
Abstract
Recently, there has been a significant increase in the use of deep learning techniques in the molecular sciences, which have shown high performance on datasets and the ability to generalize across data. However, no model has achieved perfect performance in solving all problems, and the pros and cons of each approach remain unclear to those new to the field. Therefore, this paper aims to review deep learning algorithms that have been applied to solve molecular challenges in computational chemistry. We proposed a comprehensive categorization that encompasses two primary approaches; conventional deep learning and geometric deep learning models. This classification takes into account the distinct techniques employed by the algorithms within each approach. We present an up-to-date analysis of these algorithms, emphasizing their key features and open issues. This includes details of input descriptors, datasets used, open-source code availability, task solutions, and actual research applications, focusing on general applications rather than specific ones such as drug discovery. Furthermore, our report discusses trends and future directions in molecular algorithm design, including the input descriptors used for each deep learning model, GPU usage, training and forward processing time, model parameters, the most commonly used datasets, libraries, and optimization schemes. This information aids in identifying the most suitable algorithms for a given task. It also serves as a reference for the datasets and input data frequently used for each algorithm technique. In addition, it provides insights into the benefits and open issues of each technique, and supports the development of novel computational chemistry systems.









Similar content being viewed by others
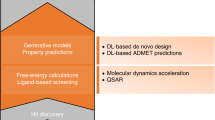

References
Zahlan A, Ranjan RP, Hayes D (2023) Artificial intelligence innovation in healthcare: literature review, exploratory analysis, and future research. Technol Soc 74:102321. https://doi.org/10.1016/j.techsoc.2023.102321
Srivastava S, Tyagi AK, Sajidha SA (2023) Chapter 3-artificial intelligence in healthcare: current situation and future possibilities. Comput Intell Med Int Things (MIoT) Appl 14:55–75. https://doi.org/10.1016/B978-0-323-99421-7.00015-5
Yazici İ, Shayea I, Din J (2023) A survey of applications of artificial intelligence and machine learning in future mobile networks-enabled systems. Eng Sci Technol Int J 44:101455. https://doi.org/10.1016/j.jestch.2023.101455
Koroteev D, Tekic Z (2021) Artificial intelligence in oil and gas upstream: trends, challenges, and scenarios for the future. Energy AI 3:100041. https://doi.org/10.1016/j.egyai.2020.100041
Zhou L, Shi X, Bao Y et al (2023) Explainable artificial intelligence for digital finance and consumption upgrading. Financ Res Lett 58:104489. https://doi.org/10.1016/j.frl.2023.104489
Gong Y (2021) Application of virtual reality teaching method and artificial intelligence technology in digital media art creation. Ecol Inform 63:101304. https://doi.org/10.1016/j.ecoinf.2021.101304
Obulesu O, Mahendra M, Thrilokreddy M (2018) Machine learning techniques and tools: a survey. Proc Int Conf Invent Res Comput Appl ICIRCA 2018:605–611. https://doi.org/10.1109/ICIRCA.2018.8597302
Goodfellow I, Bengio Y, Courville A (2016) Deep learning. MIT Press. http://www.deeplearningbook.org
Krizhevsky A, Sutskever I, Hinton GE (2017) ImageNet classification with deep convolutional neural networks. Commun ACM 60:84–90. https://doi.org/10.1145/3065386
Simonyan K, Zisserman A (2014) Very deep convolutional networks for large-scale image recognition. Am J Health-Syst Pharm 75:398–406. https://arxiv.org/abs/1409.1556
Szegedy C, Liu W, Jia Y et al (2015) Going deeper with convolutions. In: Conference on computer vision and pattern recognition (CVPR), IEEE, pp 1–9. https://doi.org/10.1109/CVPR.2015.7298594
He K, Zhang X, Ren S, Sun J (2016) Deep residual learning for image recognition. In: Conference on computer vision and pattern recognition (CVPR), IEEE, pp 770–778. https://doi.org/10.1109/CVPR.2016.90
Mehrish A, Majumder N, Bharadwaj R et al (2023) A review of deep learning techniques for speech processing. Inform Fusion 99:1566–2535. https://doi.org/10.1016/j.inffus.2023.101869
Wu Z, Pan S, Chen F et al (2021) A comprehensive survey on graph neural networks. IEEE Trans Neural Netw Learn Syst 32:4–24. https://doi.org/10.1109/TNNLS.2020.2978386
Bronstein MM, Bruna J, LeCun Y et al (2017) Geometric deep learning: going beyond euclidean data. IEEE Signal Process Mag 34:18–42. https://doi.org/10.1109/MSP.2017.2693418
Minkin VI (1999) Glossary of terms used in theoretical organic chemistry. Pure Appl Chem 71:1919–1981. https://doi.org/10.1351/pac199971101919
Nash JA, Mostafanejad M, Crawford TD, McDonald AR (2022) MolSSI education: empowering the next generation of computational molecular scientists. Comput Sci Eng 24:72–76. https://doi.org/10.1109/mcse.2022.3165607
Chan HCS, Shan H, Dahoun T et al (2019) Advancing drug discovery via artificial intelligence. Trends Pharmacol Sci 40:592–604. https://doi.org/10.1016/j.tips.2019.06.004
Pedrycz W, Chen S-M (2020) Deep learning: concepts and architectures. Stud Comput Intell. https://doi.org/10.1007/978-3-030-31756-0
Pattanayak S (2023) Introduction to deep-learning concepts and tensorflow. Pro Deep Learn TensorFlow 20:109–197. https://doi.org/10.1007/978-1-4842-8931-0_2
Alzubaidi L, Zhang J, Humaidi AJ et al (2021) Review of deep learning: concepts, CNN architectures, challenges, applications, future directions. J Big Data 8:1–74. https://doi.org/10.1186/S40537-021-00444-8
Askr H, Elgeldawi E, Aboul Ella H et al (2023) Deep learning in drug discovery: an integrative review and future challenges. Artif Intell Rev 56:5975–6037. https://doi.org/10.1007/s10462-022-10306-1
Stephenson N, Shane E, Chase J et al (2019) Survey of machine learning techniques in drug discovery. Curr Drug Metab 20:185–193. https://doi.org/10.2174/1389200219666180820112457
Melo MCR, Maasch JRMA, de la Fuente-Nunez C (2021) Accelerating antibiotic discovery through artificial intelligence. Commun Biol 4:1050. https://doi.org/10.1038/s42003-021-02586-0
Pastur-Romay LA, Cedrón F, Pazos A, Porto-Pazos AB (2016) Deep artificial neural networks and neuromorphic chips for big data analysis: pharmaceutical and bioinformatics applications. Int J Mol Sci 17:1313. https://doi.org/10.3390/ijms17081313
Elton DC, Boukouvalas Z, Fuge MD, Chung PW (2019) Deep learning for molecular design—a review of the state of the art. Mol Syst Des Eng 4:828–849. https://doi.org/10.1039/C9ME00039A
Dara S, Dhamercherla S, Jadav SS et al (2022) Machine learning in drug discovery: a review. Artif Intell Rev 55:1947–1999. https://doi.org/10.1007/s10462-021-10058-4
Mercado R, Rastemo T, Lindelöf E et al (2021) Graph networks for molecular design. Mach Learn Sci Technol 2:25023. https://doi.org/10.1088/2632-2153/abcf91
Joshi RP, Kumar N (2021) Artificial intelligence based autonomous molecular design for medical therapeutic: a perspective. https://arxiv.org/abs/2102.06045v1
Xu Y, Lin K, Wang S et al (2019) Deep learning for molecular generation. Future Med Chem 11:567–597. https://doi.org/10.4155/fmc-2018-0358
Zhou J, Cui G, Hu S et al (2020) Graph neural networks: a review of methods and applications. AI Open 1:57–81. https://doi.org/10.1016/j.aiopen.2021.01.001
Han J, Rong Y, Xu T, Huang W (2022) Geometrically equivariant graph neural networks: a survey. https://arxiv.org/abs/2202.07230v3
Lee JB, Rossi RA, Kim S et al (2019) Attention models in graphs. ACM Trans Knowl Discov Data 13:1–25. https://doi.org/10.1145/3363574
Neapolitan RE (2018) Neural networks and deep learning. Artificial intelligence. Sterling Publishing Co., Inc., New York, pp 389–411
Qian N, Sejnowski TJ (1988) Predicting the secondary structure of globular proteins using neural network models. J Mol Biol 202:865–884. https://doi.org/10.1016/0022-2836(88)90564-5
Lydia A, Francis S (2019) A survey of optimization techniques for deep learning networks. Int J Res Eng Appl Manag (IJREAM) 5:2
Yang Z, Zeng X, Zhao Y, Chen R (2023) AlphaFold2 and its applications in the fields of biology and medicine. Signal Transduct Target Ther 8:115. https://doi.org/10.1038/s41392-023-01381-z
Baek M, DiMaio F, Anishchenko I et al (1979) (2021) Accurate prediction of protein structures and interactions using a three-track neural network. Science 373:871–876. https://doi.org/10.1126/science.abj8754
Kim J, Park S, Min D, Kim W (2021) Comprehensive survey of recent drug discovery using deep learning. Int J Mol Sci 22:9983. https://doi.org/10.3390/ijms22189983
Xiong J, Xiong Z, Chen K et al (2021) Graph neural networks for automated de novo drug design. Drug Discov Today 26:1382–1393. https://doi.org/10.1016/j.drudis.2021.02.011
Ion A, Gosav S, Praisler M (2019) Artificial neural networks designed to identify NBOMe hallucinogens based on the most sensitive molecular descriptors. In: 2019 6th international symposium on electrical and electronics engineering (ISEEE). IEEE, pp 1–6
Gamidi RK, Rasmuson ÅC (2020) Analysis and artificial neural network prediction of melting properties and ideal mole fraction solubility of cocrystals. Cryst Growth Des 20:5745–5759. https://doi.org/10.1021/acs.cgd.0c00182
Bhattacharya D, Patra TK (2021) dPOLY: deep learning of polymer phases and phase transition. Macromolecules 54:3065–3074. https://doi.org/10.1021/acs.macromol.0c02655
Uzma MU, Halim Z (2023) Protein encoder: An autoencoder-based ensemble feature selection scheme to predict protein secondary structure. Expert Syst Appl 213:119081. https://doi.org/10.1016/j.eswa.2022.119081
Misiunas K, Ermann N, Keyser UF (2018) QuipuNet: convolutional neural network for single-molecule nanopore sensing. Nano Lett 18:4040–4045. https://doi.org/10.1021/acs.nanolett.8b01709
Goh GB, Siegel C, Vishnu A, Hodas N (2018) Using rule-based labels for weak supervised learning. In: Proceedings of the 24th ACM SIGKDD international conference on knowledge discovery & data mining. ACM, New York. pp 302–310
Shi T, Yang Y, Huang S et al (2019) Molecular image-based convolutional neural network for the prediction of ADMET properties. Chemom Intell Lab Syst 194:1–9. https://doi.org/10.1016/j.chemolab.2019.103853
Sharma A, Kumar R, Ranjta S, Varadwaj PK (2021) SMILES to smell: decoding the structure–odor relationship of chemical compounds using the deep neural network approach. J Chem Inf Model 61:676–688. https://doi.org/10.1021/acs.jcim.0c01288
Chollet F (2017) Xception: Deep learning with depthwise separable convolutions. In: 2017 IEEE conference on computer vision and pattern recognition (CVPR). IEEE, pp 1800–1807
Li C, Wang J, Niu Z et al (2021) A spatial-temporal gated attention module for molecular property prediction based on molecular geometry. Brief Bioinform 22:1–11. https://doi.org/10.1093/bib/bbab078
Bjerrum EJ, Threlfall R (2017) Molecular generation with recurrent neural networks (RNNs). arXiv preprint arXiv:170504612. https://doi.org/10.48550/arXiv.1705.04612
Zhumagambetov R, Molnár F, Peshkov VA, Fazli S (2021) Transmol: repurposing a language model for molecular generation. RSC Adv 11:25921–25932. https://doi.org/10.1039/D1RA03086H
Bagal V, Aggarwal R, Vinod PK, Priyakumar UD (2021) LigGPT: molecular generation using a transformer-decoder model. J Chem Inf Model 62:2064–2076
Jiang J, Zhang R, Ma J et al (2023) TranGRU: focusing on both the local and global information of molecules for molecular property prediction. Appl Intell 53:15246–15260. https://doi.org/10.1007/s10489-022-04280-y
Liu Y, Zhang R, Li T et al (2023) MolRoPE-BERT: An enhanced molecular representation with Rotary Position Embedding for molecular property prediction. J Mol Graph Model 118:108344. https://doi.org/10.1016/j.jmgm.2022.108344
Karim A, Singh J, Mishra A et al (2019) Toxicity prediction by multimodal deep learning. In: Ohara K, Bai Q (eds) Knowledge management and acquisition for intelligent systems. Springer, Cham, pp 142–152
Guo Z, Sharma PK, Du L, Abraham R (2021) MM-Deacon: multimodal molecular domain embedding analysis via contrastive learning. bioRxiv. https://doi.org/10.1101/2021.09.17.460864
Dollar OW, Horawalavithana S, Vasquez S et al (2023) MolJET: multimodal joint embedding transformer for conditional de novo molecular design and multi-property optimization. https://openreview.net/forum?id=7UudBVsIrr
Ramachandram D, Taylor GW (2017) Deep multimodal learning: a survey on recent advances and trends. IEEE Signal Process Mag 34:96–108. https://doi.org/10.1109/MSP.2017.2738401
Stahlschmidt SR, Ulfenborg B, Synnergren J (2022) Multimodal deep learning for biomedical data fusion: a review. Brief Bioinform 23:1–15. https://doi.org/10.1093/bib/bbab569
Scarselli F, Gori M, Tsoi AC et al (2008) The graph neural network model. IEEE Trans Neural Netw 20:61–80. https://doi.org/10.1109/TNN.2008.2005605
Greengard S (2021) Geometric deep learning advances data science. Commun ACM 64:13–15. https://doi.org/10.1145/3433951
Gilmer J, Schoenholz SS, Riley PF et al (2017) Neural message passing for quantum chemistry. Int Conf Mach Learn 70:1263–1272
Hao Z, Lu C, Huang Z, et al (2020) ASGN: An active semi-supervised graph neural network for molecular property prediction. In: proceedings of the 26th ACM SIGKDD international conference on knowledge discovery & data mining. ACM, New York, pp 731–752
Li Y, Li P, Yang X et al (2021) Introducing block design in graph neural networks for molecular properties prediction. Chem Eng J 414:128817. https://doi.org/10.1016/j.cej.2021.128817
Yang S, Li Z, Song G, Cai L (2021) Deep molecular representation learning via fusing physical and chemical information. Adv Neural Inf Process Syst 34:16346–16357
Li S, Zhou J, Xu T et al (2022) GeomGCL: geometric graph contrastive learning for molecular property prediction. Proc AAAI Conf Artif Intell 36:4541–4549. https://doi.org/10.1609/aaai.v36i4.20377
Dai J, Fu D, Song G et al (2022) Cross-category prediction of corrosion inhibitor performance based on molecular graph structures via a three-level message passing neural network model. Corros Sci 209:110780. https://doi.org/10.1016/j.corsci.2022.110780
Zhang S, Tong H, Xu J, Maciejewski R (2019) Graph convolutional networks: a comprehensive review. Comput Soc Netw 6:11. https://doi.org/10.1186/s40649-019-0069-y
Li Y, Zhang L, Liu Z (2018) Multi-objective de novo drug design with conditional graph generative model. J Cheminform 10:33. https://doi.org/10.1186/s13321-018-0287-6
Zhu J, Xia Y, Qin T, et al (2021) Dual-view molecule pre-training. arXiv preprint arXiv:210610234
Li G, Xiong C, Thabet A, Ghanem B (2020) Deepergcn: all you need to train deeper gcns. arXiv preprint arXiv:200607739
Liu Y, Ott M, Goyal N, et al (2019) Roberta: a robustly optimized bert pretraining approach. arXiv preprint arXiv:190711692
Lin X, Jiang Y, Yang Y (2022) Molecular distance matrix prediction based on graph convolutional networks. J Mol Struct 1257:132540. https://doi.org/10.1016/j.molstruc.2022.132540
Xiong Z, Wang D, Liu X et al (2020) Pushing the boundaries of molecular representation for drug discovery with the graph attention mechanism. J Med Chem 63:8749–8760. https://doi.org/10.1021/acs.jmedchem.9b00959
Liu Z, Lin L, Jia Q et al (2021) Transferable multilevel attention neural network for accurate prediction of quantum chemistry properties via multitask learning. J Chem Inf Model 61:1066–1082. https://doi.org/10.1021/acs.jcim.0c01224
Qian C, Xiong Y, Chen X (2021) Directed graph attention neural network utilizing 3d coordinates for molecular property prediction. Comput Mater Sci 200:110761. https://doi.org/10.1016/j.commatsci.2021.110761
Wiercioch M, Kirchmair J (2023) DNN-PP: a novel deep neural network approach and its applicability in drug-related property prediction. Expert Syst Appl 213:119055. https://doi.org/10.1016/j.eswa.2022.119055
Mansimov E, Mahmood O, Kang S, Cho K (2019) Molecular geometry prediction using a deep generative graph neural network. Sci Rep 9:20381. https://doi.org/10.1038/s41598-019-56773-5
Schütt K, Kindermans P-J, Sauceda Felix HE et al (2017) Schnet: a continuous-filter convolutional neural network for modeling quantum interactions. Adv Neural Inf Process Syst. https://doi.org/10.48550/arXiv.1706.08566
Unke OT, Meuwly M (2019) PhysNet: a neural network for predicting energies, forces, dipole moments, and partial charges. J Chem Theory Comput 15:3678–3693. https://doi.org/10.1021/acs.jctc.9b00181
Gasteiger J, Groß J, Günnemann S (2020) Directional message passing for molecular graphs. arXiv preprint arXiv:200303123. https://doi.org/10.48550/arXiv.2003.03123
Shui Z, Karypis G (2020) Heterogeneous molecular graph neural networks for predicting molecule properties. IEEE Int Conf Data Mining (ICDM) 2020:492–500. https://doi.org/10.1109/ICDM50108.2020.00058
Satorras VG, Hoogeboom E, Welling M (2021) E(n) equivariant graph neural networks. Int Conf Mach Learn. https://doi.org/10.48550/arXiv.2102.09844
Thölke P, De Fabritiis G (2022) Torchmd-net: equivariant transformers for neural network based molecular potentials. arXiv preprint arXiv:220202541. https://doi.org/10.48550/arXiv.2202.02541
Iravanizad A, Medina EIS, Stoll M (2021) RaWaNet: enriching graph neural network input via random walks on graphs. arXiv preprint arXiv:210907555
Sun M, Xing J, Wang H, et al (2021) MoCL: data-driven molecular fingerprint via knowledge-aware contrastive learning from molecular graph. Proceedings of the 27th ACM SIGKDD conference on knowledge discovery & data mining. pp. 3585–3594. https://doi.org/10.1145/3447548.3467186
Fang Y, Zhang Q, Yang H et al (2022) Molecular contrastive learning with chemical element knowledge graph. Proc AAAI Conf Artif Intell 36:3968–3976. https://doi.org/10.48550/arXiv.2112.00544
Wang Y, Wang J, Cao Z, Barati Farimani A (2022) Molecular contrastive learning of representations via graph neural networks. Nat Mach Intell 4:279–287. https://doi.org/10.1038/s42256-022-00447-x
Moon K, Im H-J, Kwon S (2023) 3D graph contrastive learning for molecular property prediction. Bioinformatics 39:1–9. https://doi.org/10.1093/bioinformatics/btad371
Fang Y, Zhang Q, Zhang N et al (2023) Knowledge graph-enhanced molecular contrastive learning with functional prompt. Nat Mach Intell 5:542–553. https://doi.org/10.1038/s42256-023-00654-0
Xu M, Powers AS, Dror RO et al (2023) Geometric latent diffusion models for 3D molecule generation. Int Conf Mach Learn 202:38592–38610
Huang L, Zhang H, Xu T, Wong K-C (2023) MDM: Molecular diffusion model for 3D molecule generation. Proc AAAI Conf Artif Intell 37:5105–5112. https://doi.org/10.1609/aaai.v37i4.25639
Hoogeboom E, Satorras VG, Vignac C, Welling M (2022) Equivariant diffusion for molecule generation in 3D. Proc Mach Learn Res 162:8867–8887
Kipf TN, Welling M (2016) Variational graph auto-encoders. arXiv preprint arXiv:161107308
Hu W, Fey M, Zitnik M et al (2020) Open graph benchmark: datasets for machine learning on graphs. Adv Neural Inf Process Syst 33:22118–22133
Li Z, Jiang M, Wang S, Zhang S (2022) Deep learning methods for molecular representation and property prediction. Drug Discov Today 27:103373. https://doi.org/10.1016/j.drudis.2022.103373
Kazerouni A, Aghdam EK, Heidari M et al (2023) Diffusion models in medical imaging: a comprehensive survey. Med Image Anal 88:102846. https://doi.org/10.1016/j.media.2023.102846
Atz K, Grisoni F, Schneider G (2021) Geometric deep learning on molecular representations. Nat Mach Intell 3:1023–1032. https://doi.org/10.1038/s42256-021-00418-8
Hancock JT, Khoshgoftaar TM (2020) Survey on categorical data for neural networks. J Big Data 7:28. https://doi.org/10.1186/s40537-020-00305-w
Zagidullin B, Wang Z, Guan Y et al (2021) Comparative analysis of molecular fingerprints in prediction of drug combination effects. Brief Bioinform 22:bbab291. https://doi.org/10.1093/bib/bbab291
Faulon J-L, Bender A (2010) Handbook of chemoinformatics algorithms. Chapman and Hall/CRC, Boca Raton
Weininger D (1988) SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J Chem Inf Comput Sci 28:31–36. https://doi.org/10.1021/ci00057a005
James CA, Weininger D, Delany J (1995) Daylight theory manual. daylight chemical information systems. In: Inc., Irvine. https://www.daylight.com/dayhtml/doc/theory/
Inc D (2018) Daylight theory: SMARTS-a language for describing molecular patterns. https://www.daylight.com/dayhtml/doc/theory/theory.smarts.html
O’Boyle N, Dalke A (2018) DeepSMILES: an adaptation of SMILES for use in machine-learning of chemical structures. chemrxiv. https://doi.org/10.26434/chemrxiv.7097960.v1
(2019) Chemical line notations for deep learning: DeepSMILES and beyond depth-first. https://depth-first.com/articles/2019/03/19/chemical-line-notations-for-deep-learning-deepsmiles-and-beyond/
Krenn M, Häse F, Nigam A et al (2020) Self-referencing embedded strings (SELFIES): A 100% robust molecular string representation. Mach Learn Sci Technol 1:045024. https://doi.org/10.1088/2632-2153/aba947
Devinyak O, Havrylyuk D, Lesyk R (2014) 3D-MoRSE descriptors explained. J Mol Graph Model 54:194–203. https://doi.org/10.1016/j.jmgm.2014.10.006
Todeschini R, Gramatica P (1997) The WHIM theory: new 3D molecular descriptors for QSAR in environmental modelling. SAR QSAR Environ Res 7:89–115. https://doi.org/10.1080/10629369708039126
Rupp M, Tkatchenko A, Müller K-R, Von Lilienfeld OA (2012) Fast and accurate modeling of molecular atomization energies with machine learning. Phys Rev Lett 108:58301. https://doi.org/10.1103/PhysRevLett.108.058301
Hansen K, Biegler F, Ramakrishnan R et al (2015) Machine learning predictions of molecular properties: accurate many-body potentials and nonlocality in chemical space. J Phys Chem Lett 6:2326–2331. https://doi.org/10.1021/acs.jpclett.5b00831
Damale M, Harke S, Kalam Khan F et al (2014) Recent advances in multidimensional QSAR (4D–6D): a critical review. Mini-Rev Med Chem 14:35–55. https://doi.org/10.2174/13895575113136660104
Grisoni F, Ballabio D, Todeschini R, Consonni V (2018) Molecular descriptors for structure-activity applications: a hands-on approach. Computational toxicology: methods and protocols. Springer, Newyork, pp 3–53
Ramakrishnan R, Hartmann M, Tapavicza E, Von Lilienfeld OA (2015) Electronic spectra from TDDFT and machine learning in chemical space. J Chem Phys. https://doi.org/10.1063/1.4928757
Ruddigkeit L, Van Deursen R, Blum LC, Reymond J-L (2012) Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. J Chem Inf Model 52:2864–2875. https://doi.org/10.1021/ci300415d
Ramakrishnan R, Dral PO, Rupp M, Von Lilienfeld OA (2014) Quantum chemistry structures and properties of 134 kilo molecules. Sci Data 1:1–7. https://doi.org/10.1038/sdata.2014.22
Chen G, Chen P, Hsieh C-Y, et al (2019) Alchemy: a quantum chemistry dataset for benchmarking ai models. arXiv preprint arXiv:190609427. https://doi.org/10.48550/arXiv.1906.09427
Sterling T, Irwin JJ (2015) ZINC 15-ligand discovery for everyone. J Chem Inf Model 55:2324–2337. https://doi.org/10.1021/acs.jcim.5b00559
Irwin JJ, Tang KG, Young J et al (2020) ZINC20—a free ultralarge-scale chemical database for ligand discovery. J Chem Inf Model 60:6065–6073. https://doi.org/10.1021/acs.jcim.0c00675
Wu Z, Ramsundar B, Feinberg EN et al (2018) MoleculeNet: a benchmark for molecular machine learning. Chem Sci 9:513–530. https://doi.org/10.1039/C7SC02664A
Delaney JS (2004) ESOL: estimating aqueous solubility directly from molecular structure. J Chem Inf Comput Sci 44:1000–1005. https://doi.org/10.1021/ci034243x
Mobley DL, Guthrie JP (2014) FreeSolv: a database of experimental and calculated hydration free energies, with input files. J Comput Aided Mol Des 28:711–720. https://doi.org/10.1007/s10822-014-9747-x
Ebenezer O, Damoyi N, Jordaan MA, Shapi M (2022) Unveiling of pyrimidindinones as potential anti-norovirus agents—a pharmacoinformatic-based approach. Molecules 27:380. https://doi.org/10.3390/molecules27020380
Richard AM, Judson RS, Houck KA et al (2016) ToxCast chemical landscape: paving the road to 21st century toxicology. Chem Res Toxicol 29:1225–1251. https://doi.org/10.1021/acs.chemrestox.6b00135
Martins IF, Teixeira AL, Pinheiro L, Falcao AO (2012) A Bayesian approach to in silico blood-brain barrier penetration modeling. J Chem Inf Model 52:1686–1697. https://doi.org/10.1021/ci300124c
Kuhn M, Letunic I, Jensen LJ, Bork P (2016) The SIDER database of drugs and side effects. Nucleic Acids Res 44:D1075–D1079. https://doi.org/10.1093/nar/gkv1075
Chmiela S, Tkatchenko A, Sauceda HE et al (2017) Machine learning of accurate energy-conserving molecular force fields. Sci Adv 3:e1603015. https://doi.org/10.1126/sciadv.1603015
Gaulton A, Bellis LJ, Bento AP et al (2012) ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res 40:D1100–D1107. https://doi.org/10.1093/nar/gkr777
Gaulton A, Hersey A, Nowotka M et al (2017) The ChEMBL database in 2017. Nucleic Acids Res 45:D945–D954. https://doi.org/10.1093/nar/gkw1074
Nakata M, Shimazaki T (2017) PubChemQC project: a large-scale first-principles electronic structure database for data-driven chemistry. J Chem Inf Model 57:1300–1308. https://doi.org/10.1021/acs.jcim.7b00083
Kim S, Cheng T, He S et al (2022) PubChem protein, gene, pathway, and taxonomy data collections: bridging biology and chemistry through target-centric views of pubchem data. J Mol Biol 434:167514. https://doi.org/10.1016/j.jmb.2022.167514
Kim S (2019) Public chemical databases. Encyclopedia of bioinformatics and computational biology. Elsevier, Amsterdam, pp 628–639
Blum LC, Reymond J-L (2009) 970 million druglike small molecules for virtual screening in the chemical universe database GDB-13. J Am Chem Soc 131:8732–8733. https://doi.org/10.1021/ja902302h
Mannhold R, Poda GI, Ostermann C, Tetko IV (2009) Calculation of molecular lipophilicity: state-of-the-art and comparison of LogP methods on more than 96,000 compounds. J Pharm Sci 98:861–893. https://doi.org/10.1002/jps.21494
Subramanian G, Ramsundar B, Pande V, Denny RA (2016) Computational modeling of β-secretase 1 (BACE-1) inhibitors using ligand based approaches. J Chem Inf Model 56:1936–1949. https://doi.org/10.1021/acs.jcim.6b00290
(2023) AIDS antiviral screen data-NCI DTP Data-NCI wiki. National Cancer Institute. https://wiki.nci.nih.gov/display/NCIDTPdata/AIDS+Antiviral+Screen+Data
Altae-Tran H, Ramsundar B, Pappu AS, Pande V (2017) Low data drug discovery with one-shot learning. ACS Cent Sci 3:283–293. https://doi.org/10.1021/acscentsci.6b00367
Gayvert KM, Madhukar NS, Elemento O (2016) A data-driven approach to predicting successes and failures of clinical trials. Cell Chem Biol 23:1294–1301. https://doi.org/10.1016/j.chembiol.2016.07.023
Artemov AV, Putin E, Vanhaelen Q et al (2016) Integrated deep learned transcriptomic and structure-based predictor of clinical trials outcomes. BioRxiv. https://doi.org/10.1101/095653
Richard AM, Huang R, Waidyanatha S et al (2021) The Tox21 10K compound library: collaborative chemistry advancing toxicology. Chem Res Toxicol 34:189–216. https://doi.org/10.1021/acs.chemrestox.0c00264
Attene-Ramos MS, Miller N, Huang R et al (2013) The Tox21 robotic platform for the assessment of environmental chemicals—from vision to reality. Drug Discov Today 18:716–723. https://doi.org/10.1016/j.drudis.2013.05.015
Schütt KT, Arbabzadah F, Chmiela S et al (2017) Quantum-chemical insights from deep tensor neural networks. Nat Commun 8:13890. https://doi.org/10.1038/ncomms13890
Chmiela S, Sauceda HE, Poltavsky I et al (2019) sGDML: constructing accurate and data efficient molecular force fields using machine learning. Comput Phys Commun 240:38–45. https://doi.org/10.1016/j.cpc.2019.02.007
Shrestha A, Mahmood A (2019) Review of deep learning algorithms and architectures. IEEE access 7:53040–53065. https://doi.org/10.1109/access.2019.2912200
Landrum G (2016) RDKit: Open-source cheminformatics. 2006. https://doi.org/10.5281/zenodo.3732262
Ramsundar B, Eastman P, Walters P, Pande V (2019) Deep learning for the life sciences: applying deep learning to genomics, microscopy, drug discovery, and more. O’Reilly Media Inc, Newton
datamol.io · GitHub https://github.com/datamol-io. Accessed 20 Oct 2023
PubChemPy · PyPI. https://pypi.org/project/PubChemPy/1.0/. Accessed 22 Oct 2023
Sun Q, Berkelbach TC, Blunt NS et al (2018) PySCF: the Python-based simulations of chemistry framework. Wiley Interdiscip Rev Comput Mol Sci 8:e1340. https://doi.org/10.1002/wcms.1340
Ochoa R, Davies M, Papadatos G et al (2014) myChEMBL: a virtual machine implementation of open data and cheminformatics tools. Bioinformatics 30:298–300. https://doi.org/10.1093/bioinformatics/btt666
Behler J, Parrinello M (2007) Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys Rev Lett 98:146401. https://doi.org/10.1103/PhysRevLett.98.146401
Schütt KT, Gastegger M, Tkatchenko A, Müller K-R (2019) Quantum-chemical insights from interpretable atomistic neural networks. Explainable AI: interpreting, explaining and visualizing deep learning. pp. 311–330. https://doi.org/10.1007/978-3-030-28954-6_17
Preuer K, Klambauer G, Rippmann F et al (2019) Interpretable deep learning in drug discovery. Explain AI Interpret Explain Vis Deep Learn. https://doi.org/10.1007/978-3-030-28954-6_18
Jumper J, Evans R, Pritzel A et al (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596:583–589. https://doi.org/10.1038/s41586-021-03819-2
Bengio Y, Simard P, Frasconi P (1994) Learning long-term dependencies with gradient descent is difficult. IEEE Trans Neural Netw 5:157–166. https://doi.org/10.1109/72.279181
Kipf TN, Welling M (2016) Semi-supervised classification with graph convolutional networks. 5th international conference on learning representations, ICLR 2017-conference track proceedings, pp. 1–14
Li G, Muller M, Thabet A, Ghanem B (2019) DeepGCNs: Can GCNs Go As Deep As CNNs? In: 2019 IEEE/CVF international conference on computer vision (ICCV). IEEE, pp 9266–9275
Wang J, Zheng S, Chen J, Yang Y (2021) Meta learning for low-resource molecular optimization. J Chem Inf Model 61:1627–1636. https://doi.org/10.1021/acs.jcim.0c01416
Guo Z, Zhang C, Yu W, et al (2021) Few-shot graph learning for molecular property prediction. In: proceedings of the web conference 2021. ACM, New York. pp 2559–2567
(2021) FS-Mol: a few-shot learning dataset of molecules. In: NeurIPS. https://github.com/microsoft/FS-Mol/
Cirq: An open source framework for NISQ algorithms. https://quantumai.google/cirq. Accessed 20 Oct 2023
McClean JR, Rubin NC, Sung KJ et al (2020) OpenFermion: the electronic structure package for quantum computers. Quantum Sci Technol 5:34014. https://doi.org/10.48550/arXiv.1710.07629
Broughton M, Verdon G, McCourt T, et al (2020) Tensorflow quantum: a software framework for quantum machine learning. arXiv preprint arXiv:200302989. https://doi.org/10.48550/arXiv.2003.02989
Google (2020) Quantum AI team and collaborators, Quantum circuit simulators (qsim). https://zenodo.org/records/5544365. Accessed 11 Nov 2023
Acknowledgements
The present study was carried out under the grant Ciencia de Frontera 2019 from CONAHCYT, CF-2019\1311317, at the Faculty of Medicine and Biomedical Sciences of the Universidad Autónoma de Chihuahua, México.
Funding
This work was funded by Consejo Nacional de Ciencia y Tecnología, CONACYT CdF-2019/1311317, CONACYT CdF-2019/1311317, CONACYT CdF-2019/1311317, CONACYT CdF-2019/1311317.
Author information
Authors and Affiliations
Contributions
A.G.P. wrote the main manuscript text and prepared figures, J.C.C wrote the main manuscript text prepared tables, All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Guzman-Pando, A., Ramirez-Alonso, G., Arzate-Quintana, C. et al. Deep learning algorithms applied to computational chemistry. Mol Divers (2023). https://doi.org/10.1007/s11030-023-10771-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11030-023-10771-y