Polymorphic DNA (RAPD) markers. In this study, yield was improved by 11% under drought and 8% under normal conditions by using five RAPD markers (Schneider et al. 1997).
Genotype by environment interactions affecting drought QTL are reported by Chavarro and Blair (2010), Asfaw et al. (2012), Asfaw and Blair (2012), Blair et al. (2012), and Mukeshimana et al. (2014). Asfaw et al. (2012) identified 9 and 69 QTLs associated with drought using mini-environment mixed model approach and composite mapping approaches, respectively. Asfaw et al. also reported that the phenotypic variation explained by QTLs is up to 37% for SPAD leaf chlorophyll and pod partitioning index traits. This result shows the importance of QTL detection for photosynthate acquisition and remobilization traits. Trapp et al. (2015) also detected two major QTLs on Pv01 and Pv02 for seed yield in several abiotic stresses and drought tolerance conditions. QTL in populations with Durango derived drought tolerance have also been analyzed (Mukeshimana et al. 2014; Briñez et al. 2017). The QTLs identified in all those studies could be important tools for MAS in bean breeding programs to select indirectly for drought tolerance traits that are difficult to screen in large populations.
Heat stress
High temperature (HT) stress is a major bean production constraint (Rainey and Griffiths 2005; De Ron et al. 2016). HT (greater than 30 °C day and/or greater than 20 °C at night) causes significant reduction in yield and quality and limits environmental adaptation. The major effect of high temperature is shown as inhibition of pollen fertility that results in blossom drop. This causes reduced seed number and quality of the seed. Researchers have identified heat-tolerant CB genotypes from diverse gene pools (Porch and Jahn 2001; Porch 2006; Porch et al. 2013). Some of these maintain pollen viability up to 5 °C higher during night temperatures compared to temperatures that are normally considered to be limiting (> 18 °C at night). Development of HT varieties under adverse environments would also increase resilience for the future global climate change threats (Porch et al. 2013; Gaur et al. 2015).
Low temperature stress
CB is sensitive to low temperatures, which can limit production in the early part of the season. Differences among genotypes for tolerance to suboptimal temperatures were reported by Dickson and Boettger (1984). The unifoliate and the first trifoliolate leaf stages were the most sensitive to freezing temperatures in CB (Meyer and Badaruddin 2001). Their estimated temperature to cause 50% mortality was − 3.25 °C, although regrowth after survival was limited, meaning few plants made it to maturity. Interspecific introgression of portions of the tepary bean genome into CB is a promising method for increasing tolerance to extreme temperatures in CB (Souter et al. 2017). Rodino et al. (2007) reported seven cultivars of P. coccineus that showed ability to germinate, emerge, and grow under cold temperature—thus showing potential for as source of cold-tolerant genes in interspecific hybridization with CB.
Low P stress
Low soil phosphorus (P) availability causes significant bean yield loss in the tropics (Ramaekers et al. 2010; Beebe 2012). About 50% of bean growing regions worldwide are affected by low soil P (Nielsen et al. 2001; Beebe 2012). More than three million hectares of bean-growing areas of Africa could suffer from P constraints (Wortmann et al. 1998). Progress has been made in developing tolerant cultivars with better P acquisition efficiency, involving higher total root length, root surface, and shallow root angle under low P (Ochoa et al. 2006; Beebe 2012; Rao et al. 2016). One of the key mechanisms identified to increase access to P is greater topsoil foraging resulting from root architectural, morphological, and anatomical traits (Lynch 2011). Shallower root growth angle of axial or seminal roots increases the topsoil foraging and thereby contributes to greater acquisition efficiency of P from low P soils.
Rao (2001) and Beebe et al. (2009) reported that greater photosynthate remobilization to the grain gives better yield under conditions of low P availability. The CIAT Bean Program has reported that bean genotype G21212 and other breeding lines identified for drought tolerance also gave better yield than poor performing lines under low soil P conditions (Beebe et al. 2008). Root QTLs associated with P acquisition in low soil P environments were reported in Beebe et al. (2006), which are linked with root parameters such as total and specific root length. QTLs associated with P use efficiency were also reported by Cichy et al. (2009a). QTL studies of P deficiency tolerance have also been conducted with other inter-genepool crosses (Ochoa et al. 2006) and within Andean genotype populations (Cichy et al. 2009a, 2009b). Mechanisms of tolerance to low soil fertility have been studied both in terms of root architecture and higher root hair density (Liao et al. 2004).
Low N stress
Low soil N affects bean production in many regions (Wortmann et al. 1998). Beans grown in marginal, generally low-nutrient and moisture-limited soils, also tend to show diminished nodulation activity (Albrecht et al. 1984). Significant genetic variability is known to exist in both the host-plant and rhizobium strains in terms of SNF, which should enable breeders to find cultivars with improved SNF ability (Bliss 1993; Snoeck et al. 2010; De Ron et al. 2015; Drevon et al. 2015; Polania et al. 2016b). Total seed N concentration could be used as selection criterion to screen advanced breeding lines for genetic variability in SNF (Miranda and Bliss 1991). Harvest index and biological yield are also considered indirect measures for genetic improvement of SNF ability in bean (Araújo et al. 2015). Farid et al. (2017) reported that selection of bean lines for their high SNF capacity could be done in both CBB-susceptible and CBB-resistant genotypes. Their work suggests that selection for SNF capacity in CBB-resistant lines of CB may have negative influence on the degree of rhizobial infection.
Aluminum toxicity and acid soil stress
Other abiotic stress factors such as aluminum (Al) and manganese toxicities associated with soil acidity are also problems to bean production, particularly in acid soil regions of Latin America and Africa (Rao 2014; Rao et al. 2016). Mechanisms of Al resistance were defined using the Al-resistant genotype ‘ICA Quimbaya’ and the Al-sensitive ‘VAX-1’ (Yang et al. 2013). The induced and sustained Al resistance of ‘Quimbaya’ was shown to be mediated by reducing the stably bound Al in the apoplast, thus allowing cell elongation and division to resume. Resistance to Al is attributed to the release of citrate by the root apex which is mediated by the multidrug and toxin extrusion (MATE) citrate transporter gene. Resistance to Al in CB was mainly dependent on the capacity to sustain citrate synthesis, thereby maintaining the cytosolic citrate pool that enables exudation. The initial Al-induced inhibition of root elongation in both Al-resistant and Al-sensitive genotypes was correlated with the expression of the 1-aminocyclopropane-1-carboxylic acid oxidase gene (Yang et al. 2013). QTLs for Al stress tolerance were first identified by López-Marín and Rao (2009). Andean genotypes have been screened for Al tolerance, with significant genetic variability identified for this trait (Blair et al. 2009).
Genetic and genomic resources for CB breeding
Germplasm collections and CB diversity
Significant national collections of CB are maintained at the USDA, in Pullman, Washington, USA (about 15,000 accessions), the Institute für Pflanzengenetik und Kulturpflanzenforschung, Germany (about 9000 accessions), in Brasilia, Brazil (CENARGEN/EMBRAPA, with about 6000 accession), in Beijing, China (CAAS, Institute of Genetic Resources with more than 5000 accessions), and the National Center for Plant Genetic Resources in Alcala de Henares, Spain (with more than 5000 bean accessions). The largest collection of CB genetic resources is maintained under the auspices of the Food and Agriculture Organization (FAO) treaty, under International Treaty on Plant Genetic Resources for Food and Agriculture (ITPGRFA), at CIAT in Cali, Colombia (around 36,000 accessions), with a backup at the Svalbard Global Seed Vault in Norway, where more than 50,000 accessions are now held. In addition to CB (P. vulgaris) and various wild Phaseolus species, these collections include four other domesticated Phaseolus species: year-long bean (Phaseolus dumosus), runner bean (Phaseolus coccineus), tepary bean (Phaseolus acutifolius), and lima bean (Phaseolus lunatus). Most of these collections were made from the centers of origin, mainly Andean and Mesoamerican regions. Smaller collections of CB accessions exist through non-governmental agencies such as Seed Savers Exchange in Decorah, Iowa or at breeding stations of national, sub-national, or multi-country regional programs (e.g., ECABREN and SABRN bean networks in East and Southern Africa, respectively). Germplasm accessibility is generally not a bottleneck for bean breeding and genetic studies.
Genetic diversity has been extensively studied in bean using different types of markers, including seed protein (e.g., phaseolin) (Gepts et al. 1986; De La Fuente et al. 2012) and isozyme analysis (Koenig and Gepts 1989). Other molecular markers used for genetic diversity in CB are DNA restriction fragment length polymorphism (RFLP) (Khairallah et al. 1990, 1992), nuclear RFLP (Becerra Velasquez and Gepts 1994), allozymes (Singh et al. 1991a; Santalla et al. 2002), and random amplified polymorphic DNA (RAPD) (Freyre et al. 1998; Beebe et al. 2000). Similar reports have also demonstrated genetic diversity through use of amplified fragment length polymorphism (AFLP) markers (Beebe et al. 2001; Papa and Gepts 2003; Zizumbo-Villarreal et al. 2005), SSR markers (Gaitán-Solís et al. 2002; Blair et al. 2006a), DNA sequencing (Gepts et al. 2008), and single nucleotide polymorphism (SNP) markers (Galeano et al. 2009a, 2009b, 2012; Blair et al. 2013). These tools help answer different questions related to evolution, domestication, and diversity of CB, which is not possible to answer with the use of phenotypic methods alone (Arif et al. 2010). For example, genes related to domestication from the Andean and Mesoamerican domestication events and evolutionary traits such as shattering have been identified (Bellucci et al. 2013; Gaut 2014) as have polymorphism in drought-related genes (Cortés et al. 2012a, 2012b). Due to their cost-effectiveness, efficiency, and simplicity, SNP, SSR, and AFLP markers have been the most commonly used markers studies on CB genetic diversity.
The world’s germplasm collections can be characterized in various ways: by genotype (i.e., marker- or sequence-based characterization), by phenotype (e.g., growth habit, seed characteristics, disease responses, photoperiod response, etc.), by pedigree or genepool or race, or by geographic origin. Ideally, these characteristics would be maintained, in combination, for all germplasm accessions, but in practice, the characterizations are incomplete and not fully correlated. Substantial phenotype data is maintained for the U.S. germplasm collection in the GRIN system. Pedigree data is generally lacking, except for selected populations, usually at institutes with long-running breeding programs (e.g., CIAT). For geographic origin, an interesting resource is the geographic information system map of germplasm origin maintained at LegumeInfo (https://legumeinfo.org). This interactive viewer displays geo-coordinates for the bean collection in GRIN, along with the phenotypic data in GRIN. The data in the viewer can then be queried by geographical location or by phenotype (e.g., photoperiod or seed size)—and then phenotypic categories or values can be displayed geographically, to look for correlations such as seed size by location—showing the Andean material generally having larger seeds than Mesoamerican landraces.
For genotypic data, several projects have generated large SNP datasets. The U.S. Bean Coordinated Agricultural Project (BeanCAP) has generated SNP calls for two diversity panels: the Mesoamerican Diversity Panel (MDP) and the Andean Diversity Panel (ADP) (Moghaddam et al. 2014). These are described and available at LegumeInfo: http://legumeinfo.org/data/public/Phaseolus_vulgaris/G19833.gnm1.div1/.
To access and utilize the highly valuable material in germplasm repositories, arguably the most common approach has been to order a subset of material of interest, on the basis of what phenotype data is available, followed by field trials to screen for particular traits (e.g., resistance to a disease of interest). Availability of genotype data (e.g., for the MDP and ADP panels) makes it possible to select for genotypic diversity as well or even for presence of particular alleles for known genes.
Genomic resources and tools for CB research and breeding
CB has a medium-sized, diploid genome of 588–637 Mbp (Arumuganathan and Earle 1991; Bennett and Leitch 2005; McClean et al. 2008). Genomic resources have advanced dramatically in recent years, with reference genome sequences, dense genetic maps, marker and genotyping sets, and many QTL and genome-wide association studies (GWAS).
Reference genome assemblies provide an important resource for organizing many other genetic and genomic features and resources. At the time of writing, there are three genome assemblies of reference quality: two versions of the Andean G19833 accession (Schmutz et al. 2014) and the Mesoamerican BAT93 assembly (Vlasova et al. 2016) [https://doi.org/10.1186/s13059-016-0883-6]. The G19833 assemblies (v1 and v2) are available for downloading, sequence searching, and browsing at both Phytozome (https://phytozome.jgi.doe.gov) and LegumeInfo (https://legumeinfo.org).
These on-line tools are typically used for basic research toward marker development and selection and for investigation of the genetic basis of traits of interest, rather than directly for day-to-day breeding work.
Together with QTL, marker, and sequence data, comparative genomic analyses are possible through the Legume Information System (LIS or LegumeInfo). The resources at LegumeInfo legume-focused gene families and gene family trees (phylogenies) also include an InterMine instance (https://mines.legumeinfo.org/beanmine) for querying regions and lists of features (e.g., genes within a QTL region, filtered for gene expression).
The legume gene families are an important tool for identifying corresponding genes (orthologs) across legume species and thereby for linking research across various legume species. For example, research on gene function in soybean is often transferrable to orthologous bean genes. A published gene in soybean (e.g., the determinacy gene Dt1) could be used to detect the gene family containing the gene (either through a BLAST search against gene families or by entering a gene name), and the family, in turn, identifies both near and more distant orthologs in CB. Expression patterns can then be checked for the bean genes in the genome browser or gene record pages at LegumeInfo.org or in the BeanMine.
To effectively use the genomic resources at LegumeInfo.org or BeanMine, it may be helpful to think of three use-cases, distinguished by starting knowledge. In the first case, one has a gene with known function in another species and wishes to find whether there is evidence for similar functions in the ortholog in common bean. In the second case, one has genetic association information, either in the form of genetic markers from a QTL studies or genomic regions from a GWAS study, and wishes to find candidate genes within that genetic or genomic region. In the third case, one starts with gene enrichment information (e.g., genes upregulated under some condition) and wishes to narrow that list to find causal genes for a trait.
For the first use-case (gene in species 1 to candidate gene in CB), the sequence for the starting gene can be used as a query in a BLAST search at legumeinfo.org—either against the reference genome sequence, which will lead to a genome browser view centered on the BLAST hits, or against gene sequences (protein or coding sequence), which will lead to pages for the respective genes. From either target location (genome browser or gene page), there are link-outs to other resources—for example, from a gene to a gene family or from a gene to gene expression profiles or from a gene to a genomic synteny in the LegumeInfo Genomic Context Viewer (GCV). Information toward validation of function can be gleaned from expression information, from phylogenetic and genomic context (from the gene tree viewer and GCV), or from overlapping QTL or GWAS regions. For GWAS and QTL regions, additional work typically needs to be done by the researcher to find the locations of flanking markers on the genome browser—which can be done by a search within the genome browser or by a search in LegumeInfo in the marker search page.
For the second use-case (from QTL or GWAS region to candidate genes), the first step is to find the region of interest in the genome browser. This can be done using a search of either flanking- or top-ranked marker, within the browser page or from the LegumeInfo in the marker search page. This needs to be done with caution, particularly for markers identified in QTL studies, because QTL regions are distributions, often with the significance region spanning many very large regions in genomic space. For either QTL or GWAS associations, it is appropriate to extend the region search to include all genes spanned by the flanking non-significant markers. For example, if there are significant markers at positions 1,000,000 and 1,100,000 and non-significant markers at 999,000 and 1,200,200, then the region that should be searched for candidate genes is from 999,000 and 1,200,200—because with greater SNP density, it is likely that significant markers would be found outside the two markers that were reported as significant. Once a set of candidate genes has been located in the genome, then functional information for predicted can be used to assess potential function in the plant, and gene lists can be further evaluated for information—either toward confirmation or elimination from candidacy.
For the third use-case (a list of genes of interest from any source to a reduced list of high-value candidates), the initial list could, in principle, come from numerous sources. Take the example of genes with significant differential expression, assayed in an RNA-Seq experiment, for response to some condition, e.g., drought response. Such a gene list can be used in a custom query at the BeanMine (available via LegumeInfo.org: https://mines.legumeinfo.org/beanmine). To be useful, the query should substantially narrow the initial gene list. This could be done by intersecting the genes with one or more genomic regions (likely determined from QTL or GWAS studies) or by another list (for example, the set of orthologs from a BLAST search from genes known in another species to mediate drought response). Both types of queries and list operations are easily conducted at the BeanMine using similar query templates available from the main page.
Linkage and association mapping resources
Linkage mapping enables identification of associations between traits and markers, for both simple Mendelian traits and quantitatively inherited traits (QTLs) (Ibarra-Perez et al. 1997; Gepts et al. 2008; De Ron et al. 2015). The first widely used genetic map in bean was developed from a backcross (BC) mapping population between Mesoamerican line ‘XR-235-1-1’ and ‘Calima’ (Andean cultivar (Vallejos et al. 1992). This linkage map included 9 seed proteins, 9 isozymes, 224 RFLP, and seed and flower color markers. These molecular markers were placed on 11 linkage groups, spanning 960 centimorgans (cM). The second genetic map was developed using RFLP markers, spanning 827 cM. These markers were placed on an F2 mapping population (cross of BAT93 by Jalo EEP558), with 142 markers being assigned to 15 linkage groups (Nodari et al. 1993).
A third genetic map was developed by Adam-Blondon et al. (1994) from the cross between Ms8EO2 and Core. This map contained 51 RFLPs, 100 RAPDs, and two sequence-characterized amplified region (SCAR) loci and spanned 567.5 cM across 12 linkage groups. These three maps were mainly based on RFLPs, though few seed protein and isozyme markers were also included (McClean et al. 2004). A consensus map was then developed utilizing these linkage maps on BAT93 × Jalo EEP558 (BJ) as a core map (McClean et al. 2004). The creation of this consensus map has provided bean breeders with the means for combining all the genetic information from multiple populations developed from diverse genetic background. It also provided the opportunity to map more loci than from single cross populations and also increased important markers over different genetic backgrounds (Rami et al. 2009).
Numerous subsequent maps have been generated, using a succession of marker types (reviewed by González et al. 2018). Although SNP markers have generally supplanted prior types of markers, important early markers and mapping populations are valuable resources for interpreting new SNP maps and crosses, sometimes through conversion of older marker types to nearby SNP markers. SSR markers (also called microsatellites), which are also typically though not exclusively PCR-based, have been extensively used in bean genetic studies. SSR markers were first reported in bean by Yu et al. (1999, 2000), with 15 different microsatellite markers included in a molecular linkage map constructed primarily using RAPD and RFLP markers. Blair et al. (2003) integrated 100 SSR markers in two linkage maps along with RFLP, AFLP, and RAPD markers. Much more saturated SSR-based maps were reported by Córdoba et al. (2010) and Blair et al. (2014). Since then, several bean genetic studies have been implemented using SSR markers and have further been employed for map comparison and integration. The sequence-characterized amplified region (SCAR) marker is another PCR-based marker that has been used for comparison of genetic map and integrating genetic maps (McClean et al. 2002). Additional types of PCR-based markers include indel-based markers, including a large set described by Moghaddam et al. (2014).
In the last decade, SNP assay methods have become far more efficient. Researchers can now inexpensively scan the whole genome to identify rare variants that are potentially associated with traits of interest. SNP discovery in maize and soybean is illustrative for many other crop species (Rafalski 2002, 2010; Hyten et al. 2010), though there are species-specific differences—for example, with the SNP frequency being roughly an order of magnitude higher in maize than soybean. In CB, SNP frequency is relatively high, with approximately one SNP per 88 bp across a genome of ~ 588 Mbp—implying more than six million SNPs are expected in the genome (Gaitán-Solís et al. 2008; Schmutz et al. 2014; Blair et al. 2018). An important recent SNP map is the high-resolution Mesoamerican × Andean cross of Stampede × Red Hawk produced by Song et al. (2015), which utilized 7276 SNP markers in an F2 mapping population of 267 RILs. This was used to anchor sequence scaffolds into pseudomolecules in the first reference genome assembly for CB (Schmutz et al. 2014). Many bean SNPs have been discovered through sequencing and genotyping by sequencing (Bhakta et al. 2015; Ariani et al. 2016; Schröder et al. 2016), and some older markers have been converted to “Kompetitive Allele Specific PCR” (KASP)-based SNP assays (Cortés et al. 2011).
Genetic maps for CB are found at LegumeInfo (https://legumeinfo.org/traits_maps#phavu) and at PhaseolusGenes (http://phaseolusgenes.bioinformatics.ucdavis.edu). Both of these websites include various genetic maps, as well as QTL features from numerous studies projected onto a reference genetic map. In the case of LegumeInfo, QTL and markers are projected onto the combined map of three populations (Fig. 1), two of which were inter-genepool, namely DOR364 × G19833 (DG) and BAT93 × Jalo EEP558 (BJ) with one Mesoamerican × Mesoamerican BAT477 × DOR364 (BD) population as described in Galeano et al. (2012). This resource has links to a high-density map for an F2 population for North American researchers based on the Mesoamerican × Andean cross of Stampede × Red Hawk (SR) (Song et al. 2015) and to an integrated map for the BJ reference population. Finally, all the maps are tied into the G19833 sequence information from LegumeInfo (Fig. 2).

Consensus genetic map, showing QTL from various studies

Reference map of the BAT93 × Jalo EEP558 population using trans-legume orthologous gene-based (TOG) markers
A great number of trait-mapping studies in CB have been produced over roughly the last 40 years, involving both mapping of qualitative and quantitative traits in biparental populations, and more recently, through genome-wide association studies in diverse germplasm collections (reviewed by González et al. 2018). A sampling of some of these important trait-mapping follows.
The primary determinacy locus, FIN, has been identified on LG01 in CB as PvTFL1y, homologous to the Arabidopsis TFL1 gene (Kwak et al. 2008; González et al. 2016). The allele conferring determinacy is the mutant (recessive) form.
Control of photoperiod has been repeatedly mapped to two loci on LG01 (Koinange et al. 1996; Kwak et al. 2008). A compelling candidate for the PPD (photoperiod) locus is an ortholog of the E3/PHYA3 gene in soybean gene (McClean et al. 2010). A candidate for the other LG01 locus, HR, is orthologous to the Flowering Time (FT) gene in Arabidopsis (Gu et al. 1998).
Traits such as seed and pod size and yield are complex, involving multiple genes and numerous epistatic interactions, but loci involved have been identified repeatedly, in various backgrounds, and with increasingly tight genetic bounds (González et al. 2018). For example, pod size and pod length QTLs have been reported in similar locations, including LG01, LG02, and LG04 (Koinange et al. 1996; Hagerty et al. 2016; Yuste-Lisbona et al. 2014).
Current and prospective methods in CB breeding
Genomics-assisted breeding
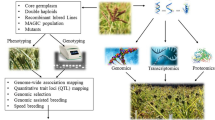
Conventional bean breeding approaches have produced many improved varieties. However, genetic progress in yield has been slow compared to crops such as soybean and maize. Improved molecular marker technologies may enable bean breeders and geneticists to speed up cultivar development. An important intermediate step in this direction is to transfer desirable QTLs (genes) into active breeding populations using MAS—and to transfer multiple traits through gene pyramiding (Das et al. 2017). Progress has been made in applying MAS toward improved resistance and it is described by Miklas et al. (2006) and Tryphone et al. (2013).
A collection of molecular breeding techniques, collectively labeled MAB by Ribaut et al. (2010), includes selection based on marker-assisted back-crossing (MABC), marker-assisted recurrent selection (MARS), and genomic selection (GS) (Fig. 3). The MABC approach is to transfer a major gene from a donor cultivar into an elite line. The MARS approach is to assemble and involve favorable alleles from various sources for the expression of quantitative traits. The GS approach relies on marker-based selection that might be performed without major testing or even prior marker × trait associations (Bernardo and Yu 2007). MABC and MARS have been effective as indirect selection techniques by selecting for traits without evaluating the trait of interest. MABC is one of the most preferred molecular approaches for transferring desirable genes into well-adapted commercial cultivars. Carneiro et al. (2010) reported that microsatellite markers linked with white mold resistance (genes) were effective in selecting individual plants with a higher resistance relative to the recurrent parent genome. Integrating MABC into bean breeding has been effective for improving traits controlled by major genes and used for stacking of few genes and QTLs (Kelly 2004; Carneiro et al. 2010; Varshney et al. 2010). It is also efficient for gene pyramiding (e.g., combining two or more strains of the same pathogen).

An early example of marker-assisted selection was used to determine and select anthracnose resistance genes Co-5 and Co-42. The markers SAB3 and SAS13 are associated with the anthracnose resistance genes Co-5 and Co-42, respectively, in the donor parent G2333 (the source of resistance genes to anthracnose). This resistant parent was crossed with susceptible commercial cultivars, and resistance was selected for by selecting for these markers (SAB3 and SAS13) in the backcross progeny. The resistance was effectively transferred to the BC1 population (Garzón et al. 2008). In that study, the Co-5 and Co-42 anthracnose resistance genes associated with the markers (SAB3 and SAS13) can be stacked to increase the level of resistance to anthracnose. Subsequent studies have also selected for bruchid and virus resistance (Blair et al. 2010b).
MAS in bean breeding and the progress that has been made are well explained by Miklas et al. (2006), who describe success toward several diseases of CB. MABC for quantitatively inherited traits including yield and drought tolerance has not yet been well developed in CB (O’Boyle et al. 2007). Markers linked to QTL for disease and insect resistance have been identified and are being utilized to introgress genes into elite varieties (Briñez et al. 2017).
The MARS approach helps breeders identify superior alleles for complex traits such as drought resistance and yield and to develop superior breeding lines from both parents. Bernardo and Charcosset (2006) reported that MARS is effective for identifying multiple genomic regions and to detect both minor and major QTLs. Thus, MARS may be able to achieve greater genetic gain compared to MABC.
GS, another emerging molecular approach, is a form of molecular MAS that enables breeders to increase genetic gain in a short period of time for quantitatively inherited traits (Heffner et al. 2009). GS is different from MABC and MARS in that it directly identifies better genotypes via predicted breeding value (BV), using markers with genome-wide distribution. GS methods use a training population and a validation population. The training population consists of elite lines that are both phenotyped and genotyped with genome-wide markers. These markers are treated as random instead of fixed effects.
In GS, the molecular marker effects on the phenotypes of elite materials are assessed concurrently in a model. It is assumed that one or more markers are in linkage disequilibrium with corresponding QTL associated with the trait. In GS, the model for prediction is fitted to detect the entire additive genetic variances based on totality of the effects of the molecular markers, to estimate breeding values of individual markers. This model is also applied to the genomic data of a validation population in which the individuals are genotyped but not phenotyped. The model produces genomic estimated breeding values (GEBV), which captures the effects of markers in the training population toward phenotypes of interest. After predicting the breeding value (GEBV) for each genotype in the breeding program, genotypes with higher breeding values are either recycled into the crossing program or dropped. The advantage of using GS over conventional breeding is that it has the potential to reduce the number of breeding cycles and reduce the need for phenotyping in every cycle, while maintaining genetic diversity. GS should be efficient than traditional MAS at selecting for complex traits with low heritability, since models comprised of many markers are able to pick up low-effect genes.
Genomic resources in bean have also enabled the use of GWAS to identify marker-trait associations. A significant advantage of GWAS over QTL studies is that the marker-based associations (typically SNPs) can be integrated with other GWAS, as long as the markers are placed on a common genomic reference assembly. The markers acquire position by virtue of the genomic sequence rather than through genetic mapping and recombination-counting (Schmutz et al. 2014). Significant GWAS research has been conducted in CB to find genetic associations with some traits such as agronomic performance and SNF ability (Kamfwa et al. 2015a, 2015b), anthracnose and angular leaf spot resistance (Perseguini et al. 2016), cooking time (Cichy et al. 2015), anthracnose resistance (Zuiderveen et al. 2016), and drought tolerance (Hoyos-Villegas et al. 2017). More research and a better understanding of different biotic and abiotic stress tolerance traits in the context of GWAS in CB are still needed.
High-throughput phenotyping approaches for CB breeding
Progress in high-throughput phenotyping (HTP) in CB has generally lagged genomic progress. It remains difficult and costly to do precise phenotyping of simple and complex traits such as plant height, biomass, flowering, and yield for a large breeding population with replicated tests across different environments, requiring large number of plant measurements—many of which are time-sensitive and growth-stage dependent. This bottleneck has led to some new HTP approaches, unlocking prospects for non-destructive field and lab-based phenotyping (Cobb et al. 2013; D’Agostino and Tripodi 2017; Varshney et al. 2018).
Currently, most plant trait phenotyping is heavily dependent on visual observation and manual measurements, which are time consuming, labor intensive, costly, error-prone, and liable to miss subtle phenotypic variations (Kumar et al. 2015). Some HTP imaging techniques are non-invasive and accurate, reducing dependence on invasive or destructive methods (Bhat et al. 2015)—employing, for example, multi-spectral imaging for different traits such as phenology, leaf disease (chlorosis, necrosis), plant structure, and biomass accumulation. For example, near infra-red cameras are used for measuring tissue water content and chlorophyll fluorescence analysis to assess photosynthetic efficiency (Kumar et al. 2015). A combination of new imaging techniques and robotic and conveyer belt systems in greenhouse could be used for bean HTP (McDonald et al. 2016). At a smaller plot level, ground-based HTP including tractor-based system or modified vehicles such as phenomobiles and phenocarts equipped with global positioning system (GPS) and sensors could be easily applied to bean phenotyping.
Demand-led breeding
Although many aspects of bean improvement depend on technical factors, we would also like to highlight an important sociological approach that has demonstrated success in CB breeding. CB has steadily evolved from primarily a smallholder subsistence crop (Katungi et al. 2009) to market-oriented production (Buruchara et al. 2011). This shift in focus has necessitated a revision in the varietal development process and seed system. The hands-on nature of participatory variety selection (PVS) has evolved from more contractual and consultative to demand-led breeding (Persley and Anthony 2017) where multidisciplinary researchers work closely with bean value chain actors to develop bean varieties that meet the needs of farmers and others in the value chain. This paradigm shift in bean breeding has been toward a value chain-focused approach, with relatively less emphasis placed on the farmer-focused approach of a few farmers engaged in selecting varieties for ecological suitability. Currently, nearly all the bean breeders in PABRA are employing demand-led breeding approaches. A variety of factors that include different users groups (women/men, more market-oriented/home consumption), a range of agroecological zones, and preference information (from participants including households, farmers, traders, and processors) are resulting in fine-tuning of formal breeding programs. Conventional plant breeding has been successful in developing bean cultivars that can be used in environments that are fairly homogenous and stable, but it has been less effective in developing cultivars in complex and marginalized drought affected environments (Ceccarelli et al. 2000; Fufa et al. 2010; Assefa et al. 2014) Conventional plant breeding is also framed to accommodate limited requirements and the particular needs of farmers and particular growing environments (Assefa et al. 2014). Through demand-led breeding (DLB), bean breeders can also enhance varietal diversity through involvement of actors throughout the value chain, as well as minimizing effort that might be invested in developing varieties that are unacceptable to farmers and local communities, and traders/processors. DLB is also able to exploit genotype by environment interaction by taking advantage of specific adaptations to particular locations and growing conditions, such as periodic drought or soil mineral toxicity.
Tremendous gains have been made in getting conventionally selected varieties released through the formal cultivar release system. Through collaborative efforts, PABRA has succeeded in fast-tracking the release of bean varieties selected for a range of preferred traits. The number of varieties released rose from 73 in 1990–2000 to 130 in 2001–2008 to 340 in 2009–2016, including seven PABRA members with less resourced breeding programs: Burundi, Cameroon, Democratic Republic of Congo, Swaziland, Congo Brazzaville, Lesotho, and Guinea (http://www.pabra-africa.org/pabra-annual-report-20162017-20th-anniversary-special-edition). As result of use wider impact seed systems approach (Rubyogo et al. 2010), within the PABRA region, improved bean varieties occupy 56% of the bean-growing area. More importantly, yields have nearly doubled in Ethiopia (Berhanu et al. 2018), while they have increased by 55% in Uganda, 20% in Burundi, and 12% in Rwanda over the last decade (PABRA 2017). This significant increase of yield for improved bean cultivars is found to be related to the positive impacts (CIAT 2013; Larochelle et al. 2013).
Gaps in common bean improvement and potential future developments
Although a great deal of work is underway globally toward bean improvement, we see several areas of current weakness—and, concomitantly, areas for opportunity.
Integration of robust, high-value markers into breeding programs
There remains a lack of some key information for CB improvement, including easily assayed markers tightly linked to important traits and more complete understanding of the mechanisms underlying quantitative traits. In the absence of that mechanistic understanding, single markers will generally be of limited use, particularly given the complexity and heterogeneity of genetic backgrounds in CB. Further, even the best markers for high-value traits are of limited utility in a breeding program unless the breeder has access to efficient, low-cost assays, integrated as a regular part of the annual breeding cycle.
Better characterization of strengths and weaknesses of genomic selection and related methods
Although GS methods have been in use for more than a decade (Bernardo and Yu 2007) and have been shown to be useful in multiple species (being particularly helpful in avoiding the need to do expensive phenotyping during each selection round), the method remains challenging to do well—generally requiring careful phenotyping in the first generations in for distinct populations and breeding projects. It works better in some germplasm collections than others and can be highly affected by population structure, which may not be straightforward to identify or correct for.
Better characterization of germplasm resources
Complete genome sequence assemblies are available for several bean accessions, and there are available methods for high-throughput genotyping, e.g., genotyping-by-sequencing (Schröder et al. 2016; Ariani et al. 2016) or SNP chips (Song et al. 2015). Nevertheless, no complete catalog of variants across the global germplasm collections exists for CB. This would be helpful in identifying both unique germplasm as well as redundancies across collections.
Solutions for physiological weak points in CB
CB remains a vulnerable crop in several ways. It has limited tolerance to high temperatures, particularly during flowering. It also generally has poor tolerance to cold temperatures and to drought and is vulnerable to many diseases and pests. Improvements in any of these areas remain a daunting challenge, but there are opportunities for improvements through introgressions, both from wild P. vulgaris accessions with valuable traits and by interspecific crosses to gain traits for tolerance to broader environmental ranges from species such as P. acutifolius and P. coccineus.
Combining marker-assisted breeding methods with demand-led breeding
While DLB and MAB have each been highly selective where applied, DLB has typically been an approach used in developing countries and MAB (summarized in Fig. 3) more often used in resource-wealthy breeding situations. Merging the two should have promise—although the technical requirements of MAB are significant enough to require substantial coordination by experienced organizations (e.g., CIAT or ICRISAT or focused international grant-led projects). The types of methods to incorporate into DLB include MAB and GS (Fig. 3). MAB or GS can further improve the efficiency of selection for biotic and abiotic stress resistance/tolerance traits that typically have a low heritability and also help to increase the initial frequency of favorable alleles in bulk populations. Then, the farmers select the elite lines in their own field (Steele et al. 2004; Kanbar and Shashidhar 2010), helping reduce program costs in the breeding project while also helping build buy-in by farmers.
Summary
Conventional plant breeding and a collection of worldwide germplasm have been the primary source of improvements in CB. However, enormous opportunities still exist to improve the efficiency and accuracy of bean breeding and to increase genetic gain with the use of genomic tools, improved phenotyping methods, and well-coordinated DLB projects. Genomic data facilitate the identification of traits and regions for introgression by direct selection of specific alleles. Genomic approaches are also used for diversity analysis, germplasm characterization, and identification of tightly linked markers for important traits. Linkage maps and identification of QTLs for important traits have enabled MAS, which is now commonly used, and is important for simply inherited traits, sometimes several at a time, through pyramiding. GWAS and genomic selection are also poised for broader use. Reference genome sequences are now available in CB, which will help breeders identify genes involved in major traits. Sociological insights related to DLB outcomes include participation of local value chains, from farmers to traders to retailers and consumers.
References
Adam-Blondon AF, Sevignac M, Bannerot MD (1994) SCAR, RAPD and RFLP markers linked to a dominant gene (Are) conferring resistance to anthracnose in common bean. Theor Appl Genet 88:865–870
Albrecht SL, Bennett JM, Boote KJ (1984) Relationship of nitrogenase activity to plant water stress in field-grown soybeans. Field Crop Res 8:61–71
Ambachew D, Mekbib F, Asfaw A, Beebe SE, Blair MW (2015) Trait relations in common bean genotypes grown under managed-stress for drought and field infestation of bean fly. The Crop Journal 3:305–316
Araújo SS, Beebe SE, Crespi M, Delbreil B, González EM, Grube V et al (2015) Abiotic stress responses in legumes: strategies used to cope with environmental challenges. Crit Rev Plant Sci 34(3):237–280. https://doi.org/10.1080/07352689.2014.898450
Ariani A, Teran Y, Berny JC , Gepts P (2016) Genome-wide identification of SNPs and copy number variation in common bean (Phaseolus vulgaris L.) using genotyping-by-sequencing (GBS). Mol Breed 36:87
Arif IA, Bakir MA, Khan AH, Farhan AH, Al Homaidan AA, Bahkali AH, Al Sadoon M, Shbrak M (2010) A brief review of molecular techniques to assess plant diversity. Int J Mol Sci 11:2079–2096
Arumuganathan EK, Earle D (1991) Nuclear DNA content of some important plant species. Plant Mol Biol Report 9:208–218
Asfaw A, Blair MW, Struik PC (2012) Multienvironment quantitative trait loci analysis for photosynthate acquisition, accumulation, and remobilization traits in common bean under drought stress. G3 2:579–595
Asfaw A, Blair MW (2012) Quantitative trait loci for rooting pattern traits of common beans grown under drought stress versus non-stress conditions. Mol Breed 30:681–695
Asfaw A, Blair MW (2014) Quantification of drought tolerance in Ethiopian common bean varieties. Agric Sci 5:124–139
Assefa T, Wu J, Beebe SE, Rao MI, Marcomin D, Rubyogo JC (2015) Improving adaptation to drought stress in small red common bean: phenotypic differences and predicted genotypic effects on grain yield, yield components and harvest index. Euphytica 203:477–489
Assefa T, Beebe SE, Rao IM, Cuasquer JB, Duque E, Myriam C, Rivera M, Battisti A, Lucchin M (2013) Pod harvest index as a selection criterion to improve drought resistance in white pea bean. Field Crop Res 148:24–33
Assefa T, Sperling L, Dagne B, Argaw W, Tessema D, Beebe SE (2014) Participatory plant breeding with traders and farmers for white pea bean in Ethiopia. J Agric Educ Exten 20:497–512
Assefa T, Rao IM, Cannon SB, Wu J, Gutema Z, Blair MW, Paul O, Alemayehu A, Dagne B (2017) Improving adaptation to drought stress in white pea bean (Phaseolus vulgaris L.): genotypic effects on grain yield, yield components and pod harvest index. Plant Breed 136:548–561
Beaver JS, Arnaud-Santana E, Coyne DP (1996) Yield stability of determinate and indeterminate red mottled beans. J Agri University of Puerto Rico 80:187–189
Becerra Velasquez VL, Gepts P (1994) RFLP diversity of common bean (Phaseolus vulgaris) in its centres of origin. Genome 37:256–263
Beebe SE, Corrales M (1991) Breeding for disease resistance. In: Schoonhoven A, Voysest O (eds) Common beans: research for crop improvement. CAB Int, CIAT, Cali Wallingford, pp 561–610
Beebe SE, Skroch PW, Tohme J, Duque MC, Pedraza F, Nienhuis J (2000) Structure of genetic diversity among common bean landraces of middle American origin based on correspondence analysis of RAPD. Crop Sci 40:264–273
Beebe SE, Rojas-Pierce M, Yan X, Blair MW, Pedraza F, Muñoz F, Tohme F, Lynch JP (2006) Quantitative trait loci for root architecture traits correlated with phosphorus acquisition in common bean. Crop Sci 46:413–423
Beebe SE (2012) Common bean breeding in the tropics. Plant Breed Rev 36:357–426
Beebe SE, Rao IM, Blair MW, Acosta-Gallegos JA (2013) Phenotyping common beans for adaptation to drought. Front Physiol doi 4. https://doi.org/10.3389/fphys.2013.00035
Beebe SE, Rao IM, Cajiao C, Grajales M (2008) Selection for drought resistance in common bean also improves yield in phosphorus limited and favorable environments. Crop Sci 48:582–592. https://doi.org/10.2135/cropsci2007.07.0404
Beebe SE, Rao IM, Blair MW, Butare L (2009) Breeding for abiotic stress tolerance. In: common bean: present and future challenges. Proceedings of the 14th Australian plant breeding and 11th SABRAO conference, Brisbane. pp 10–14
Beebe S, Rengifo J, Gaitan E, Duque MC, Tohme J (2001) Diversity and origin of Andean landraces of common bean. Crop Sci 41:854–862
Bellucci E, Elena E, Rau D, Rodriguez M, Biagetti E, Giardini A, Nanni L, Papa R (2013) Genomics of origin, domestication and evolution of Phaseolus vulgaris. In: Tuberosa R, Graner A, Frison E (eds) (springer), pp 483-507
Bennett M, Leitch I (2005) Plant DNA C-Values database
Berhanu A, Kassaye N, Tigist S, Kidane T, Dagmawit T, Rubyogo JC, Mukankusi CM (2018) Ethiop J of Crop Sc 6(1):1–14
Bernardo R, Yu J (2007) Prospects for genomewide selection for quantitative traits in maize. Crop Sci 47:1082–1090
Bernardo R, Charcosset A (2006) Usefulness of gene information in marker-assisted recurrent selection: a simulation appraisal. Crop Sci 614–621. doi: https://doi.org/10.2135/cropsci2005.05-0088
Bezaweletaw K, Belete B, Sripichitt P (2006) Genetic gain in grain yield potential and associated agronomic traits in haricot bean (Phaseolus vulgaris L.). Kasetsart J. (Nat. Sci.) 40: 835–847
Bhakta MS, Jones VA, Vallejos CE (2015) Punctuated distribution of recombination hotspots and demarcation of pericentromeric regions in Phaseolus vulgaris L. PLoS One 10:e0116822
Bhat JA, Salgotra RK, Dar MY (2015) Phenomics: a challenge for crop improvement in genomic era. Mol Plant Breed 6:1–11
Bitocchi E, Bellucci E, Giardini A, Rau D, Rodriguez M, Biagetti E, Santilocchi R, Spagnoletti Zeuli P, Gioia T, Logozzo G, Attene G, Nanni L, Papa R (2013) Molecular analysis of the parallel domestication of the common bean in Mesoamerican and the Andes. New Phytol 197:300–331. https://doi.org/10.1111/j.1469-8137.2012.04377.x
Blair MW, Córdoba JM, Muñoz C, Yuyo DK (2014) BAC-end microsatellites from intra and inter-genic regions of the common bean genome and their correlation with cytogenetic features. PLoS One 9:e101873
Blair MW, Cortés AJ, Farmer AD, Huang W, Ambachew D, Penmetsa RV, Carrasquilla-Garcia N, Assefa T, Cannon SB (2018) Uneven recombination rate and linkage disequilibrium across a reference SNP map for common bean (Phaseolus vulgaris L.). PLoS ONE 13:e0189597. https://doi.org/10.1371/journal.pone.0189597
Blair MW, Cortés A, Penmetsa RV, Carrasquilla-Garcia N, Farmer A, Cook D (2013) Development of high throughput SNP markers for parental polymorphism screening and diversity analysis in common bean (Phaseolus vulgaris L.). Theor Appl Genet 126:535–548. https://doi.org/10.1007/s00122-012-1999-z
Blair MW (2013) Mineral biofortification strategies for food staples: the example of common bean. J Agric Food Chem 61:8287–8294
Blair MW, Galeano CH, Tovar E, Muñoz-Torres MC, Velasco A, Beebe SE, Rao IM (2012) Development of a Mesoamerican intra-genepool genetic map for QTL detection in a drought tolerant x susceptible common bean (Phaseolus vulgaris L.) cross. Mol Breed 29:71–88
Blair MW, Prieto S, Diaz LM, Buendía HF, Cardona C (2010a) Linkage disequilibrium at the APA-Arcelin insecticidal seed storage protein locus of common bean (Phaseolus vulgaris L.). BMC Plant Bio 10:79
Blair MW, Muñoz C, Buendía HF, Flower J, Bueno JM, Cardona C (2010b) Genetic mapping of microsatellite markers around the arcelin bruchid resistance locus in common bean. Theor Appl Genet 121:393–402
Blair MW, López-Marín HD, Rao IM (2009) Identification of aluminum resistant Andean common bean (Phaseolus vulgaris L.) genotypes. Brazil J Plant Physiol 21:291–300
Blair MW, Rodriguez LM, Pedraza F, Morales F, Beebe SE (2007) Genetic mapping of the bean golden yellow mosaic geminivirus resistance gene bgm-1 and linkage with potyvirus resistance in common bean (Phaseolus vulgaris L.). Theor Appl Genet 114:261–271
Blair MW, Giraldo MC, Buendia HF, Tovar E, Duque MC et al (2006a) Microsatellite marker diversity in common bean (Phaseolus vulgaris L). Theor Appl Genet 113:100–109
Blair MW, Muñoz C, Garza R, Cardona C (2006b) Molecular mapping of genes for resistance to the bean pod weevil (Apion godmani Wagner) in common bean. Theor Appl Genet 112:913–923
Blair MW, Pedraza F, Buendia HF, Gaitán-Solís E, Beebe SE, Gepts P, Tohme J (2003) Development of a genome-wide anchored microsatellite map for common bean (Phaseolus vulgaris L.). Theor Appl Genet 107:1362–1374. https://doi.org/10.1007/s00122-003-1398-6
Bliss F (1993) Breeding common bean for improved nitrogen fixation. Plant Soil 15:71–79
Brisco EI (2012) Empoasca leafhopper species resistance in common bean, Phaseolus vulgaris: Field Screening and QTL Identification. Dissertation, Michigan State University, Plant Breeding, Genetics and Biotechnology Program/Crop and Soil Sciences
Briñez B, Perseguini JM, Santa RJ, Bassi D, Ribeiro GG et al (2017) Mapping QTLs for drought tolerance in a SEA 5 x AND 277 common bean cross with SSRs and SNP markers. Genet Mol Biol 40:813–823
Broughton WJ, Hernandez G, Blair MW, Beebe SE, Gepts P, Vanderleyden J (2003) Bean (Phaseolus spp.)—model food legumes. Plant Soil 252:55–128
Buruchara R, Chirwa R, Sperling L, Mukankusi C, Rubyogo JC, Mutonhi R et al (2011) Development and delivery of bean varieties in Africa: the Pan-Africa Bean Research Alliance (PABRA) model. African Crop Sci J 19:227–245
Campa A, Pérez-Vega E, Pascual A, Ferreira JJ (2010) Genetic analysis and molecular mapping of quantitative trait loci in common bean against Pythium ultimum. Phytopathology 100:315–1320
Carneiro FF, Dos Santos JB, Leite ME (2010) Marker-assisted backcrossing using microsatellites and validation of SCAR Phs marker for resistance to white mold in common bean. Electron J Biotechnol doi. https://doi.org/10.2225/vol13
Ceccarelli S, Grando S, Tutwiler R, Baha J, Martini AM, Salahieh H, Goodchild A, Michael M (2000) A methodological study on participatory barley breeding I. Selection phase Euphytica 111:91–104
Chavarro MC, Blair MW (2010) QTL analysis of heat and drought tolerance in an inter-genepool common bean population. Tropical Plant Biol 3:204–218
CIAT 2013 A bean revolution in sub-Saharan Africa. International Center for Tropical Agriculture (CIAT). http://www.ard-europe.org/fileadmin/SITE_ MASTER/content/eiard/documents/Impact_case_studies_2013/CIAT_-_A_ bean_revolution_in_sub-Saharan_Africa.pdf
Cichy KA, Wiesinger JA, Mendoza FA (2015) Genetic diversity and genome-wide association analysis of cooking time in dry bean (Phaseolus vulgaris L.). Theor Appl Genet 128:1555–1567. https://doi.org/10.1007/s00122-015-2531-z
Cichy KA, Blair MW, Galeano CH, Snapp SS, Kelly JD (2009a) QTL analysis of root architecture traits and low phosphorus tolerance in an Andean bean population. Crop Sci 49:59–68
Cichy KA, Snapp SS, Blair MW (2009b) Plant growth habit, root architecture traits and tolerance to low soil phosphorus in an Andean bean population. Euphytica 165:257–268
Cobb JN, DeClerck G, Greenberg A, Clark R, McCouch S (2013) Next-generation phenotyping: requirements and strategies for enhancing our understanding of genotype–phenotype relationships and its relevance to crop improvement. Theor Appl Genet 126:867–887
Córdoba JM, Chavarro C, Rojas F, Muñoz C, Blair BM (2010) Identification and mapping of simple sequence repeat markers from common bean (Phaseolus vulgaris L.) bacterial artificial chromosome end sequences for genome characterization and genetic–physical map integration. Plant Genome 3:154–165. https://doi.org/10.3835/plantgenome2010.06.0013
Cortés AJ, Blair MW (2017) Lessons from common bean on how wild relatives and landraces can make tropical crops more resistant to climate change. In: Grillo O (ed) Rediscovery of landraces as a resource for the future, InTech. ISBN 978-953-51-5806-6
Cortés A, Chavarro C, Blair MW (2011) SNP marker diversity in common bean (Phaseolus vulgaris L.). Theor Appl Genet 123:827–845
Cortés AJ, Chavarro M, Madriñan S, This D, Blair MW (2012a) Molecular ecology and selection of drought related Asr gene polymorphisms in wild and cultivated common bean (Phaseolus vulgaris L.). BMC Genetics 13:58. doi: https://doi.org/10.1186/1471-2156-13-58
Cortés AJ, This D, Chavarro MC, Madriñan S, Blair MW (2012b) Nucleotide diversity patterns at the drought related DREB encoding genes in wild and cultivated common bean (Phaseolus vulgaris L.). Theor Appl Genet 125(5):1069–1085. https://doi.org/10.1007/s00122-012-1896-5
Darkwa K, Ambachew D, Mohammed H, Asfaw A, Blair MW (2016) Evaluation of common bean (Phaseolus vulgaris L.) genotypes for drought stress adaptation in Ethiopia. Crop J 4:367–376
D’Agostino N, Tripodi P (2017) NGS-based genotyping, high-throughput phenotyping and genome-wide association studies laid the foundations for next-generation breeding in horticultural crops. Diversity 9:38
Das G, Patra JK, Baek KH (2017) Insight into MAS: a molecular tool for development of stress resistant and quality of rice through gene stacking. Front Plant Sci 9:985. https://doi.org/10.3389/fpls.2017.00985
De La Fuente M, López-Pedrouso M, Alonso J, Santalla M, De Ron AM, Álvarez G et al (2012) In-depth characterization of the phaseolin protein diversity of common bean (Phaseolus vulgaris L.) based on two-dimensional electrophoresis and mass spectrometry. Food Tech Biotechnol 50:315
De Ron AM, Papa R, Bitocchi E, González AM, Debouck DG, Brick MA et al (2015) Common bean: grain legumes. In: De Ron AM (ed) Handbook of plant breeding. New York, NY; Dordrecht; Heidelberg; London: Springer, pp 1-36
De Ron AM, Rodiño AP, Santalla M, González AM, Lema MJ, Martín I et al (2016) Seedling emergence and phenotypic response of common bean germplasm to different temperatures under controlled conditions and in open field. Front Plant Sci 7:1087. https://doi.org/10.3389/fpls.2016.01087
Dickson MH, Boettger MA (1984) Emergences, growth, and blossoming of bean (Phaseolus vulgaris L.) at suboptimal temperatures. J Am Soc Hortic Sci 109:257–260
Drevon JJ, Alkama N, Bargaz A, Rodi-o AP, Sungthongwises K, Zaman-Allah M (2015) The legume-rhizobia symbiosis: grain legumes. In: De Ron AM (ed) Handbook of plant breeding. New York, NY; Dordrecht; Heidelberg; London: Springer, 267–290
Duc G, Agrama H, Bao S, Berger J, Bourion V, De Ron AM et al (2015) Breeding annual grain legumes for sustainable agriculture: new methods to approach complex traits and target new cultivar ideotypes. Crit Rev Plant Sci 34:381–411. https://doi.org/10.1080/07352689.2014.898469
Farid M, Earl HJ, Pauls KP, Navabi A (2017) Response to selection for improved nitrogen fixation in common bean (Phaseolus vulgaris L.). Euphytica 213:99
Freyre R, Skroch P, Geffroy V, Adam-Blondon AF, Shirmohamadali A et al (1998) Towards an integrated linkage map of common bean. 4. Development of a core linkage map and alignment of RFLP maps. Theor Appl Genet 97:847–856
Fufa F, Grando S, Kafawin O, Shakhatreh Y, Ceccarelli S (2010) Efficiency of farmers' selection in a participatory barley breeding programme in Jordan. Plant Breed 129:156–161
Gaitán-Solís E, Choi IY, Quigley C, Cregan P, Tohme J (2008) Single nucleotide polymorphisms in common bean: their discovery and genotyping using a multiplex detection system. Plant Genome 1:125–134
Gaitán-Solís E, Duque MC, Edwards KJ, Tohme J (2002) Microsatellite repeats in common bean (Phaseolus vulgaris): isolation, characterization, and cross-species amplification in Phaseolus ssp. Crop Sci 42:2128–2136
Galeano CH, Gomez M, Rodriguez LM, Blair MW (2009a) CEL I nuclease digestion for SNP discovery and marker development in common bean (Phaseolus vulgaris L.). Crop Sci 49:381–394. https://doi.org/10.2135/cropsci2008.07.0413
Galeano CH, Fernández AC, Gómez M, Blair MW (2009b) Single strand conformation polymorphism-based SNP and Indel markers for genetic mapping and synteny analysis of common bean (Phaseolus vulgaris L.). BMC Genomics 10:629. doi: https://doi.org/10.1186/1471-2164-10-629
Galeano CH, Cortés AJ, Fernández AC, Soler A, Franco-Herrera N, Makunde G, Vanderleyden J, Blair MW (2012) Gene-based single nucleotide polymorphism markers for genetic and association mapping in common bean. BMC Genet 13:48. https://doi.org/10.1186/1471-2156-13-48
Gaur PM, Samineni S, Krishnamurthy L, Kumar S, Ghanem ME, Beebe S, Rao I, Chaturvedi SK, Basu PS, Nayyar H, Jayalakshmi V, Babbar A, Varshney RK (2015) High temperature tolerance in grain legumes. Legume Perspectives 7:23–24
Garzón LN, Ligarreto GA, Blair MW (2008) Molecular marker-assisted backcrossing of anthracnose resistance into Andean climbing beans (Phaseolus vulgaris L.). Crop Sci 48:562–570. https://doi.org/10.2135/cropsci2007.08.0462
Gaut BS (2014) The complex domestication history of the common bean. Nat Genet 46:663–664
Gepts P, Aragão F, de Barros E, Blair MW, Broughton W, Galasso I, Herna’ndez G et al (2008) Genomics of Phaseolus beans, a major source of dietary protein and micronutrients in the tropics. In: Moore P, Ming R (eds) Genomics of tropical crop plants. Springer, Berlin, pp 113–143
Gepts P, Osborn TC, Rashka K, Bliss FA (1986) Phaseolin-protein variability in wild forms and landraces of the common bean (Phaseolus vulgaris): evidence for multiple centers of domestication. Econ Bot 40:451–468
González A, Yuste-Lisbona F, Rodiño A, Saburido S, Bretones S, De Ron AM, Lozano R, Santalla M (2016) Major contribution of flowering time and vegetative growth to plant production in common bean as deduced from a comparative genetic mapping. Front Plant Sci 7:1940
González AM, Yuste-Lisbona FJ, Fernández-Lozano A, Lozano R, Santalla M (2018) Genetic mapping and QTL analysis in common bean. In: Pérez de la Vega M, Santalla M, Marsolais F (eds) The common bean genome. Springer International Publishing AG 2017, pp 69-107. doi.org/10.1007/978-3-319-63526-2
Gu W, Zhu J, Wallace D, Singh SP, Weeden NF (1998) Analysis of genes controlling photoperiod sensitivity in common bean using DNA markers. Euphytica 102:125–132
Hagerty CH, Cuesta-Marcos A, Cregan P, Song Q, McClean P, Myers JR (2016) Mapping snap bean pod and color traits, in a dry bean × snap bean recombinant inbred population. J J Am Soc Hortic Sci 141(2):131–138
Hanson PM, Pastor-Corrales MA, Kornegay JL (1993) Heritability and sources of Ascochyta blight resistance in common bean. Plant Dis 77:711–711
Heffner EL, Sorrells ME, Jannink JL (2009) Genomic selection for crop improvement. Crop Sci 49:1–12
Hoyos-Villegas V, Song Q, Kelly DJ (2017) Genome-wide association analysis for drought tolerance and associated traits in common bean. Plant Genome 10:1–17. https://doi.org/10.3835/plantgenome2015.12.0122
Hurtado-Gonzales OP, Valentini G, Gilio TS, Martins AM, Song Q, Pastor-Corrales MA (2017) Fine mapping of Ur-3, a historically important rust resistance locus in common bean. G3 7:557-569
Hyten DL, Song Q, Fickus EW, Quigley CV, Lim J, Choi I, Hwang E, Pastor-Corrales M, Cregan PB (2010) High-throughput SNP discovery and assay development in common bean. BMC Genomics 11:475–482
Ibarra-Perez FJ, Ehdaie B, Waines JG (1997) Estimation of outcrossing rate in common bean. Crop Sci 37:60–65
Johnson WC, Guzmán P, Mandala D, Mkandawire AC, Temple S, Gilbertson RL et al (1997) Molecular tagging of the bc-3 gene for introgression into Andean common bean. Crop Sci 37:248–254
Jung G, Ariyarathne HM, Coyne DP, Nienhuis J (2003) Mapping QTL for bacterial brown spot resistance under natural infection in field and seedling stem inoculation in growth chamber in common bean. Crop Sci 43:350–357
Kamfwa K, Mwala M, Okori P, Gibson P, Mukankusi C (2013) Identification of QTL for Fusarium root rot resistance in common bean. J crop improv 27:406–418
Kamfwa K, Cichy KA, Kelly JD (2015a) Genome-wide association study of agronomic traits in common bean. Plant Genome 8:1–12
Kamfwa K, Cichy K, Kelly JD (2015b) Genome-wide association analysis of symbiotic nitrogen fixation in common bean. Theor Appl Genet 128:1999–2017
Kanbar A, Shashidhar HE (2010) Participatory selection assisted by DNA markers for enhanced drought resistance and productivity in rice (Oryza sativa L.). Euphytica 178:137–150
Katungi E, Farrow A, Chianu J, Sperling L, Beebe SE (2009) Common bean in eastern and southern Africa: a situation and outlook analysis. International Centre for Tropical Agriculture 61
Keller B, Manzanares C, Jara C, Lobaton JD, Studer B, Raatz B (2015) Fine-mapping of a major QTL controlling angular leaf spot resistance in common bean (Phaseolus vulgaris L.). Theor Appl Genet 128(5):813–826
Kelly JD, Miklas PN (1998) The role of RAPD markers in breeding for disease resistance in common bean. Mol Breed 4:1–11
Kelly JD, Vallejo VA (2004) A comprehensive review of the major genes conditioning resistance to anthracnose in common bean. Hort Sci 39:1196–1207
Kelly JD (2004) Advances in common bean improvement: some case histories with broader applications. Acta Hortic 637:99–122
Khairallah MM, Adams MW, Sears BB (1990) Mitochondrial DNA polymorphisms of Malawian bean lines: further evidence for two major gene pools. Theor Appl Genet 80:753–761
Khairallah MM, Sears BB, Adams MW (1992) Mitochondrial restriction fragment length polymorphism in wild Phaseolus vulgaris L: insights of the domestication of common bean. Theor Appl Genet 84:915–922
Koenig R, Gepts P (1989) Allozyme diversity in wild Phaseolus vulgaris: further evidence for two major centers of diversity. Theor Appl Genet 78:809–817
Koinange E, Singh S, Gepts P (1996) Genetic control of the domestication syndrome in common bean. Crop Sci 36:1037–1045
Kumar J, Pratap A, Kumar S (2015). Plant phenomics: an overview: Phenomics in crop plants. In: eds Kumar J, Pratap a, Kumar S trends, options and limitations. Pp 1-10
Kwak M, Velasco D, Gepts P (2008) Mapping homologous sequences for determinacy and photoperiod sensitivity in common bean (Phaseolus vulgaris). J Hered 99:283–291
Liao H, Yan X, Rubio G, Beebe SE, Blair MW, Lynch JP (2004) Genetic mapping of basal root gravitropism and phosphorus acquisition efficiency in common bean. Funct Plant Biol 31:959–970
López-Marín HD, Rao IM, Blair MW (2009) Quantitative trait loci for root morphology traits under aluminum stress in common bean (Phaseolus vulgaris L) Theor Appl Genet 119:449–458
Lynch JP (2011) Root phenes for enhanced soil exploration and phosphorus acquisition: tools for future crops. Plant Physiol 156:1041–1049. https://doi.org/10.1104/pp.111.175414
Larochelle C., Alwang J., Norton G.W., Katungi E., and Labarta R. A. 2013. Ex post impact of adopting improved bean varieties on poverty and food security in Uganda and Rwanda. Diffusion and impact of improved varieties in Africa (DIIVA)—Objective 3 Report. Virginia Tech: Blacksburg, VA and CIAT: Cali, Colombia
McClean PE, Mamidi S, McConnell M, Chikara S, Lee R (2010) Synteny mapping between common bean and soybean reveals extensive blocks of shared loci. BMC Genomics 11:184
McClean PE, Matt L, Gepts P, Jackson SA (2008) Phaseolus vulgaris: a diploid model for soybean. In: eds Stacey G Genetics and genomics of soybean. pp 55–76
McClean P, Kami J, Gepts P (2004) Genomics and genetic diversity in common bean. Legume Crop Genomics 4:60–82
McClean PE, Lee RK, Otto C, Gepts P, Bassett MJ (2002) Molecular and phenotypic mapping of genes controlling seed coat pattern and color in common bean (Phaseolus vulgaris L.). J Hered 93:148–152. https://doi.org/10.1093/jhered/93.2.148
McDonald LS, Panozzo JF, Salisbury PA, Ford R (2016) Discriminant analysis of defective and non-defective field pea (Pisum sativum L.) into broad market grades based on digital image features. PloS ONE 11: e0155523
Meyer DW, Badaruddin M (2001) Frost tolerance of ten seedling legume species at four growth stages. Crop Sci 41:1838–1842. https://doi.org/10.2135/cropsci2001.1838
Miklas PN, Pastor-Corrales MA, Jung G, Coyne DP, Kelly JD, McClean PE, Gepts P (2002) Comprehensive linkage map of bean rust resistance genes. Ann Rep Bean Improv Coop 45:125–129
Miklas PN, Kelly JD, Beebe SE, Blair WM (2006) Common bean breeding for resistance against biotic and abiotic stresses: from classical to MAS breeding. Euphytica 147:106–131
Miklas PN, Fourie D, Trapp J, Larsen RC, Chavarro C, Blair MW, Gepts P (2011) Genetic characterization and molecular mapping gene for resistance to halo blight in common bean. Crop Sci 51:2439–2448
Miklas PN, Fourie D, Chaves B, Chirembe C (2017) Common bacterial blight resistance QTL BC420 and SU91 effect on seed yield, seed weight, and canning quality in dry bean. Crop Sci 57:802–811
Mir RR, Zaman-Allah M, Sreenivasulu N, Trethowan R, Varshney RK (2012) Integrated genomics, physiology and breeding approaches for improving drought tolerance in crops. Theor Appl Genet 125:625–645. https://doi.org/10.1007/s00122-012-1904-9
Miranda BD, Bliss FA (1991) Selection for increased seed nitrogen accumulation in common bean: implications for improving dinitrogen fixation and seed yield. Plant Breed 106:301–311
Moghaddam SM, Song Q, Mamidi S, Schmutz J, Lee R, Cregan P, Osorno JM, McClean PE (2014) Developing market class specific InDel markers from next generation sequence data in Phaseolus vulgaris L. Front Plant Sci 5:185. https://doi.org/10.3389/fpls.2014.00185
Montoya CA, Beaver JS, Rodriguez R, Miklas PN, Godoy-Lutz G (1997) Heritability of resistance to web blight in five common bean populations. Crop Sci 37:780–783
Muedi HH, Fourie D, McLaren NW (2015) Distribution and severity of bacterial brown spot on dry beans in South Africa: an update. South African J Sci 111:1–6
Mukeshimana G, Butare L, Cregan PB, Blair MW, Kelly JD (2014) Quantitative trait loci associated with drought tolerance in common bean. Crop Sci 54:923–938. https://doi.org/10.2135/cropsci2013.06.0427
Murray JD, Michaels TE, Cardona C, Schaafsma AW, Pauls KP (2004) Quantitative trait loci for leafhopper (Empoasca fabae and Empoasca kraemeri) resistance and seed weight in the common bean. Plant Breed 123:474–479
Nielsen KL, Eshel A, Lynch JP (2001) The effect of phosphorus availability on the carbon economy of contrasting common bean (Phaseolus vulgaris L.) genotypes. J Exp Bot 52:329–339
Nodari RO, Tsai SM, Gilbertson RL, Gepts P (1993) Towards an integrated linkage map of common bean. 2. Development of an RFLP-based linkage map. Theor Appl Genet 85:513–520. https://doi.org/10.1007/BF00220907
Oblessuc PR, Baroni RM, Garcia AF, Chioratto AF, Carbonell SM, Camargo LA et al (2012) Mapping of angular leaf spot resistance QTL in common bean (Phaseolus vulgaris L.) under different environments. BMC Genet 13:50
Oblessuc PR, Matiolli CC, Chiorato AF, Camargo LA, Benchimol-Reis LL, Melotto M (2015) Common bean reaction to angular leaf spot comprises transcriptional modulation of genes in the ALS10.1 QTL. Front Plant Sci 6:152
O’Boyle PD, Kelly JD, Kirk WW (2007) Use of marker-assisted selection to breed for resistance to common bacterial blight in common bean. J Amer Soc Hort Sci 132:381–386
Ochoa IE, Blair MW, Lynch JP (2006) QTL analysis of adventitious root formation in common bean (Phaseolus vulgaris L.) under contrasting phosphorus availability. Crop Sci 46:1609–1621
PABRA (2017) Annual report 2016/2017—20th Anniversary. Special Edition. PABRA–Nairobi Kenya
Papa R, Gepts P (2003) Asymmetry of gene flow and differential geographical structure of molecular diversity in wild and domesticated common bean (Phaseolus vulgaris L.) from Mesoamerica. Theor Appl Genet 106:239–250
Perseguini JM, Oblessuc PR, Rosa JR, Gomes KA, Chiorato AF, Carbonell SM, Garcia AF, Vianello RP, Benchimol -Reis L (2016) Genome-wide association studies of anthracnose and angular leaf spot resistance in common bean (Phaseolus vulgaris L.). PLoS One doi: https://doi.org/10.1371/journal.pone.0150506, 2016
Persley GJ, Anthony VM (2017) The business of plant breeding: market-led approaches to new variety design in Africa. CABI Wallingford, Oxon. UK www.cabi.org
Polania J, Rao IM, Cajiao C, Rivera M, Raatz B, Beebe S (2016a) Physiological traits associated with drought resistance in Andean and Mesoamerican genotypes of common bean (Phaseolus vulgaris L.). Euphytica 210:17–29. https://doi.org/10.1007/s10681-016-1691-5
Polania J, Poschenrieder C, Rao IM, Beebe SE (2016b) Estimation of phenotypic variability in symbiotic nitrogen fixation ability of common bean under drought stress using 15N natural abundance in grain. European J Agron 79:66–73. https://doi.org/10.1016/j.eja.2016.05.014
Polania J, Rao IM, Cajiao C, Grajales M, Rivera M, Velasquez F, Raatz B, Beebe SE (2017) Shoot and root traits contribute to drought resistance in recombinant inbred lines of MD 23–24 × SEA 5 of common bean. Front Plant Sci 8(296):1–18. https://doi.org/10.3389/fpls.2017.00296
Porch TG (2006) Application of stress indices for heat tolerance screening of common bean. Journal of Agron Crop Sci 192:390–394
Porch TG, Beaver JS, Debouck DG, Jackson SA, Kelly JD, Dempewolf H (2013) Use of wild relatives and closely related species to adapt common bean to climate change. Agronomy 3(2):433–461
Porch TG, Jahn M (2001) Effects of high-temperature stress on microsporogenesis in heat-sensitive and heat-tolerant genotypes of Phaseolus vulgaris. Plant Cell Env 24:723–731. https://doi.org/10.1046/j.1365-3040.2001.00716.x
Rafalski AJ (2002) Novel genetic mapping tools in plants. Plant Sci 162:329–333
Rafalski AJ (2010) Association genetics in crop improvement. Curr Opin Plant Biol 13:174–180. doi: 1016/j.pbi.2009.12.004
Rainey KM, Griffiths PD (2005) Differential response of common bean genotypes to high temperature. J Amer Soc Hort Sci 130:18–23
Ramaekers L, Remans R, Rao IM, Blair MW, Vanderleyden J (2010) Strategies for improving phosphorus uptake efficiency of crop plants. Field Crops Res 117:169–176
Rami ME, Bouchet S, Klein P, Klein R et al (2009) A consensus genetic map of sorghum that integrates multiple component maps and high-throughput Diversity Array Technology (DArT) markers. BMC Plant Biol doi 9:13. https://doi.org/10.1186/1471-2229-9-13
Rao IM (2014) Advances in improving adaptation of common bean and Brachiaria forage grasses to abiotic stress in the tropics. In: Pessarakli M (ed) Handbook of plant and crop physiology. FL pp, Boca Raton, pp 847–889
Rao IM, Miles JW, Beebe SE, Horst WJ (2016) Root adaptations to soils with low fertility and aluminium toxicity. Ann Bot 118:593–605. https://doi.org/10.1093/aob/mcw073
Rao IM (2001) Role of physiology in improving crop adaptation to abiotic stresses in the tropics: the case of common bean and tropical forages. In: Pessarakli M (ed) Handbook of plant and crop physiology. Marcel Dekker Inc., New York, pp 583–613
Rao IM, Beebe SE, Polani J, Ricaurte J, Cajiao C, Garcia RM (2013) Can tepary bean be a model for improvement of drought resistance in common bean. Afr Crop Sci J 85:203–211
Rao IM, Beebe SE, Polania J, Grajales M, Cajiao C, Ricaurte J, Garcia R, Rivera M (2017) Evidence for genotypic differences among elite lines of common bean in their ability to remobilize photosynthate to increase yield under drought. J Agric Sci 155:857–875. https://doi.org/10.1017/S0021859616000915
Ribaut JM, De Vicente MC, Delannay X (2010) Molecular breeding in developing countries: challenges and perspectives. Curr Opin Plant Biol 13:213–218
Ribeiro ND, Filho AC, Poersch NL, Jost E, Rosa SS (2008) Genetic progress in traits of yield, phenology and morphology of common bean. Crop Breed & Appl Biotech 8:232–238
Rodino AP, Lema EM, Marlene PB, Santalla M, De Ron AM (2007) Assessment of runner bean (Phaseolus coccineus L.) germplasm for tolerance to low temperature during early seedling growth. Euphytica 155:63–70. https://doi.org/10.1007/s10681-006-9301-6
Román-Avilés B, Kelly JD (2005) Identification of quantitative trait loci conditioning resistance to Fusarium root rot in common bean. Crop Sci 45:1881–1890
Rubyogo JC, Sperling L, Muthoni R. Buruchara R. (2010) Bean seed delivery for small farmers in sub-Saharan Africa: the power of partnerships. Society and Natural Resources 23(4)285–302
Rubyogo JC, Sperling L, Teshale A (2007) A new approach for facilitating farmers’ access to bean seed. LEISA Magazine 23(2):2007
Santalla M, Rodiño A, De Ron A (2002) Allozyme evidence supporting southwestern Europe as a secondary center of genetic diversity for common bean. Theor Appl Genet 104:934–944
Schmutz J, McClean PE, Mamidi S, Wu GA, Cannon SB, Grimwood J et al (2014) A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet 46,707–713. 46,707–713. doi: https://doi.org/10.1038/ng.3008
Schneider KA, Rosales-Serna R, Ibarra-Perez F, Cazares-Enriquez B, Acosta-Gallegos JA, Ramirez-Vallejo P, Wassimi N, Kelly JD (1997) Improving common bean performance under drought stress. Crop Sci 37:43–50. https://doi.org/10.2135/cropsci1997.0011183X003700010007x
Schröder S, Mamidi S, Lee R, McKain MR, McClean PE, Osorno JM (2016) Optimization of genotyping by sequencing (GBS) data in common bean (Phaseolus vulgaris L.). Molecular breed 36:1–9
Singh SP, Nodari R, Gepts P (1991a) Genetic diversity in cultivated common bean. 1. Allozymes. Crop Sci 31:19–23
Singh SP, Gepts P, Debouck DG (1991b) Races of common bean (Phaseolus vulgaris, Fabaceae). Econ Bot 45:79–396
Singh SP (2001) Broadening the Genetic Base of common bean cultivars: a review. Crop Sci 41:1659–1675
Singh SP (1991) Breeding for seed yield. In: Schoonhoven A, Voysest O (eds) Common beans: research for crop improvement. CAB Int, CIAT, Cali, Wallingford, pp 383–443
Singh SP (1992) Common bean improvement in the tropics. Plant Breed Rev 10:199–269
Snoeck C, Vanderleyden J, Beebe SE (2010) Strategies for genetic improvement of common bean and rhizobia towards efficient interactions. Plant Breed Rev pp 21–72
Song Q, Jia G, Hyten DL, Jenkins J, Hwang EY, Schroeder SG et al (2015) SNP assay development for linkage map construction, anchoring whole-genome sequence, and other genetic and genomic applications in common bean. G3 5: 2285-2290
Souter JR, Gurusamy V, Porch TG, Bett KE (2017) Successful introgression of abiotic stress tolerance from wild tepary bean to common bean. Crop Sci 57:1–12
Steele KA, Virk DS, Prasad SC, Kumar R, Singh DN, Gangwar JS, Witcombe JR (2004) Combining PPB and marker-assisted selection: strategies and experiences with rice. Crop Sci 44:1560–1571
Strausbaugh CA, Myers JR, Forster RL, McClean PE (1999) Bc-1 and Bc-u-two loci controlling bean common mosaic virus resistance in common bean are linked. J Amer Soc Hort Sci 124:644–648
Takegami JC, Beaver JS, Godoy-Lutz G, Echávez-Badel R, Steadman JR (2004) Inheritance of web blight resistance in common bean. J Agri University of Puerto Rico 88:45–54
Tock AJ, Fourie D, Walley PG, Holub EB, Soler A, Cichy KA, Pastor-Corrales MA, Song Q, Porch TG, Hart JP, Vasconcellos RCC, Vicente JG, Barker GC, Miklas PN (2017) Genome-wide linkage and association mapping of halo blight resistance in common bean to race 6 of the globally important bacterial pathogen. Front Plant Sci 8:1170
Trapp JJ, Urrea CA, Cregan PB, Miklas PN (2015) Quantitative trait loci for yield under multiple stress and drought conditions in a dry bean population. Crop Sci 55:1596–1607
Tryphone GM, Chilagane LA, Protas D, Kusolwa PM, Nchimbi-Msolla S, Tryphone GM et al (2013) Marker assisted selection for common bean diseases improvements in Tanzania: prospects and future needs. In: Anderson SB (ed) Plant breeding from laboratories to fields. https://doi.org/10.5772/52823
Vallejos CE, Sakiyama NS, Chase CD (1992) A molecular marker-based linkage map of Phaseolus vulgaris L. Genetics 131:733–740
Varshney RK, Glaszmann JC, Leung HRJ (2010) More genomic resources for less-studied crops. Trends Biotechnology 28:452–460. https://doi.org/10.1016/j.tibtech.2010.06.007
Varshney RK, Thudi M, Pandey MK, Tardieu F, Ojiewo C, Vadez V, Whitbread AM, Siddique KM, Nguyen HT, Carberry PS, Bergvinson D (2018) Accelerating genetic gains in legumes for the development of prosperous smallholder agriculture: integrating genomics, phenotyping, systems modelling and agronomy. J Exp Bot 69:3293–3312. https://doi.org/10.1093/jxb/ery088
Viteri DM, Cregan PB, Trapp JJ, Miklas P, Singh SP (2014) A new common bacterial blight resistance QTL in VAX 1 common bean and interaction of the new QTL, SAP6, and SU91 with bacterial strains. Crop Sci 54:1598–1608
Vlasova A, Capella-Gutiérrez S, Rendón-Anaya M, Hernández-Oñate M, Minoche AE, Erb I, Câmara F, Prieto-Barja P, Corvelo A, Sanseverino W, Westergaard G, Dohm JC, Pappas GJ, Saburido-Alvarez S, Kedra D, Gonzalez I, Cozzuto L, Gómez-Garrido J, Aguilar-Morón MA, Andreu N, Aguilar OM, Garcia-Mas J, Zehnsdorf M, Vázquez MP, Delgado-Salinas A, Delaye L, Lowy E, Mentaberry A, Vianello-Brondani RP, García JL, Alioto T, Sánchez F, Himmelbauer H, Santalla M, Notredame C, Gabaldón T, Herrera-Estrella A, Guigó R (2016) Genome and transcriptome analysis of the Mesoamerican common bean and the role of gene duplications in establishing tissue and temporal specialization of genes. Genome Biol 17:32
Voysest O, Dessert M (1991) Bean cultivars: classes and commercial: In: Schoonhoven A, Voysest O (eds) Common beans: research for crop improvement. CAB Int, CIAT, Cali, Wallingford, pp 119–159
Wortmann CS, Kirkby RA, Eledu CA, Allen DJ (1998) Atlas of common bean (Phaseolus vulgaris L.) production in Africa. CIAT Pan African Bean Research Alliance, CIAT publication No. 297, Cali, p 131
Xu Y, Crouch JH (2008) Marker-assisted selection in plant breeding: from publications to practice. Crop Sci 48:391–407
Yang Z, Rao IM, Horst WJ (2013) Interaction of aluminium and drought stress on root growth and crop yield on acid soils. Plant Soil 372:3–25. https://doi.org/10.1007/s11104-012-1580-1
Yu K, Park SJ, Poysa V, Gepts P (2000) Integration of simple sequence repeat (SSR) markers into a molecular linkage map of common bean (Phaseolus vulgaris L.). Heredity 91:429–434
Yu K, Park JS, Poysa V (1999) Abundance and variation of microsatellite DNA sequences in beans (Phaseolus and Vigna). Genome 42:27–34
Yuste-Lisbona F, González A, Capel C, García-Alcázar M, Capel J, De Ron AM, Santalla M, Lozano R (2014) Genetic variation underlying pod size and color traits of common bean depends on quantitative trait loci with epistatic effects. Mol Breed 33:939–952
Zizumbo-Villarreal D, Colunga-GarcíaMarín P, Payró de la Cruz E, Delgado-Valerio PG (2005) Population structure and evolutionary dynamics of wild–weedy–domesticated complexes of common bean in a Mesoamerican region. Crop Sci 35:1073–1083
Zuiderveen GH, Padder BA, Kamfwa K, Song Q, Kelly JD (2016) Genome-wide association study of anthracnose resistance in Andean beans (Phaseolus vulgaris). PLoS One 11:e0156391. https://doi.org/10.1371/journal.pone.0156391
Acknowledgments
This review was supported in part by an appointment to the Agricultural Research Service (ARS) Postdoctoral Research Program administered by the Oak Ridge Institute for Science and Education (ORISE) through an interagency agreement between the U.S. Department of Energy (DOE). This research was funded by the United States Department of Agriculture Agricultural Research Service (USDA-ARS) project 5030-21000-062-00D, the Iowa State University Department of Agronomy, and the Home Economics Agricultural Experiment Station, Iowa State University. The USDA is an equal opportunity provider and employer.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Assefa, T., Assibi Mahama, A., Brown, A.V. et al. A review of breeding objectives, genomic resources, and marker-assisted methods in common bean (Phaseolus vulgaris L.). Mol Breeding 39, 20 (2019). https://doi.org/10.1007/s11032-018-0920-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-018-0920-0