
Abstract
The World Health Organization (WHO) recently declared the monkeypox outbreak ‘A public health emergency of international concern’. The monkeypox virus belongs to the same Orthopoxvirus genus as smallpox. Although smallpox drugs are recommended for use against monkeypox, monkeypox-specific drugs are not yet available. Drug repurposing is a viable and efficient approach in the face of such an outbreak. Therefore, we present a computational drug repurposing study to identify the existing approved drugs which can be potential inhibitors of vital monkeypox virus proteins, thymidylate kinase and D9 decapping enzyme. The target protein structures of the monkeypox virus were modelled using the corresponding protein structures in the vaccinia virus. We identified four potential inhibitors namely, Tipranavir, Cefiderocol, Doxorubicin, and Dolutegravir as candidates for repurposing against monkeypox virus from a library of US FDA approved antiviral and antibiotic drugs using molecular docking and molecular dynamics simulations. The main goal of this in silico study is to identify potential inhibitors against monkeypox virus proteins that can be further experimentally validated for the discovery of novel therapeutic agents against monkeypox disease.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The World Health Organization (WHO) declared the monkeypox outbreak ‘A public health emergency of international concern’ on 23rd July 2022 [1]. Monkeypox symptoms are similar to smallpox, and the disease affects the skin, mucous membranes, tonsils, spleen and lymph nodes [2, 3]. In 1970, the first human case of monkeypox infection was reported in a 9-month-old baby in the Democratic Republic of Congo [4, 5]. Since then, the virus has been endemic to Africa, eventually developing into the Central African and West African clades [6,7,8]. The current outbreak of monkeypox is attributed to the West African clade, which also caused the 2003 outbreak in countries like the UK, Singapore and Israel [9]. Between 1 January 2022 and 22 June 2022, 3413 laboratory confirmed cases of monkeypox have been reported across 50 countries in 5 WHO regions [10]. Conspicuously, the virus can spread rapidly among humans and is considered a potential biological weapon [11, 12].
The monkeypox virus is a zoonotic orthopoxvirus that belongs to the Poxviridae family and Chordopoxvirinae subfamily [13]. In particular, monkeypox belongs to the same family as causative agents for cowpox, mousepox and smallpox [14, 15]. Notably, the genomes of orthopoxviruses are among the largest known for animal viruses with around 200 distinct genes [16, 17]. Hence, several studies have focused on the genome, proteome, structure and morphogenesis of the poxviruses [16, 18, 19]. Poxviruses are double-stranded DNA viruses which replicate in the cytoplasm of the host cell [18, 20]. In case of monkeypox virus, the replication begins by the attachment of the virus with the host cell which is mediated by virion proteins and the host cell surface glycosaminoglycans [21]. The fusion of the viral particles with the host cell takes place with the help of non-glycosylated transmembrane viral proteins [21, 22]. After the entry of the virus into the host cell, the viral core is released into the cytoplasm. The viral DNA forms compact structures wrapped by membrane from the rough endoplasmic reticulum of the host cell. These compact structures are known as factories where the DNA replication occurs [18, 22]. As the replication progresses, these factories enlarge to produce mature virions which can further infect other host cells [21, 22].
The monkeypox virus genome is 96.3% similar to the smallpox causative variola virus of the same family [23]. Further, there is significant overlap between the proteomes of monkeypox virus and vaccinia virus [24, 25]. Vaccinia virus is used in the smallpox vaccine and is a model poxvirus in laboratory settings [16, 19]. Smallpox vaccines have been reported to be 85% effective against monkeypox [26,27,28]. However, smallpox vaccination programs have been discontinued since the eradication of the disease in 1980 [29]. Even though drugs for smallpox are recommended for use against monkeypox [3, 30, 31], their safety and efficacy in human subjects is yet to be established [32]. Therefore, the current re-emergence of monkeypox suggests that the development of monkeypox-specific drugs and therapeutics is the critical need of the hour.
Since new drug discovery involves several steps such as initial lead identification, animal studies and clinical trials, any such endeavor can usually take at least a decade. Therefore, drug repurposing is an attractive alternative in the face of an epidemic. The drug repurposing approach has several benefits including a significant reduction in testing time. In particular, previously approved drugs for other diseases have undergone rigorous toxicity analysis, and thus, can be safely administered to the public [33, 34]. During the ongoing COVID-19 pandemic, drug repurposing has been extensively employed to accelerate the search for new therapeutics. A prominent example of such repurposing is Remdesivir, approved by the US Food and Drug Administration (FDA) to treat COVID-19 [35, 36]. Interestingly, Remdesivir was initially developed for treating hepatitis C infection, and then, was under investigation for treating Ebola virus and Marburg virus infections [37, 38], before the investigational drug was repurposed for treating COVID-19. As the reported monkeypox cases are increasing worldwide, drug repurposing studies can be crucial to combat the disease. In this direction, we present an in silico drug repurposing study to predict approved drugs which can be used against monkeypox. Specifically, we consider two monkeypox virus proteins as drug targets namely, thymidylate kinase (TMPK) and D9 decapping enzyme, which are important for the viral replication cycle. Further, previous studies have emphasized that both TMPK and D9 decapping enzyme of viruses can be effective antiviral targets [39,40,41,42,43]. While this manuscript was under review, we note that four other computational drug repurposing studies have appeared which consider different target proteins of monkeypox virus [44,45,46,47], including one of the proteins considered in this study.
Unlike other viruses, orthopoxviruses encode their own TMPK [39]. TMPK catalyzes the ATP-dependent phosphorylation of thymidine 5′-phosphate (dTMP) to thymidine 5′-diphosphate (dTDP) in the presence of magnesium [48]. Subsequently, dTDP is converted into thymidine 5′-triphosphate (dTTP) which is a crucial building block in DNA synthesis. Thus, disrupting dTTP metabolism can block the development of viral infection [43]. Also, TMPK plays a key role in the activation of antiviral drugs which are nucleoside analogues [48]. The decapping enzymes in orthopoxviruses play a crucial role in suppressing the host protein synthesis and restricting the accumulation of viral double-stranded RNA. Poxviruses encode two decapping enzymes, D9 expressed early in infection and D10 expressed after viral DNA replication. Poxvirus mRNAs contain 5′-7-methylguanosine (m7G) cap and poly(A) tail. The D9 decapping enzyme can remove the protective m7G cap from mRNAs producing an m7GDP and 5′-monophosphate RNA promoting mRNA degradation [49]. Due to these reasons, TMPK and D9 decapping enzyme are attractive drug targets to design inhibitors against monkeypox.
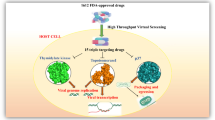
Computational approaches have become an integral part of drug discovery pipeline, enabling screening of small molecule libraries to identify lead molecules which can be optimized to develop candidate drugs for clinical trials [50]. In this in silico drug repurposing study, we have followed a detailed workflow (Fig. 1) to identify potential antivirals against monkeypox. We have virtually screened a manually curated library of 202 US FDA approved small molecule drugs against two important proteins for the viral replication cycle, namely, TMPK and D9 decapping enzyme. Importantly, the 202 US FDA approved drugs selected for this virtual screening study are known antivirals or antibiotics. Since crystallized structures for proteins in the monkeypox virus are not yet available, we employed homology modelling to construct the protein structures of TMPK and D9 decapping enzyme. Thereafter, we performed molecular docking of the 202 approved drugs against the prepared protein structures to identify potential inhibitors for the two chosen drug targets. Subsequently, we performed molecular dynamics (MD) simulations of the protein–ligand complexes for the top inhibitors predicted from the docking studies of the two drug targets, to computationally assess the stability of the protein–ligand complexes. In a nutshell, our in silico study predicts already approved drugs as promising candidates for repurposing against monkeypox.

Computational drug repurposing workflow to identify US FDA approved drugs as potential inhibitors of monkeypox proteins namely, TMPK and D9 decapping enzyme which are vital for the viral life cycle
Methods
Compilation of the ligand library of approved antivirals or antibiotics
Drug repurposing attempts to find an alternative use for an already approved drug outside the scope of its original medication [51]. Efforts to repurpose the already approved drugs can expedite the drug discovery process, especially for emerging viral diseases [52]. Previous studies have indicated that it may be more effective to repurpose an antiviral or antibiotic drug against other viral infections [53]. Specifically, antimicrobial agents are known to inhibit the viral replication cycle [54, 55]. In this context, we have manually curated a library of drugs approved by US FDA which are in use as antivirals or antibiotics to computationally predict the potential inhibitors of monkeypox proteins. To curate a library of antivirals or antibiotics, we considered the US FDA approved drugs from the FDA orange book (https://www.accessdata.fda.gov/scripts/cder/ob/index.cfm) and DrugBank [56]. Further, we manually checked the therapeutic class of these drugs in multiple sources including published literature [57, 58] and DrugBank [56], and considered only drugs which are reported to be used as antivirals or antibiotics (Supplementary Table S1). Finally, we curated a list of 202 drugs approved by US FDA which are in use as antivirals or antibiotics (Supplementary Table S1). During the compilation of this list of 202 approved drugs, we omitted drugs which were first approved by US FDA and later withdrawn or discontinued, and such information was obtained from the FDA orange book (https://www.accessdata.fda.gov/scripts/cder/ob/index.cfm) and Withdrawn database [59].
Homology modelling of drug target proteins in monkeypox virus
The 3D structures for the two proteins, TMPK and D9 decapping enzyme, of monkeypox have not been experimentally determined to date. Therefore, we performed homology modelling using the SWISS-MODEL [60] webserver (https://swissmodel.expasy.org/interactive). For this, the sequences of the two proteins, TMPK and D9 decapping enzyme, in the Israel strain (GenBank MN648051.1) of the monkeypox virus were retrieved from the NCBI database (https://www.ncbi.nlm.nih.gov/nuccore/MN648051). As vaccinia virus belongs to the same family as monkeypox virus, we used the experimentally determined 3D structures of the two proteins, TMPK and D9 decapping enzyme, in vaccinia virus available in Protein Data Bank (PDB) (https://www.rcsb.org/) as template to create the modelled structures.
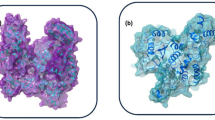
For TMPK of monkeypox virus, the crystal structure of TMPK of vaccinia virus (PDB 2V54) was used as the reference with sequence identity of 98.53%. For the modelled structure of TMPK in monkeypox virus, we find that 98.51% of the amino acid residues are in the favoured regions of the Ramachandran plot (Fig. S1), and the model structure has a MolProbity [61] score of 1.46. The root mean square deviation (RMSD) of the modelled structure of TMPK in monkeypox virus with the TMPK in vaccinia virus was found to be 0.117 Å (Fig. S1). For D9 decapping enzyme of monkeypox virus, the crystal structure of D9 decapping enzyme of vaccinia virus (PDB 7SEZ) was used as the reference with sequence identity of 97.65%. For the modelled structure of D9 decapping enzyme in monkeypox virus, we find that 95.19% of the amino acid residues are in the favoured regions of the Ramachandran plot (Fig. S1), and the model structure has a MolProbity [61] score of 1.50. The RMSD of the modelled structure of D9 decapping enzyme in monkeypox virus with the D9 decapping enzyme in vaccinia virus was found to be 0.076 Å (Fig. S1). The modelled protein structures and the respective active sites are shown in Fig. 2.

Cartoon representation of the modelled structures for monkeypox virus proteins namely, TMPK and D9 decapping enzyme. a The key amino acid residues in the binding site of TMPK are highlighted as sticks and colored in blue. The key binding site residues, D13, K17, R41, L53, N65, F68, R72, R93 and Y101, are shown in an expanded view. b The key amino acid residues in the binding site of D9 decapping enzyme are highlighted as sticks and colored in red. The key binding site residues, E16, R50, F54, D151 and Y158, are shown in expanded view
Molecular dynamics simulation
To assess the stability of the modelled protein structures for TMPK and D9 decapping enzyme, we performed MD simulations of the free protein using GROMACS 5.1.5 [62] and the GROMOS96 53a6 [63] force field. For the MD simulations of the two proteins, the modelled protein structures were placed at the center of a dodecahedron box with periodic boundary conditions and a minimum distance of 12 Å from the box edge. SPC water model was used to solvate the system. The TMPK system was neutralized using 7 Na+ ions, and the D9 system was neutral. Both protein systems were energy minimized using the steepest descent algorithm, with energy minimization tolerance set at 100 kJ mol−1 nm−1. Thereafter, the free protein systems were subjected to a 1 ns NVT simulation with 2 fs time step at 300 K temperature and with a position restraint. Subsequently, a 1 ns NPT simulation with 2 fs time step was performed to equilibrate the pressure to 1 bar. The bond lengths were constrained using the LINCS [64] algorithm during the NVT and NPT simulations. A final equilibration simulation was performed for 1 ns with 2 fs time step after removing the position restraint. Finally, the equilibrated systems were simulated for 100 ns in triplicate (3 replicas) for each protein. During the 100 ns MD simulations, the v-rescale [65] temperature and Parrinello-Rahman [66] pressure coupling method were used for maintaining the system temperature at 300 K and pressure at 1 bar during the 100 ns simulation.
For the MD simulation of a protein–ligand complex, the ligand topology parameter was generated using the Automated Topology Builder (ATB) version 3.0 [67] (https://atb.uq.edu.au/). The MD simulations of the protein–ligand docked complexes were carried out with the same parameters as described above for the free protein simulations. Using GROMACS, we computed root mean square deviation (RMSD), radius of gyration (Rg), and root mean square fluctuation (RMSF) of the free protein and protein–ligand complexes.
Molecular docking of the compiled drug library against monkeypox virus proteins
Molecular docking of the compiled drug library against the monkeypox virus proteins, TMPK and D9 decapping enzyme, was carried out using AutoDock Vina [68]. For each of the two proteins, the last frame of the MD trajectory for the corresponding free protein MD simulation was used for docking. The 3D structures of the ligands and the proteins in .pdb file format were converted to .pdbqt file format using the scripts, prepare_ligand4.py and prepare_receptor4.py, from AutoDock tools [69], respectively. The search space centre and dimensions of the grid box were manually determined by considering the key binding site residues in the two target proteins (Fig. 2). The grid centre was determined using PyMOL [70] by computing the centre of mass of the key binding site residues in each protein. Finally, protein–ligand docking was performed with the exhaustiveness parameter in AutoDock Vina set to 24 [71].
From the output of AutoDock Vina, the best docked pose or conformation of the ligand (with the lowest binding energy) was selected, and thereafter, the corresponding protein–ligand complex was generated using custom python scripts and pdb-tools [72]. Further, we used custom scripts described in our previous publication [73] to analyze the protein–ligand docked complexes including identification of the ligand binding site residues and non-covalent interactions between protein residues and the ligand. When we started this work, there were no known ligands reported in the published experimental literature that could inhibit TMPK or D9 decapping enzyme in monkeypox or other poxviruses. Therefore, we could not compare the potential inhibitors identified in this study with any reference biological ligand.
MM-PBSA calculation
We used Molecular Mechanics Poisson-Boltzmann Surface Area (MM-PBSA) method to compute the binding energy of the top inhibitors predicted for each of the two target proteins considered in this study. For each inhibitor, we extracted 51 snapshots from the MD simulation trajectory between 100 to 150 ns at a 1 ns interval for the protein–ligand complex. The binding free energy of the protein–ligand complexes were computed using g_mmpbsa [74, 75].
Results and discussion
Virtual screening workflow
In this in silico drug repurposing study, we identified potential inhibitors of TMPK and D9 decapping enzyme of monkeypox virus using the virtual screening workflow shown in Fig. 1. First, we prepared a library of 202 US FDA approved drugs which are either antivirals or antibiotics (Methods; Supplementary Table S1). Second, the three-dimensional (3D) structures of the TMPK and D9 decapping enzyme in monkeypox virus were constructed using homology modelling based on published crystal structures (PDB 2V54, 7SEZ) of the corresponding proteins in vaccinia virus (Methods; Fig. 2).
Third, to assess the stability of the modelled protein structures, the free protein structures for TMPK and D9 decapping enzyme were subjected to MD simulations of 100 ns in triplicate (Methods). For TMPK and D9 decapping enzyme, the root mean square deviation (RMSD) of the Cα atoms show fewer fluctuations after 20 ns (Fig. 3a, d). The root mean square fluctuation (RMSF) shows the dynamics of the amino acid residues in a protein. For TMPK and D9 decapping enzyme, the RMSF of the amino acid residues show fluctuations primarily in the loop region indicating that the secondary structures of the two proteins are stable (Fig. 3b, e). The radius of gyration (Rg) of a protein in the MD trajectory shows the compactness of the protein. For TMPK and D9 decapping enzyme, the Rg show little variation indicating that the two protein structures are compact during the MD simulation (Fig. 3c, f). Subsequently, we used the stable structures of TMPK and D9 decapping enzyme at the end of their MD simulation trajectories at 100 ns for molecular docking. Note that clustering of the protein trajectories from the free protein MD simulation can also be performed [76,77,78] to select representative protein structures for further analysis, however, we only considered the stable structure of both TMPK and D9 decapping enzyme at the end of the free protein MD simulation in this study to expedite the drug repurposing against monkeypox.

Analysis of MD trajectories from free protein simulations of TMPK and D9 decapping enzyme. In each case, MD simulation of 100 ns was performed in triplicate. a Root mean square deviation (RMSD) of Cα atoms of residues in TMPK. b Radius of gyration (Rg) of TMPK structure. c Root mean square fluctuation (RMSF) of Cα atoms of residues in TMPK. d RMSD of Cα atoms of residues in D9 decapping enzyme. e Rg of D9 decapping enzyme structure. f RMSF of Cα atoms of residues in D9 decapping enzyme
Fourth, we performed molecular docking of the 202 US FDA approved drugs in the compiled ligand library of antivirals or antibiotics against the stable protein structures of TMPK and D9 decapping enzyme (Methods). The binding site residues important for the activity of TMPK in monkeypox were determined by comparing the modelled protein structure with the crystallized protein structure of TMPK from the vaccinia virus (PDB 2V54). TMPK belongs to the Nucleotide monophosphate kinase (NMP) family and contains 9 important binding site residues (Fig. 2a). TMPK binds to Thymidine diphosphate (TDP) at its NMP binding site. In particular, the base binds to Arg72 (R72) and Phe68 (F68), and the sugar group is bound by Tyr101 (Y101), Leu53 (L53) and Asp13 (D13), and the phosphate group is bound by Arg93 (R93), Arg41 (R41) and Lys17 (K17). Asn65 (N65) is present in the binding pocket and is also important for the interaction [39]. The binding site residues important for the decapping activity of the D9 decapping enzyme in monkeypox were determined by comparing the modelled protein structure with the crystallized protein structure of D9 decapping enzyme from the vaccinia virus (PDB 7SEZ). The D9 decapping enzyme contains 5 important residues in the m7GDP binding pocket (Fig. 2b). The m7GDP is sandwiched between the two aromatic residues Phe54 (F54) and Tyr158 (Y158) which are important for its recognition. The Asp151 (D151) and Glu16 (E16) interact with the guanine base of m7GDP. The phosphate chain of the m7GDP is stabilized by interactions with Arg50 (R50) [49].
Fifth, considering these important binding site residues, an interaction cutoff of 4 or more non-covalent interactions between ligand and important binding sites in the target protein in the best docked pose was used to filter top hits. After imposing the interaction cutoff, the top 2 drugs were selected based on the lowest binding (docking) energy. For TMPK, the drugs Tipranavir (T1) and Cefiderocol (T2) were the top hits, and for D9 decapping enzyme, the drugs Tipranavir (D1), Doxorubicin (D2) and Dolutegravir (D3) were the top hits (Fig. 4; Table 1). Sixth, to examine the stability of the protein–ligand complexes for the top hits, we performed MD simulations of the docked complexes for both target proteins. The free energy of the protein–ligand complexes was computed using g_mmpbsa [74, 75].

Two-dimensional (2D) structure and drug name of the top predicted inhibitors of the monkeypox virus proteins, TMPK and D9 decapping enzyme. T1 and T2 were predicted to be the top inhibitors for TMPK whereas D1, D2 and D3 were predicted to be the top inhibitors for D9 decapping enzyme
Top potential inhibitors of TMPK and D9 decapping enzyme of monkeypox virus among approved drugs
Supplementary Table S1 gives the drug name, drug identifier and therapeutic class, of the 202 US FDA approved drugs, which are either antivirals or antibiotics, and are part of the ligand library in this virtual screening study. The binding site residues and residues involved in non-covalent interactions in the best docked poses for the top 2 inhibitors for TMPK and D9 decapping enzyme are given in Table 2. In case of TMPK, the top 2 predicted inhibitors are Tipranavir and Cefiderocol, and in case of D9 decapping enzyme, the top 2 predicted inhibitors are Tipranavir, Doxorubicin and Dolutegravir. Since Doxorubicin and Dolutegravir have the same docking-based binding energy for the target protein D9 decapping enzyme, both drugs are considered in the list of top 2 predicted inhibitors for the protein (Table 1). A three-dimensional (3D) and two-dimensional (2D) visualization of the non-covalent interactions in the best docked pose for these top inhibitors with residues of the drug target proteins, TMPK and D9 decapping enzyme, are shown in Figs. 5 and 6, respectively.

Cartoon representation of the non-covalent interactions in the best docked poses for the top predicted inhibitors with residues of the target proteins namely, TMPK and D9 decapping enzyme in monkeypox virus. a Interactions between the top predicted inhibitors with the residues of TMPK. The amino acid residues involved in hydrogen bond or halogen bond interactions are shown as red sticks. Drugs T1 and T2 are shown as blue and light grey sticks, respectively. b Interactions between the top predicted inhibitors with the residues of D9 decapping enzyme. The amino acid residues involved in hydrogen or halogen bond interactions are shown as dark grey sticks. Drugs D1, D2 and D3 are shown as pink, orange and red sticks, respectively. In both parts, the yellow dashed line represents the hydrogen bond whereas the blue dashed line represents the halogen bond. Further, the oxygen, nitrogen, fluorine and sulfur atoms in the ligands are shown in red, blue, light blue and yellow, respectively

2D representation of the non-covalent interactions in the best docked poses for the top predicted inhibitors with residues of the target proteins namely, TMPK and D9 decapping enzyme in monkeypox virus. a Interactions between the top predicted inhibitors with the residues of TMPK. b Interactions between the top predicted inhibitors with the residues of D9 decapping enzyme. In both parts, the protein residues involved in hydrogen bond or halogen bond interactions with the ligand are shown as green ovals. Yellow dashed lines represent hydrogen bond interactions while the violet dashed lines represent halogen bond interactions. Protein residues involved in hydrophobic interactions with the ligand are shown as blue short circle segments with spikes. Protein residues involved in the aromatic interactions with the ligand are shown as brown ovals
Drug T1 Tipranavir [79], has a docking binding energy with TMPK of − 7.6 kcal mol−1. It is a nonpeptidic HIV protease inhibitor [80]. The nonpeptidic nature offers molecular flexibility to Tipranavir, and makes it easier to fit into the active site [81] of TMPK. In the best docked pose, Tipranavir binds with the TMPK residues E142, N37, P39, R41, D50, I49, L53, R93, T18, Y35, E141, K17, D92, S15 and Q40. Further, Tipranavir forms hydrogen bonds with the TMPK residues R41, P39, D50 and N37, and trifluoromethyl group of Tipranavir forms halogen bond interactions with the TMPK residues S15, K17, T18 and D92.
Drug T2 Cefiderocol [82], has a docking binding energy with TMPK of − 7.6 kcal mol−1. It is a siderophore cephalosporin antibiotic which has been approved by the US FDA for the treatment of complicated urinary tract infections. Moreover, the drug is also included in the WHO list of essential medicines [83, 84]. Cefiderocol contains a pyrrolidinium group on the C-3 side chain and an aminothiazole group in the C-7 side chain which improve its antibacterial activity [82]. In the best docked pose, Cefiderocol binds with the TMPK residues E142, Q40, Y35, P39, R41, L53, T54, E141, T18, R93, S15, N37, D92 and K17. In the best docked pose, the pyrrolidinium group forms hydrogen bonds with the TMPK residues T54 and L53, the chlorocatechol group forms hydrogen bonds with the TMPK residues S15, K17 and N37, the aminothiazole group forms hydrogen bonds with the TMPK residues Q40 and Y35, and the carboxylic acid group forms hydrogen bond with the TMPK residue N37. Further, a hydrogen bond is also seen between the drug and TMPK residue R41. Moreover, the chlorine in the chlorocatechol group of Cefiderocol forms halogen bond interactions with the TMPK residue N37.
Drug D1 Tipranavir also has a docking binding energy with D9 decapping enzyme of − 11 kcal mol−1. In the best docked pose, Tipranavir binds with the D9 residues R15, F54, Y158, F35, T149, E16, K198, E105, A58, F154, I159, G160, T149 and Y201. In the best docked pose, Tipranavir forms hydrogen bonds with the D9 residues R15, T149, Y158, I159, Y201, K198 and C162. Further, halogen bond interactions are observed between the trifluoromethyl group of Tipranavir and D9 residues Q62 and Q63. In particular, we highlight that Tipranavir is a top hit for both drug target proteins considered in this study.
Drug D2 Doxorubicin [85], also called Adriamycin, was originally isolated from Streptomyces peucetius var. caesius, and has a glycosidic structure [86]. Doxorubicin is an anthracycline antibiotic with antitumour activity, and is used for the treatment of breast cancer, AIDS-related Kaposi’s sarcoma and other cancers [85, 87]. The drug is also included in the WHO list of essential medicines [83]. Doxorubicin has a docking binding energy with D9 decapping enzyme of − 9.9 kcal mol−1. In the best docked pose, Doxorubicin binds with the D9 residues E183, F35, Y158, T149, E16, F54, D151, F154, H33, Q62, L147, I159 and G160. In the best docked pose, the chromophore moiety in Doxorubicin forms hydrogen bonds with the D9 residues T149 and D151.
Drug D3 Dolutegravir [88], has a docking binding energy with D9 decapping enzyme of − 9.9 kcal mol−1. Dolutegravir is a HIV integrase inhibitor [88, 89], which is also included in the WHO list of essential medicines [83]. As per WHO recommendation, Dolutegravir can be used as first and second line treatment for HIV infection across all populations [90]. In the best docked pose, Dolutegravir binds with the D9 residues C162, D151, E16, F154, F35, F54, G160, K198, L147, T149 and Y158. In the best docked pose, the electron withdrawing aromatic ring of Dolutegravir forms hydrogen bonds with D9 residues K198 and C162, and further, the ligand forms hydrogen bonds with D9 residues T149, Y158 and Q62. Fluorine atom of Dolutegravir forms halogen bond interactions with D9 residue L147, and Dolutegravir forms aromatic interactions with D9 residues F35 and Y158.
The above-mentioned 4 US FDA approved drugs which are identified as top potential inhibitors of TMPK and D9 decapping enzyme in monkeypox, have also previously been reported in the literature as promising candidates for repurposing against other viral diseases. Tipranavir, which shows binding with both TMPK and D9 decapping enzyme, has been reported previously as a promising candidate for repurposing against SARS-CoV-2 [91, 92] and flaviviruses like West Nile virus and Zika virus [93]. Cefiderocol has also been shown to be affective against melioidosis [94], ventilator associated bacterial pneumonia, and other Gram-negative bacterial infections [95]. Doxorubicin and Dolutegravir have also been reported previously as a promising candidate for repurposing against SARS-CoV-2 [92, 96]. These studies suggest that the approved drugs predicted as top inhibitors in this study are promising candidates for repurposing against monkeypox virus infections. Lastly, the docking-based binding energies for an expanded list of top inhibitors predicted in this study for TMPK and D9 decapping enzyme are given in Supplementary Tables S2 and S3, respectively.
MD-based stability analysis of protein–ligand complexes of top inhibitors
We performed 150 ns MD simulations for the protein–ligand docked complexes of the top 2 inhibitors for TMPK and D9 decapping enzyme (Methods). We observed that all the protein–ligand complexes for the top 2 inhibitors for TMPK were stable during these MD simulations. The average RMSD of Cα atoms in TMPK for TMPK-T1 = 1.95 ± 0.39 Å and for TMPK-T2 = 1.62 ± 0.21 Å (Fig. 7a). The RMSF of the amino acid residues in TMPK-T1 and TMPK-T2 complexes closely followed the RMSF values of apo TMPK residues (Figs. 3c and 7b). The Rg of the TMPK shows considerably small deviation in both protein–ligand complexes with 16.68 ± 0.09 Å for TMPK-T1 and 16.57 ± 0.08 Å for TMPK-T2 (Fig. 7c). This suggests that TMPK was stable and compact during the MD simulation. However, we observed some deviation in the stability of the ligand in the binding site of the protein during the MD simulations of the protein–ligand complexes. Both the ligands were stable after 100 ns with RMSD values of ligand heavy atoms for TMPK-T1 = 3.64 ± 0.98 Å, and TMPK-T2 = 7.47 ± 0.54 Å (Fig. 7d).

Analysis of the trajectories from 150 ns of MD simulations of protein–ligand complexes for top inhibitors (T1 and T2) of TMPK. a RMSD of Cα atoms of amino acid residues in TMPK. b RMSF of Cα atoms of amino acid residues in TMPK. c Rg of the TMPK protein structure. d RMSD of the heavy atoms of the ligands T1 and T2
Similar to the protein–ligand complexes of TMPK, we also analyzed the MD trajectories of the protein–ligand complexes for top inhibitors of D9 decapping enzyme. We observed that all the protein–ligand complexes for the top 2 inhibitors for D9 decapping enzyme were stable during the MD simulation. The average RMSD of Cα atoms in D9 decapping enzyme for D9–D1 = 2.28 ± 0.34 Å, D9–D2 = 2.82 ± 0.44 Å, and D9–D3 = 2.02 ± 0.3 Å (Fig. 8a). The RMSF of the amino acid residues in D9–D1, D9–D2 and D9-D3 complexes closely followed the RMSF values of apo D9 decapping enzyme residues (Figs. 3f and 8b). The Rg of the D9 decapping enzyme shows considerably small deviation in all three protein–ligand complexes with 18.56 ± 0.13 Å for D9–D1, 18.22 ± 0.14 Å for D9–D2 and 18.53 ± 0.14 Å for D9–D3 (Fig. 8c). This suggests that D9 decapping enzyme was stable and compact during the MD simulation. We also observed that the three ligands are stable in the binding site of the D9 decapping enzyme during the MD simulations of the protein–ligand complexes. In particular, the RMSD values of the ligand heavy atoms for D9–D1 = 5.06 ± 0.4 Å, D9–D2 = 2.7 ± 0.62 Å, and D9–D3 = 3.32 ± 0.44 Å (Fig. 8d).

Analysis of the trajectories from 150 ns of MD simulations of protein–ligand complexes for top inhibitors (D1, D2 and D3) of D9 decapping enzyme. a RMSD of Cα atoms of amino acid residues in D9 decapping enzyme. b RMSF of Cα atoms of amino acid residues in D9 decapping enzyme. c Rg of the D9 decapping enzyme structure. d RMSD of the heavy atoms of the ligands D1, D2 and D3
The Molecular Mechanics Poisson-Boltzmann Surface Area (MM-PBSA) method is routinely used to better estimate the binding energy of a ligand in a protein–ligand complex. We also computed the MM-PBSA-based binding energy of the top 2 inhibitors of TMPK and D9 decapping enzyme in this study (Methods; Table 1). For TMPK, the MM-PBSA-based binding energy for Tipranavir (T1) in complex TMPK-T1 = − 9.04 ± 6.37 kcal mol−1, and Cefiderocol (T2) in complex TMPK-T2 = − 18.74 ± 4.53 kcal mol−1. Similarly, for D9 decapping enzyme, the MM-PBSA-based binding energy for Tipranavir (D1) in complex D9-D1 = − 26.65 ± 4.29 kcal mol−1, Doxorubicin (D2) in complex D9–D2 = − 32.57 ± 4.17 kcal mol−1, and Dolutegravir (D3) in complex D9–D3 = − 9.87 ± 3.98 kcal mol−1.
Conclusion
Monkeypox virus, belonging to the same Orthopoxvirus genus as smallpox causative agent, has been endemic to African regions for some time. After the eventual eradication of smallpox disease, vaccination against Orthopoxviruses have been halted for the past four decades. The current re-emergence of the monkeypox virus in a largely unprepared unvaccinated population is an international emergency demanding immediate attention by the scientific community [9]. Instead of infeasible mass vaccination campaigns within a short time span, monkeypox-specific therapeutics may yield sustainable solutions for patients affected with monkeypox disease. Drug repurposing is an efficient way of identifying potential therapeutics against the monkeypox virus, reducing the time for the initial testing and screening costs. To this end, in this in silico drug repurposing study, we identified US FDA approved antiviral and antibiotic drugs as potential inhibitors of vital monkeypox virus proteins. We modelled the 3D structures of TMPK and D9 decapping enzyme of monkeypox virus using the published crystal structures of corresponding vaccinia virus proteins. The modelled protein structures were subjected to MD simulations to evaluate their stability, and thereafter, docked against a manually curated library of 202 US FDA approved antivirals and antibiotics. Afterward, by employing the interaction and binding energy cutoffs, we show that the drugs Tipranavir, Cefiderocol, Doxorubicin and Dolutegravir have significant binding to the two target proteins in monkeypox considered here. Finally, the stability of the protein–ligand complexes of the top predicted inhibitors were assessed using 150 ns MD simulations of the complexes. To the best of our knowledge, this is one of the first virtual screening and in silico drug repurposing study carried out for monkeypox virus. We would also underline that further in vivo and in vitro experimental evaluations are needed to validate the four promising inhibitors of monkeypox proteins predicted by this study.
Data availability
Relevant data associated with this study is contained within the article or in supplementary material.
Code availability
Not applicable.
References
WHO (2022) WHO Director-General declares the ongoing monkeypox outbreak a Public Health Emergency of International Concern. https://www.who.int/europe/news/item/23-07-2022-who-director-general-declares-the-ongoing-monkeypox-outbreak-a-public-health-event-of-international-concern. Accessed 4 Aug 2022
Huhn GD, Bauer AM, Yorita K et al (2005) Clinical characteristics of human monkeypox, and risk factors for severe disease. Clin Infect Dis 41:1742–1751. https://doi.org/10.1086/498115
Adler H, Gould S, Hine P et al (2022) Clinical features and management of human monkeypox: a retrospective observational study in the UK. Lancet Infect Dis 22:1153–2116. https://doi.org/10.1016/S1473-3099(22)00228-6
Ladnyj ID, Ziegler P, Kima E (1972) A human infection caused by monkeypox virus in Basankusu Territory, Democratic Republic of the Congo. Bull World health Organ 46:593–597
Jezek Z, Khodakevich LN, Wicketti JF (1987) Smallpox and its post-eradication surveillance. Bull World Health Organ 65:425–434
Likos AM, Sammons SA, Olson VA et al (2005) A tale of two clades: monkeypox viruses. J Gen Virol 86:2661–2672. https://doi.org/10.1099/vir.0.81215-0
Nakazawa Y, Mauldin M, Emerson G et al (2015) A phylogeographic investigation of African Monkeypox. Viruses 7:2168–2184. https://doi.org/10.3390/v7042168
Yinka-Ogunleye A, Aruna O, Dalhat M et al (2019) Outbreak of human monkeypox in Nigeria in 2017–18: a clinical and epidemiological report. Lancet Infect Dis 19:872–879. https://doi.org/10.1016/S1473-3099(19)30294-4
Otu A, Ebenso B, Walley J et al (2022) Global human monkeypox outbreak: atypical presentation demanding urgent public health action. Lancet Microbe 3:e554–e555. https://doi.org/10.1016/S2666-5247(22)00153-7
WHO (2022) Multi-country monkeypox outbreak: situation update. https://www.who.int/emergencies/disease-outbreak-news/item/2022-DON396. Accessed 1 Jul 2022
Lane HC, Montagne JL, Fauci AS (2001) Bioterrorism: a clear and present danger. Nat med 7:1271–1273. https://doi.org/10.1038/nm1201-1271
Henderson DA, Inglesby TV, Bartlett JG et al (1999) Smallpox as a biological weapon: medical and public health management. JAMA 281:2127–2137. https://doi.org/10.1001/jama.281.22.2127
Bunge EM, Hoet B, Chen L et al (2022) The changing epidemiology of human monkeypox—a potential threat? A systematic review. PLOS Negl Trop Dis 16:e0010141. https://doi.org/10.1371/journal.pntd.0010141
McCollum AM, Damon IK (2014) Human monkeypox. Clin Infect Dis 58:260–267. https://doi.org/10.1093/cid/cit703
Parker S, Nuara A, Buller RML, Schultz DA (2007) Human monkeypox: an emerging zoonotic disease. Future Microbiol 2:17–34. https://doi.org/10.2217/17460913.2.1.17
Harrison SC, Alberts B, Ehrenfeld E et al (2004) Discovery of antivirals against smallpox. Proc Natl Acad Sci USA 101:11178–11192. https://doi.org/10.1073/pnas.0403600101
Liu L, Cooper T, Howley P, Hayball J (2014) From crescent to mature virion: vaccinia virus assembly and maturation. Viruses 6:3787–3808. https://doi.org/10.3390/v6103787
Moss B (2013) Poxvirus DNA replication. Cold Spring Harb Perspect Biol 5:a010199. https://doi.org/10.1101/cshperspect.a010199
Condit RC, Moussatche N, Traktman P (2006) A nutshell: structure and assembly of the vaccinia virion. Academic Press, London
Reyes ED, Kulej K, Pancholi NJ et al (2017) Identifying host factors associated with DNA replicated during virus infection. Mol Cell Proteom 16:2079–2097. https://doi.org/10.1074/mcp.M117.067116
Kaler J, Hussain A, Flores G et al (2022) Monkeypox: a comprehensive review of transmission, pathogenesis, and manifestation. Cureus 14:e26531. https://doi.org/10.7759/cureus.26531
Kmiec D, Kirchhoff F (2022) Monkeypox: a new threat? Int J Mol Sci 23:7866. https://doi.org/10.3390/ijms23147866
Di Giulio DB, Eckburg PB (2004) Human monkeypox: an emerging zoonosis. Lancet Infect Dis 4:15–25. https://doi.org/10.1016/S1473-3099(03)00856-9
Manes NP, Estep RD, Mottaz HM et al (2008) Comparative proteomics of human monkeypox and vaccinia intracellular mature and extracellular enveloped virions. J Proteome Res 7:960–968. https://doi.org/10.1021/pr070432+
Kim VV, Leiliang Z et al (2009) Poxvirus proteomics and virus-host protein interactions. Microbiol Mol Biol Rev 73:730–749. https://doi.org/10.1128/MMBR.00026-09
Reynolds MG, Damon IK (2012) Outbreaks of human monkeypox after cessation of smallpox vaccination. Trends Microbiol 20:80–87. https://doi.org/10.1016/j.tim.2011.12.001
Grant R, Nguyen L-BL, Breban R (2020) Modelling human-to-human transmission of monkeypox. Bull World health Organ 98:638–640. https://doi.org/10.2471/BLT.19.242347
Fine PEM, Jezek Z, Grab B, Dixon H (1988) The transmission potential of monkeypox virus in human populations. Int J Epedemiol 17:643–650. https://doi.org/10.1093/ije/17.3.643
Belongia EA, Naleway AL (2003) Smallpox vaccine: the good, the bad, and the ugly. Clin Med Res 1:87–92. https://doi.org/10.3121/cmr.1.2.87
Rizk JG, Lippi G, Henry BM et al (2022) Prevention and treatment of monkeypox. Drugs 82:957–963. https://doi.org/10.1007/s40265-022-01742-y
Guarner J, del Rio C, Malani PN (2022) Monkeypox in 2022—what clinicians need to know. JAMA 328:139–140. https://doi.org/10.1001/jama.2022.10802
Sherwat A, Brooks JT, Birnkrant D, Kim P (2022) Tecovirimat and the treatment of monkeypox—past, present, and future considerations. N Engl J Med 387:579–581. https://doi.org/10.1056/NEJMp2210125
Pushpakom S, Iorio F, Eyers PA et al (2019) Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov 18:41–58. https://doi.org/10.1038/nrd.2018.168
Scherman D, Fetro C (2020) Drug repositioning for rare diseases: knowledge-based success stories. Therapies 75:161–167. https://doi.org/10.1016/j.therap.2020.02.007
Beigel JH, Tomashek KM, Dodd LE et al (2020) Remdesivir for the treatment of covid-19—final report. N Engl J Med 383:1813–1826. https://doi.org/10.1056/NEJMoa2007764
Rubin D, Chan-Tack K, Farley J, Sherwat A (2020) FDA approval of remdesivir—a step in the right direction. N Engl J Med 383:2598–2600. https://doi.org/10.1056/NEJMp2032369
Malin JJ, Suárez I, Priesner V et al (2020) Remdesivir against COVID-19 and other viral diseases. Clin Microbiol Rev 34:e00162-e220. https://doi.org/10.1128/CMR.00162-20
Tchesnokov EP, Feng JY, Porter DP, Götte M (2019) Mechanism of inhibition of ebola virus RNA-dependent RNA polymerase by remdesivir. Viruses 11:326. https://doi.org/10.3390/v11040326
Caillat C, Topalis D, Agrofoglio LA et al (2008) Crystal structure of poxvirus thymidylate kinase: an unexpected dimerization has implications for antiviral therapy. Proc Natl Acad Sci USA 105:16900–16905. https://doi.org/10.1073/pnas.0804525105
Guimarães AP, Ramalho TC, França TCC (2014) Preventing the return of smallpox: molecular modeling studies on thymidylate kinase from Variola virus. J Biomol Struct Dyn 32:1601–1612. https://doi.org/10.1080/07391102.2013.830578
Guimarães AP, de Souza FR, Oliveira AA et al (2015) Design of inhibitors of thymidylate kinase from Variola virus as new selective drugs against smallpox. Eur J Med Chem 91:72–90. https://doi.org/10.1016/j.ejmech.2014.09.099
Bednarczyk M, Peters JK, Kasprzyk R et al (2022) Fluorescence-based activity screening assay reveals small molecule inhibitors of vaccinia virus mRNA decapping enzyme D9. ACS Chem Biol 17:1460–1471. https://doi.org/10.1021/acschembio.2c00049
Cui Q, Shin W, Luo Y et al (2013) Thymidylate kinase: an old topic brings new perspectives. Curr Med Chem 20:1286–1305. https://doi.org/10.2174/0929867311320100006
Altayb HN (2022) Fludarabine, a potential DNA-dependent RNA polymerase inhibitor, as a prospective drug against monkeypox virus: a computational approach. Pharmaceuticals 15:1129. https://doi.org/10.3390/ph15091129
Lam HY, Guan JS, Mu Y (2022) In silico repurposed drugs against monkeypox virus. Molecules 27:5277. https://doi.org/10.3390/molecules27165277
Abduljalil JM, Elfiky AA (2022) Repurposing antiviral drugs against the human monkeypox virus DNA-dependent RNA polymerase; in silico perspective. J Infect. https://doi.org/10.1016/j.jinf.2022.09.002
Li D, Liu Y, Li K, Zhang L (2022) Targeting F13 from monkeypox virus and variola virus by tecovirimat: Molecular simulation analysis. J Infect 85:e99–e101. https://doi.org/10.1016/j.jinf.2022.07.001
Topalis D, Collinet B, Gasse C et al (2005) Substrate specificity of vaccinia virus thymidylate kinase. FEBS J 272:6254–6265. https://doi.org/10.1111/j.1742-4658.2005.05006.x
Peters JK, Tibble RW, Warminski M et al (2022) Structure of the poxvirus decapping enzyme D9 reveals its mechanism of cap recognition and catalysis. Structure 30:721-732.e4. https://doi.org/10.1016/j.str.2022.02.012
Sliwoski G, Kothiwale S, Meiler J, Lowe EW (2014) Computational methods in drug discovery. Pharmacol Rev 66:334–395. https://doi.org/10.1124/pr.112.007336
Ashburn TT, Thor KB (2004) Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov 3:673–683. https://doi.org/10.1038/nrd1468
Rajput A, Kumar A, Megha K et al (2021) DrugRepV: a compendium of repurposed drugs and chemicals targeting epidemic and pandemic viruses. Brief Bioinform 22:1076–1084. https://doi.org/10.1093/bib/bbaa421
Mercorelli B, Palù G, Loregian A (2018) Drug repurposing for viral infectious diseases: how far are we? Trends Microbiol 26:865–876. https://doi.org/10.1016/j.tim.2018.04.004
Colson P, Raoult D (2016) Fighting viruses with antibiotics: an overlooked path. Int J Antimicrob Agents 48:349–352. https://doi.org/10.1016/j.ijantimicag.2016.07.004
Strating JRPM, van der Linden L, Albulescu L et al (2015) Itraconazole inhibits enterovirus replication by targeting the oxysterol-binding protein. Cell Rep 10:600–615. https://doi.org/10.1016/j.celrep.2014.12.054
Wishart DS, Feunang YD, Guo AC et al (2017) DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 46:D1074–D1082. https://doi.org/10.1093/nar/gkx1037
Newman DJ, Cragg GM (2020) Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J Nat Prod 83:770–803. https://doi.org/10.1021/acs.jnatprod.9b01285
Chong CR, Sullivan DJ (2007) New uses for old drugs. Nature 448:645–646. https://doi.org/10.1038/448645a
Siramshetty VB, Nickel J, Omieczynski C et al (2016) WITHDRAWN–a resource for withdrawn and discontinued drugs. Nucleic Acids Res 44:D1080–D1086. https://doi.org/10.1093/nar/gkv1192
Waterhouse A, Bertoni M, Bienert S et al (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46:W296–W303. https://doi.org/10.1093/nar/gky427
Chen VB, Arendall WB 3rd, Headd JJ et al (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr 66:12–21. https://doi.org/10.1107/S0907444909042073
Abraham MJ, Murtola T, Schulz R et al (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1:19–25. https://doi.org/10.1016/j.softx.2015.06.001
Oostenbrink C, Villa A, Mark AE, Van Gunsteren WF (2004) A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J Comput Chem 25:1656–1676. https://doi.org/10.1002/jcc.20090
Hess B, Bekker H, Berendsen HJ, Fraaije JG (1997) LINCS: a linear constraint solver for molecular simulations. J Comput Chem 18:1463–1472. https://doi.org/10.1002/(SICI)1096-987X(199709)18:12%3c1463::AID-JCC4%3e3.0.CO;2-H
Berendsen HJ, van Postma J, Van Gunsteren WF et al (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81:3684–3690. https://doi.org/10.1063/1.448118
Parrinello M, Rahman A (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys 52:7182–7190. https://doi.org/10.1063/1.328693
Stroet M, Caron B, Visscher KM et al (2018) Automated topology builder version 3.0: prediction of solvation free enthalpies in water and hexane. J Chem Theory Comput 14:5834–5845. https://doi.org/10.1021/acs.jctc.8b00768
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461. https://doi.org/10.1002/jcc.21334
Morris GM, Huey R, Lindstrom W et al (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791. https://doi.org/10.1002/jcc.21256
DeLano WL (2002) The PyMOL molecular graphics system
Forli S, Huey R, Pique ME et al (2016) Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc 11:905–919. https://doi.org/10.1038/nprot.2016.051
Rodrigues JPGLM, Teixeira JMC, Trellet M, Bonvin AMJJ (2018) pdb-tools: a swiss army knife for molecular structures. F1000Res 7:1961. https://doi.org/10.12688/f1000research.17456.1
Vivek-Ananth RP, Rana A, Rajan N et al (2020) In silico identification of potential natural product inhibitors of human proteases key to SARS-CoV-2 infection. Molecules 25:3822. https://doi.org/10.3390/molecules25173822
Kumari R, Kumar R, Lynn A (2014) g_mmpbsa–a GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model 54:1951–1962. https://doi.org/10.1021/ci500020m
Baker NA, Sept D, Joseph S et al (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA 98:10037–10041. https://doi.org/10.1073/pnas.181342398
Vivek-Ananth RP, Sahoo AK, Srivastava A, Samal A (2022) Virtual screening of phytochemicals from Indian medicinal plants against the endonuclease domain of SFTS virus L polymerase. RSC Adv 12:6234–6247. https://doi.org/10.1039/D1RA06702H
Vivek-Ananth RP, Krishnaswamy S, Samal A (2022) Potential phytochemical inhibitors of SARS-CoV-2 helicase Nsp13: a molecular docking and dynamic simulation study. Mol Divers 26:429–442. https://doi.org/10.1007/s11030-021-10251-1
Santiago Á, Razo-Hernández RS, Pastor N (2020) The TATA-binding Protein DNA-binding domain of eukaryotic parasites is a potentially druggable target. Chem Biol Drug Des 95:130–149. https://doi.org/10.1111/cbdd.13630
King JR, Acosta EP (2006) Tipranavir. Clin Pharmacokinet 45:665–682. https://doi.org/10.2165/00003088-200645070-00003
Turner SR, Strohbach JW, Tommasi RA et al (1998) Tipranavir (PNU-140690): a potent, orally bioavailable nonpeptidic HIV protease inhibitor of the 5,6-dihydro-4-hydroxy-2-pyrone sulfonamide class. J Med Chem 41:3467–3476. https://doi.org/10.1021/jm9802158
Kandula VR, Khanlou H, Farthing C (2005) Tipranavir: a novel second-generation nonpeptidic protease inhibitor. Expert Rev Anti-Infect Ther 3:9–21. https://doi.org/10.1586/14787210.3.1.9
Sato T, Yamawaki K (2019) Cefiderocol: discovery, chemistry, and in vivo profiles of a novel siderophore cephalosporin. Clin Infect Dis 69:S538–S543. https://doi.org/10.1093/cid/ciz826
WHO (2021) World Health Organization model list of essential medicines: 22nd list
FDA (2020) FDA approves new antibacterial drug to treat complicated urinary tract infections as part of ongoing efforts to address antimicrobial resistance. https://www.fda.gov/news-events/press-announcements/fda-approves-new-antibacterial-drug-treat-complicated-urinary-tract-infections-part-ongoing-efforts. Accessed 29 Jun 2022
Patel AG, Kaufmann SH (2012) How does doxorubicin work? Elife 1:e00387. https://doi.org/10.7554/eLife.00387
Arcamone F, Cassinelli G, Franceschi G, et al (1972) Structure and physicochemical properties of adriamycin (doxorubicin). In: International symposium on adriamycin; Springer. International symposium on adriamycin; Springer, pp 9–22
Weiss RB (1992) The anthracyclines: will we ever find a better doxorubicin? In: Seminars in Oncology. Seminars in Oncology, pp 670–686
Osterholzer DA, Goldman M (2014) Dolutegravir: a next-generation integrase inhibitor for treatment of HIV infection. Clin Infect Dis 59:265–271. https://doi.org/10.1093/cid/ciu221
Mesplède T, Wainberg MA (2013) Integrase strand transfer inhibitors in HIV therapy. Infect Dis Ther 2:83–93. https://doi.org/10.1007/s40121-013-0020-8
WHO (2019) WHO recommends dolutegravir as preferred HIV treatment option in all populations. https://www.who.int/news/item/22-07-2019-who-recommends-dolutegravir-as-preferred-hiv-treatment-option-in-all-populations. Accessed 7 Jul 2022
Arshad U, Pertinez H, Box H et al (2020) Prioritization of anti-SARS-Cov-2 drug repurposing opportunities based on plasma and target site concentrations derived from their established human pharmacokinetics. Clin Pharmacol Ther 108:775–790. https://doi.org/10.1002/cpt.1909
Indu P, Rameshkumar MR, Arunagirinathan N et al (2020) Raltegravir, indinavir, tipranavir, dolutegravir, and etravirine against main protease and RNA-dependent RNA polymerase of SARS-CoV-2: a molecular docking and drug repurposing approach. J Infect Public Health 13:1856–1861. https://doi.org/10.1016/j.jiph.2020.10.015
Stefanik M, Valdes JJ, Ezebuo FC et al (2020) FDA-approved drugs efavirenz, tipranavir, and dasabuvir inhibit replication of multiple flaviviruses in vero cells. Microorganisms 8:599. https://doi.org/10.3390/microorganisms8040599
Delaney B, Gemma R, Andrew H et al (2021) Burkholderia pseudomallei clinical isolates are highly susceptible in vitro to cefiderocol, a siderophore cephalosporin. Antimicrob Agents Chemother 65:e00685-e720. https://doi.org/10.1128/AAC.00685-20
Karlowsky JA, Hackel MA, Tsuji M et al (2019) In vitro activity of cefiderocol, a siderophore cephalosporin, against Gram-negative bacilli isolated by clinical laboratories in North America and Europe in 2015–2016: SIDERO-WT-2015. Int J Antimicrob Agents 53:456–466. https://doi.org/10.1016/j.ijantimicag.2018.11.007
Sajid Jamal QM, Alharbi AH, Ahmad V (2021) Identification of doxorubicin as a potential therapeutic against SARS-CoV-2 (COVID-19) protease: a molecular docking and dynamics simulation studies. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2021.1905551
Acknowledgements
The authors acknowledge the use of the supercomputing machine Nandadevi at The Institute of Mathematical Sciences, Chennai for molecular dynamics simulations. Areejit Samal would like to thank funding from the Department of Atomic Energy (DAE) India, and the Max Planck Society Germany (through the award of a Max Planck Partner Group).
Funding
The authors did not receive any specific funding to carry out this research.
Author information
Authors and Affiliations
Contributions
AKS, PDA, IM and AS designed research. AKS, PDA, IM, RPV, KK, GK, GR and AS compiled and analyzed data. AKS, IM and RPV performed computations. AKS, PDA, IM, RPV and AS wrote the manuscript. AS supervised the project.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
This is a purely computational study and ethics approvals are not applicable.
Consent to participate
Not applicable.
Consent for publication
All co-authors have read and approved the manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file1 (PDF 584 KB)
Figure S1: Ramachandran plots for the modelled structures of monkeypox virus proteins, alignment of the monkeypox virus protein sequence with the vaccinia virus protein sequence, and superimposition of the amino acid residues of the monkeypox virus protein structure with vaccinia virus protein structure. The Ramachandran plots display the torsional angles, phi (φ) and psi (ψ) of the amino acid residues in the modelled proteins. (a) Ramachandran plot for the modelled structure of TMPK. (b) Alignment of the protein sequence of monkeypox virus TMPK with vaccinia virus TMPK (PDB 2V54) which was used as a template to model the 3D structure of the monkeypox virus TMPK. (c) Superimposition of the protein structure of monkeypox virus TMPK (in green color) with the protein structure of vaccinia virus TMPK (in wheat color; PDB 2V54) which was used as a template to model the 3D structure of the monkeypox virus TMPK. (d) Ramachandran plot for the modelled structure of D9 decapping enzyme. (e) Alignment of the protein sequence of monkeypox virus D9 decapping enzyme with vaccinia virus D9 decapping enzyme (PDB 7SEZ) which was used as a template to model the 3D structure of monkeypox virus D9 decapping enzyme. (f) Superimposition of the protein structure of monkeypox virus D9 decapping enzyme (in cyan color) with the protein structure of vaccinia virus D9 decapping enzyme (in salmon color; PDB 7SEZ) which was used as a template to model the 3D structure of the monkeypox virus D9 decapping enzyme.
Supplementary file2 (XLSX 26 KB)
Table S1: Compiled list of 202 US FDA approved antiviral and antibiotic drugs. For each drug, the PubChem compound identifier, DrugBank accession number and therapeutic class information is provided. We also provide the PubMed identifiers of published literature which support the therapeutic class of the drug. Table S2: Top drug inhibitors predicted for the TMPK of monkeypox virus. For each drug, we provide the docking based binding energy in kcal mol-1. Table S3: Top drug inhibitors predicted for the D9 decapping enzyme of monkeypox virus. For each drug, we provide the docking based binding energy in kcal mol-1.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sahoo, A.K., Augusthian, P.D., Muralitharan, I. et al. In silico identification of potential inhibitors of vital monkeypox virus proteins from FDA approved drugs. Mol Divers 27, 2169–2184 (2023). https://doi.org/10.1007/s11030-022-10550-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11030-022-10550-1