
Abstract
Direct air capture (DAC) is a developing technology for removing carbon dioxide (CO2) from the atmosphere or from low-CO2-containing sources. In principle, it could be used to remove sufficient CO2 from the atmosphere to compensate for hard-to-decarbonize sectors, such as aviation, or even for polishing gas streams containing relatively low CO2 concentrations. In this paper, the performance of lime-based sorbents for CO2 capture from air in a fixed bed was investigated. The effects of sorbent type, particle diameter, air flow rate, and relative humidity on the breakthrough time, breakthrough shape, and global reaction rate over a series of capture and regeneration cycles were examined. The greatest reaction rates and conversions were obtained when the sorbents were pre-hydrated and inlet air was humidified to 55% relative humidity. Humidifying the air alone leads to axial carbonation gradients since there is competition between CO2 and water with the available CaO. Negligible conversion, over the duration of the experiment, is obtained in a dry system without pre-hydration and humid air. A shrinking-core gas–solid reaction model was fitted to the breakthrough curves in order to estimate the surface reaction and effective diffusion constants. Although the surface reaction constants of the two sorbents were similar, the pelletized limestone had a greater effective diffusivity due to its greater porosity. At mild calcination conditions with air at 850 °C, the pelletized particles maintained their activity over nine carbonation–calcination cycles with a conversion drop of only 9% points. However, calcination under oxy-fuel conditions (CO2 at 920 °C) reduced the pellet carbonation conversion from 81 to 59% and pore surface area from 12.01 to 3.20 m2/g after only 4 cycles. This research clearly shows that DAC using lime-based sorbents is technically feasible, and that regeneration schemes compatible with technologies like calcium looping (CaL) are applicable for the air capture option. Finally, this study demonstrates that DAC using lime-based materials can be in the future a strategy to address emissions from transportation and distributed CO2 sources and to mitigate climate change.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
It is clear that anthropogenic emission of greenhouse gases (GHGs) is causing significant climate change (IPCC 2007; Lüthi et al. 2008; Petit et al. 1999). According to the 2010 annual report of the International Energy Agency (IEA), two thirds of the global carbon dioxide (CO2) emissions are from the electricity/heat generation and transport sectors (IEA 2011). Since fossil fuels are still the least expensive source of energy, it is predicted that GHG emissions due to combustion of fossil fuels (e.g., coal, oil, and natural gas) will rise by 25–90% between 2000 and 2030 (Ranjan and Herzog 2011). Carbon capture and storage (CCS) is the process of capturing CO2 from large point sources and producing a concentrated and pressurized stream of CO2 for storage. However, about 60% of CO2 emissions is attributed to dilute, distributed sources (IEA 2013) and while oceans absorb much of the emitted CO2, 20–40% of it remains in the atmosphere (Bottoms 1930).
Direct air capture (DAC) is a developing, flexible technology for removing CO2 from the atmosphere that can, in principle, be built anywhere. Unlike other CO2 removal processes, it does not require a relatively concentrated source of CO2 such as provided by flue gas or process streams from carbon-intensive industries. The most important benefit of DAC is to remove sufficient CO2 from the environment, to compensate for otherwise difficult-to-decarbonize sectors such as aviation or transportation. Given that DAC is always going to be more expensive than decarbonizing concentrated gas streams from fossil fuel conversion, its most likely applications are either to polish dilute gas streams (residual CO2 from CCS processes) or as a method to compensate for the use of fossil fuels in transportation options.
Capturing CO2 with a partial pressure p0 from a mixture of gases and separating it as a concentrated CO2 stream with a pressure P, needs kTln(P/P0) energy, where k and T are the Boltzman constant and working temperature, respectively. Therefore, capturing CO2 (partial pressure ~ 4 × 10−4 bar) from air and compressing it to 101.3 bar, which is required for sequestration, needs an energy input of almost 4 GJ/t carbon. Comparing this value with the carbon-specific energy content of fossil fuels, such as coal, oil, and natural gas (40–70 GJ/t carbon), suggests that part of the energy from carbon fuels could be used for CO2 capture rather than the CO2 being emitted to the atmosphere (Keith et al. 2006). The environmental advantages of CO2 capture from air are undeniable. For instance, CO2 capture plants could be constructed near the final storage sites, in locations where land is cheap, and energy for the separation process could be obtained from renewable sources (e.g., wind or solar) and, additionally, transportation costs are marginal. DAC could also be used for the tailing CO2 treatment of power generation plants or oil and gas refineries, where 100% CO2 capture is not normally achieved (Nikulshina et al. 2006).
One method for DAC using hydroxide solutions has been explored by Carbon Engineering Ltd. in Canada (Carbon Engineering 2019). The process consists of two main cycles: an air contactor (CO2 capture cycle) and a sorbent regeneration cycle. In the first cycle, CO2 from air is absorbed by means of a chemical solution to form a carbonate (e.g., K2CO3). The carbonate solution then reacts with calcium hydroxide (Ca(OH)2) to form calcium carbonate (CaCO3). In the next cycle, the CaCO3 is separated from solution and calcined to lime (CaO) in an oxy-fired fluidized bed that operates at approximately 900 °C. During this stage, CO2 is released while the CaO is recycled and rehydrated to Ca(OH)2. Finally, this hydroxide is used to react with the carbonate solution (Mahmoudkhani and Keith 2009). However, since the process uses a solution, good contact between the liquid and ambient air is critical when a spray-based system and a packed tower are employed (Mahmoudkhani et al. 2009). Furthermore, these methods have some drawbacks, due to the high cost of sorbent regeneration, solvent losses, and the presence of alkali metal salt impurities in any calcium loop that enhances sintering and loss of sorption capacity. Therefore, eliminating the use of alkali metal hydroxides altogether and using lime-based sorbents is inherently simpler, and takes advantage of the fact that lime is, after water, arguably the cheapest and most commonly available industrial chemical, and has numerous uses, including as will be discussed below, in the cement and steel industries.
By contrast, amine-based scrubbing is a mature technology that has been used for more than 80 years in the oil and gas industry for acid gas (H2S and CO2) removal (Astarita et al. 1993). However, for DAC, solutions such as diethanolamine (DEA) and monoethanolamine (MEA) are unsuitable since their CO2 capture capacity drops drastically at low CO2 partial pressures. Therefore, amine-based solid sorbents have been proposed instead. Supported amine adsorbents are classified into two categories. In class 1 adsorbents, amine molecules are loaded into polymeric or silica supports. These amine molecules are physisorbed on the surface and in the pores of the support. For class 2 adsorbents, amine molecules are bound to the surface of the support by covalent bonds. Studies show that class 2 adsorbents have better performance than other adsorbents such as zeolites and mesoporous silica at low CO2 partial pressure (Choi et al. 2011). Despite the relatively low regeneration energy requirement of amine-based sorbents, their relatively high costs, poor CO2 capture capacity from air, and degradation are issues that make them less attractive options (Choi et al. 2011; Sculley and Zhou 2012).
Finally, CO2 capture at high temperature via new lime-based sorbents, which are enhanced with calcium aluminate cement binders and pelletized, has been investigated by CanmetENERGY-Ottawa. Thermogravimetric analyzer (TGA) results show improved sorption capacity relative to natural limestone (Manovic and Anthony 2009a, 2010; Manovic et al. 2012) due to the formation of mayenite (Ca12Al14O33), which reduces sintering during CO2 capture cycles (Manovic and Anthony 2009a, b, 2010; Manovic et al. 2012).
Limestone is extensively used in the cement industry as a source of CaO and is responsible for 7–10% of anthropogenic CO2 emissions (Zheng et al. 2016). Calcium looping (CaL) has been proposed as an option for the cement industry to capture CO2 (Erans et al. 2018). Here, the CaO from CaL can be used in DAC, or if DAC is carried out with a lime-based sorbent, then the spent lime can be fed back into the cement industry. Recently, it has also been demonstrated that lime can be very efficiently used in the steel industry for decarbonization as well, using a variant of the CaL option (Tian et al. 2018). The basic philosophy of this work is that limestone and lime-based materials are ubiquitous, cheap, nontoxic and are compatible with two of the most important industrial sources of anthropogenic CO2 (namely cement and steel), and a lime-based process, if it can be made to perform at a comparable level to DAC processes using sodium or potassium hydroxide, is inherently preferable. Thus, while to date, experiments on capturing CO2 focused on concentrations relevant to coal flue gases (10–15%) at elevated temperatures (Zheng et al. 2016; Erans et al. 2018), here we seek to demonstrate that these sorbents are suitable for DAC.
Significant carbonation of calcium hydroxide at ambient temperature can be achieved when the relative humidity (RH) of air is above 40% (Beruto and Botter 2000). Dheilly et al. (2002) suggest that the H2O rather than CO2 partial pressure dominates the carbonation reaction at low temperature. Furthermore, Yang et al. (2003) observed via atomic force microscopy (AFM) that the carbonation of calcium hydroxide at ambient conditions occurs when nano-droplets of water are formed on the surface of Ca(OH)2. The carbonation of lime at low temperature can then be said to occur in several steps (Kalinkin et al. 2005). First, gaseous CO2 dissolves in the water droplets and forms H2CO3 (Eq. 1) and then calcium hydroxide from the surface of the sorbent also dissolves in the water droplets. Here, H2CO3 dissociates to H+, HCO3−, and CO32− ions (Eqs. 2 and 3), and Ca(OH)2 to Ca2+ and OH− ions in solution (Eq. 4).
The overall carbonation reaction of Ca(OH)2 can then be written as follows:
The formation of CaCO3 on the surface of Ca(OH)2 impedes diffusion of CO2. However, studies show high carbonation conversion of Ca(OH)2 is possible at ambient conditions and, therefore, it can be concluded that the new solid phase on the surface of the sorbent particle is non-protective (Nakicenovic et al. 2000). This can be explained by the formation of calcium bicarbonate (Ca(HCO3)2), (Pontiga et al. 2013):
The higher solubility of Ca(HCO3)2 enhances CO2 diffusion through the bulk of the particle. Finally, Ca(HCO3)2 reacts with Ca(OH)2 and forms CaCO3:
Since these carbonation reactions (Eqs. 5 and 7) occur in parallel, the formed solid phase can be considered as a non-protective layer. As the solubility of gases in water is greater at lower temperatures, the carbonation of Ca(OH)2 at a lower temperature is enhanced (Dheilly et al. 2002; Pontiga et al. 2013).
The objective of this work was to investigate the use of natural lime and pelletized lime-cement particles for the direct capture of CO2 from air at ambient conditions, with the sorbents being regenerated via temperature swing. Two particle sizes were considered and experiments were performed in a fixed bed. Using these data, a model was developed to simulate the breakthrough curves and to extract the carbonation kinetics data for various operating conditions.
2 Materials and methodology
2.1 Materials
The composition of the natural Cadomin lime, calcined at 850 °C, from X-ray fluorescence (XRF) spectroscopy is presented in Table 1. The pelletized lime particles were made from powdered lime (dp < 45 μm) and calcium aluminate cement (CA-14, ~ 71%wt. Al2O3; ~ 28%wt. CaO; ~ 1%wt. impurities). The lime and cement were mixed in a mass ratio of 9:1 in a mechanical pelletizer (Glatt GmbH). Water was then sprayed into the vessel continuously. The resulting pellet size was controlled by the speed of an agitator and chopper attached to the vessel. Table 2 shows some physical properties of the natural and pelletized lime sorbents. The samples with desired particle size range were obtained by sieving. Then, the samples were calcined at 850 °C in air and their pore surface area was measured by the Brunauer–Emmett–Teller (BET) technique with a Micromeritics-TriStar II apparatus. The results presented in Table 2 show that the natural lime particles have a greater bulk density while the pellets have a greater pore surface area, primarily due to the formation of mayenite during calcination (Manovic and Anthony 2009a, b).
2.2 Methodology
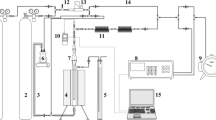
The schematic of the experimental setup is shown in Fig. 1. A compressor was used to circulate atmospheric air through a water bubbler and then a packed bed of sorbent. The average CO2 concentration of the inlet air, Co, was 415 ppm and its relative humidity, as measured by a Vaisala RH-Transmitter, was maintained at ~ 55% during all experiments. It should be noted that experiments without the presence of humidified air were also performed to show the effect of humidity for air capture with lime-based sorbents at low temperature. The air stream exiting the bed was sent to an online gas analyzer to determine the residual CO2 content, C, of the stream leaving the system.

Experimental setup
The column containing the bed of particles is a stainless steel tube with a height of 78.6 mm and inner diameter of 7.8 mm, as shown in Fig. 2. A uniform bed porosity can be assumed when the column diameter is 10× to 15× greater than the mean particle diameter (Akehata and Sato 1985; Gunn 1968; Gunn and Pryce 1969; Stephenson and Stewart 1986). The fixed bed height was set at 68 mm, yielding an aspect ratio of around 8 that will mitigate the impact of entrance effects. Moreover, the column lower section contains glass beads of 200–300 μm in diameter filled to a height of 10 mm in order to minimize fluid channeling. Finally, a paper filter with pore size of 8 μm was also inserted at the bottom of the column. Gas flow rates of 0.5 and 1.0 LPM (at 20 °C and 1 barg) were chosen to ensure that the flow regime remained constant in all experiments. The particle Reynolds number varied between 0.37 and 1.12, well below the upper limit value of 10 for laminar flow (Ranade et al. 2011).

Fixed bed reactor
Before each experiment, the bed material was pre-hydrated for 3 h by passing nitrogen through the water bubbler and then the fixed bed. An experiment was typically conducted for only 1 carbonation cycle. For the experiments performed in series of cycles, after each carbonation cycle, sorbents were calcined and prepared for the next cycle. After complete breakthrough, the conversions of sorbent samples from the top, middle, and bottom of the bed were measured by TGA (METTLER TOLEDO-SDTA581) to determine the carbonation profile throughout the bed.
3 Results and discussion
3.1 Importance of system moisture
The carbonation conversion, X′, is defined in Eq. 8:
where m2 and m1 are the mass of carbonated and calcined sample after dehydration, respectively, m0 is the initial mass of the calcined sample, A is the fraction of CaO in the sorbent, and MCaO and MCO2 are the molecular weight of calcium oxide and carbon dioxide, respectively.
Figures 3 and 4 show that, although the natural limestone or pelletized limestone particles are pre-hydrated for 3 h, the absence of humidity in the inlet air led to a relatively short breakthrough time (Fig. 3) and the sorbent carbonation was low, at less than 20% for pelletized limestone and less than 10% for natural limestone (Figs. 4 a, b). The conditions for all experiments are as given in Fig. 3 unless noted otherwise. These results are in agreement with those in the literature, showing that negligible carbonation occurs when there is insufficient humidity in the reactive system (Beruto and Botter 2000; Dheilly et al. 2002; Nikulshina et al. 2007).

Breakthrough curves in the absence of humidity in air: pre-hydrated sorbent, dp = 250–425 μm, Q = 1 LPM (1 barg, 20 °C)

TGA analysis of post-run sorbents in the absence of humidity in air: pre-hydrated sorbent, dp = 250–425 μm, dry inlet air, and Q = 1 LPM (1 barg, 20 °C). a Pelletized limestone. b Natural limestone
On the other hand, if the inlet air is humidified up to 50–55% relative humidity, but sorbents are not pre-hydrated, an axially non-uniform carbonated bed is observed (Table 3). This phenomenon is due to the partial carbonation of sorbents in the first layers of the bed. While there is competition between CO2 and water to react with CaO, the partial carbonation reaction on the surface of the sorbents prevents further hydration and decreases the reaction rate at the surface. However, in comparison with a dry system where relative humidity was negligible and sorbents were not pre-hydrated, the observed carbonation conversions were more than 50% higher.
3.2 Effect of particle size and flow rate
Figure 5 shows the effect of particle size and gas flow rate on the breakthrough time and breakthrough curve for both the pelletized and natural limestone. As smaller particles have a greater specific surface area (m2/g), it is expected (and observed) that their overall CO2 capture in comparison with larger particles is better. As the slopes of the breakthrough curves are similar for two different gas flow rates, the external mass transfer resistance from bulk to the surface of the particles is, as expected, negligible. The breakthrough time using the pelletized limestone particles was shorter primarily due to the lower content of CaO as a result of the binder as the total mass loading into the bed was kept constant.

Impact of particle size and gas flow rate on the breakthrough curve. a Pelletized limestone. b Natural limestone. Flow rates at 1 barg and 20 °C
Table 4 presents the carbonation conversion at the top, middle, and bottom of the bed. The axial variation between the lowest and highest values was always less than 8% points for a given operating condition. This shows that a relatively uniform carbonation conversion along the bed was obtained, confirming the necessity of both moist air and pre-hydration (see axial conversion variation in Table 3).
Moreover, a mass balance on CO2 in the gas phase was performed to confirm the sorbent carbonation conversion measured by TGA:
where n is the number of CO2 moles that are adsorbed in the bed, P is the pressure of the system, Q is volumetric flow rate, R is the ideal gas constant, η is the ratio of the area above the breakthrough curve to the total area at time t where C/C0 = 0.9, and T is temperature. To find the surface area above the breakthrough curve, the break point is defined as C/C0 = 0.1 and the end of the breakthrough curve is where C/C0 = 0.9. The difference between estimates was always less than 10% (average of 5.6%).
In the next stage of the experimental program, the pelletized lime and natural lime particles with a diameter range of 425–600 μm were used in a series of nine carbonation/calcination cycles. These cycles consisted of CO2 capture from air at ambient conditions (carbonation) and regeneration of sorbents (calcination) at 850 °C in air for 75 min. After each carbonation period, the sorbent conversion was measured by TGA and after each calcination period, the sorbent BET surface area was measured. This series of experiments was then repeated for pelletized lime at the more realistic calcination conditions of 920 °C in pure CO2 for 12 min (representing oxy-fuel combustion), while the rest of the operating conditions remained constant.
3.3 Performance of sorbents in series of carbonation/calcination cycles
Table 5 shows the carbonation conversion and BET surface area after selected cycles for pelletized and natural limestone particles. Carbonation conversion of pellets decreased from 80 to 71% after 9 cycles (10 calcinations) due to sintering and loss of porosity during calcination. Although the natural limestone sorbents have an initial carbonation conversion greater than that of the pelletized limestone sorbents, after the same number of cycles, their net decay due to sintering is greater since the pellets have mayenite, which helps better maintain the structural morphology of the particle.
Here, carbonation conversions are relatively high due to the mild calcination conditions of air at 850 °C and for comparison the performance of pelletized limestone calcined for 5 cycles using CO2 at 920 °C is presented in Table 6. The higher calcination temperature results in much greater reduction in pore surface area such that the carbonation conversion dropped below 60% after 5 carbonation cycles.
3.4 Modeling carbonation kinetics
Previous investigations on DAC via lime-based sorbents (Beruto and Botter 2000; Dheilly et al. 2002; Nikulshina et al. 2007) used small particles that did not present significant transport limitations. This study explores performance of larger particles (250–425 μm and 425–600 μm) that are suitable for use in fixed or fluidized beds. Hence, fitting the breakthrough curves from this study with the previously developed surface reaction rate-limiting models is not suitable since diffusional resistance plays a more pronounced role. For non-catalytic gas–solid reactions, the unreacted shrinking core model is one of the simplest models as it does not require structural parameters of the reacting solid. Usually, three major steps are considered in such models:
Diffusion of the gaseous reactant (CO2) through the gaseous film surrounding the particle to the particle surface; not a rate-limiting step in the present setup.
Diffusion of the gaseous reactant through the product layer to the unreacted core; characterized by the effective diffusivity, De.
Reaction of the gaseous reactant with the solid at the unreacted core surface; characterized by Ks, the surface reaction rate constant.
In this research, the model accounts for reactions given in Eqs. 1–7. Moreover, as explained above, the formation of the Ca(HCO3)2 on the product layer enhances the diffusion of CO2 to the bulk of the particle. However, continued formation of CaCO3 impedes further CO2 diffusion. The model predicts the time required to reach a given conversion, based on the following equations, as detailed by Levenspiel (1972):



where ƮDP, ƮMT, and ƮR,SC are the characteristic times for diffusion of the gaseous reactant through the product layer of the particles, external mass transfer from the bulk gas to the surface of the particle, and chemical reaction at the interface between the unreacted core of the pellet and the product layer, respectively, and XB is conversion, calculated as:
To find the relationship between C0 and XB at any point on the breakthrough curve, a mass balance on a differential axial cross-section element of the bed based on the method proposed by Fenouil and Lynn (1996) was used:
where ut is the moving velocity of the transition zone, calculated by an overall CO2 mass balance over the bed:
Initially, there is no protective layer (CaCO3) and carbonation rate is controlled by the kinetics of the chemical reaction. After the formation of the product layer (CaCO3), diffusion through this layer becomes the CO2 capture controlling step. Results from fitting the data presented in Fig. 5 are summarized in Table 7. Figures 6 and 7 compare experimental data and model predictions for conversion (XB in Eqs. 14 and 15). As it can be seen, although there are slight variations, the agreement between model and experimental measurements is generally very good.

Modeling of the breakthrough curves of pelletized limestone. adp = 250–425 μm and Q = 0.5 L/min. bdp = 250–425 μm and Q = 1.0 L/min. cdp = 425–600 μm and Q = 0.5 L/min. ddp = 425–600 μm and Q = 1.0 L/min. Flow rates at 1 barg and 20 °C

Modeling of the breakthrough curve of natural limestone. adp = 250–425 μm and Q = 0.5 L/min. bdp = 250–425 μm and Q = 1.0 L/min. cdp = 425–600 μm and Q = 0.5 L/min. ddp = 425–600 μm and Q = 1.0 L/min. Flow rates at 1 barg and 20 °C
From Fig. 5, it can be seen that the impact of gas flow rate was marginal in terms of affecting the shape of the breakthrough curve. The mass transfer resistance from the bulk gas to the surface of the particles was thus assumed in the model to be negligible, which was validated by the fact that the gas flow rate does not affect the fitting model parameters, Ks and De. For the pelletized limestone particles, the average effective diffusivity is 6.30 × 10−7 and 5.38 × 10−7 m2/s for particle diameters of 250–425 μm and 425–600 μm, respectively, whereas the surface reaction rate constant is similar for all conditions with an average value of 2.36 × 10−3 m/s. Effective diffusivity for smaller particles is higher than for larger particles, while the surface reaction rate constant is similar for both particle ranges.
Similar results are obtained with the natural limestone particles where smaller particles had a greater effective diffusivity and the surface reaction constant is similar for all conditions as well as for the average value obtained for the pelletized limestone. However, effective diffusivity of pelletized limestone is greater than for natural limestone. For instance, the average value of De is 5.38 × 10−7 m2/s for pelletized limestone with a particle diameter range of 425–600 μm, while its average value is 8.41 × 10−8 m2/s for natural limestone with the same particle size. Similarly, for the particle diameter range of 250–425 μm, the average value of De is 6.30 × 10−7 m2/s and 4.91 × 10−7 m2/s for the pelletized and natural limestone, respectively. This arises because the binder increases the pore surface area of the sorbent.
4 Conclusions
The performance of the natural limestone from Cadomin and the pelletized limestone (90% natural limestone and 10% calcium aluminate cement (CA-14)) was compared as sorbent for direct CO2 capture from air at ambient conditions in a fixed bed reactor. Results show that pre-hydration of the sorbents leads to a uniformly carbonated bed. Moreover, relative humidity is an important parameter on the carbonation of sorbents. In a dry system, the breakthrough time was very short and the carbonation conversion of the sorbents was low in comparison with a system with 55% relative humidity.
Comparing the performance of two sorbents in series of 9 carbonation/calcination cycles at 850 °C under air shows that, due to the presence of the binder in pelletized limestone sorbents, their net decay in carbonation conversion (from 80 to 71%) was less than for natural limestone (from 93 to 76%). Moreover, the decay in pore surface area (from 12.01 to 7.92 m2/g) compared with that of natural limestone (from 11.54 to 5.93 m2/g) shows that pelletized limestone does not sinter as much as natural limestone. Hence, pelletized limestone sorbents were chosen for further study under simulated oxy-fuel calcination conditions (920 °C in pure CO2). However, their carbonation conversion fell (from 81 to 59%) and pore surface area drastically decreased (from 12.01 to 3.20 m2/g) after 5 cycles. The presented results on sorbent performance are promising and show that DAC employing lime-based sorbents can be considered as a feasible strategy to mitigate climate change taking CO2 directly from the atmosphere.
The shrinking core model was used for mathematical modeling of breakthrough curve data. Based on preliminary observations, changing the flow rate does not have an effect on the overall kinetics of the reaction. Hence, the mass transfer resistance from bulk gas to the particle surface was assumed negligible. The carbonation of sorbents can be divided into two regimes: reaction-controlled and diffusion-controlled. Initially, the carbonation rate is controlled by the chemical reaction kinetics on the surface of the particle. As the reaction proceeds, the formation of the product layer on the surface of the particles moves the reaction into a diffusion-controlled regime. Results from mathematical modeling also confirm these hypotheses. Changing the flow rate does not affect the fitting values of effective diffusivity or the surface reaction constant of the sorbents. Furthermore, the surface reaction constant of smaller particles is similar to that of larger particles, while their effective diffusivity was greater. The average effective diffusivity, as expected, was greater for pelletized limestone than for natural limestone. This can be attributed to their higher porosity. Therefore, the main difference between the pelletized limestone and natural limestone in CO2 capture at ambient conditions arises from the higher effective diffusivity of pelletized limestone.
Abbreviations
- A:
-
Calcium oxide fraction in the particle (–)
- C 0 :
-
CO2 concentration in inlet air (ppm)
- C :
-
CO2 concentration (ppm)
- C Ag :
-
Inlet gas concentration (kg/m3)
- d p :
-
Particle diameter (m)
- D :
-
Column diameter (m)
- D e :
-
Effective diffusivity (m2/s)
- k :
-
Boltzmann constant (J/K)
- K s :
-
Surface reaction constant (m/s)
- K g :
-
External mass transfer coefficient (m/s)
- m :
-
Mass (g)
- n :
-
Number of moles (mol)
- M :
-
Molecular weight (g/mol)
- P :
-
Pressure (Pa)
- Q :
-
Volumetric flow rate (m3/s)
- r c :
-
Unreacted core radius (m)
- R :
-
Particle radius (m)
- R′:
-
Gas constant (J/(K·mol))
- Re :
-
Reynolds number (–)
- t :
-
Time (s)
- T :
-
Temperature (K)
- u t :
-
Moving velocity of the transition zone (m/s)
- u g :
-
Superficial velocity of the gas into the reactor (m/s)
- X :
-
Fractional conversion (–)
- X′:
-
Carbonation conversion (Eq. 8) (–)
- X B :
-
Carbonation conversion (Eq. 14) (–)
- η :
-
Adsorption ratio (–)
- ρs :
-
Density (kg/m3)
- ρB :
-
Bulk density (kg/m3)
- Ʈ MT :
-
Characteristic time for external mass transfer (Eq. 11) (s)
- Ʈ DP :
-
Characteristic time for diffusion (Eq. 12) (s)
- Ʈ R,SC :
-
Characteristic time for chemical reaction (Eq. 13) (s)
- ε fb :
-
Bed void fraction (–)
References
Akehata T, Sato K (1985) Flow distribution in packed beds. J Chem Eng Japan 27:430–436
Astarita G, Savage DW, Bisio A (1993) Gas treating with chemical solvents. Wiley, New York
Beruto DT, Botter R (2000) Liquid like H2O adsorption layers to catalyze the Ca(OH)2/CO2 solid-gas reaction and to form a non-protective solid product layer at 20 °C. J Eur Ceram Soc 20:497–503
Bottoms RR (1930) U.S. Patent No. 1,783,901
Carbon Engineering (2019) Direct air capture. http://carbonengineeringcom/about-dac/. Accessed 15 Jan 2019
Choi S, Drese JH, Eisenberger PM, Jones CW (2011) Application of amine-tethered solid sorbents for direct CO2 capture from the ambient air. Environ Sci Technol 45:2420–2427
Dheilly RM, Tudo J, Sebaı̈bi Y, Quéneudec M (2002) Influence of storage conditions on the carbonation of powdered Ca(OH)2. Constr Build Mater 16:155–161
Erans M, Jeremias M, Zheng L, Yao JG, Blamey J, Manovic V, Fennell PS, Anthony EJ (2018) Pilot testing of enhanced sorbents for calcium looping with cement production. Appl Energy 225:392–401
Fenouil LA, Lynn S (1996) Design of entrained-flow and moving, packed, and fluidized-bed sorption systems: grain-model kinetics for hot coal-gas sulfurization with limestone. Ind Eng Chem Res 35:1024–1043
Gunn DJ (1968) Mixing in packed and fluidized beds. Chem Eng J CE153–172
Gunn DJ, Pryce C (1969) Dispersion in packed beds. Trans IChemE 47:T341–T350
International Energy Agency (IEA) (2011) CO2 emissions from fuel combustion
International Energy Agency (IEA) (2013) CO2 emissions from fuel combustion
International Panel on Climate Change (IPCC) (2007) Fourth assessment report of the Intergovernmental Panel on Climate Change “climate change 2007 - the physical science basis”
Kalinkin AM, Kalinkina EV, Zalkind OA, Makarova TI (2005) Chemical interaction of calcium oxide and calcium hydroxide with CO2 during mechanical activation. Inorg Mater 41:1073–1079
Keith DW, Ha-Duong M, Stolaroff JK (2006) Climate strategy with CO2 capture from the air. Climate Change 7474:17–45
Levenspiel O. (1972) Chemical reaction engineering, 2nd ed, chapter 13, pp 357–371
Lüthi D, Floch ML, Bereiter B, Blunier T, Barnola JM, Siegenthaler U, Raynaud D, Jouzel J, Fischer H, Kawamura K, Stocker TF (2008) High-resolution carbon dioxide concentration record 650,000-800,000 years before present. Nature 453:379–382
Mahmoudkhani M, Keith DW (2009) Low energy sodium hydroxide recovery for CO2 capture from atmospheric air - thermodynamic analysis. Int J Greenh Gas Contr 3:376–384
Mahmoudkhani M, Heidel KR, Ferreira JC, Keith DW, Cherry RS (2009) Low energy packed tower and caustic recovery for direct capture of CO2 from air. Energy Procedia 1:1535–1542
Manovic V, Anthony EJ (2009a) CaO-based pellets supported by calcium aluminate cements for high-temperature CO2 capture. Environ Sci Technol 43:7117–7122
Manovic V, Anthony EJ (2009b) Long-term behavior of CaO-based pellets supported by calcium aluminate cements in a long series of CO2 capture cycles. Ind Eng Chem Res 48:8906–8912
Manovic V, Anthony EJ (2010) CO2 carrying behavior of calcium aluminate pellets under high temperature/high-CO2 concentration calcination conditions. Ind Eng Chem Res 49:6916–6922
Manovic V, Wu Y, He I, Anthony EJ (2012) Spray water reactivation pelletization of spent CaO-based sorbents from calcium looping cycles. Environ Sci Technol 46:12720–12725
Nakicenovic N. et al. (2000) Emissions scenarios. A special report of working group III of the IPCC, Cambridge University Press: pp. 599
Nikulshina V, Hirsch D, Mazzotti M, Steinfield A (2006) CO2 capture from air and co-production of H2 via the Ca(OH)2-CaCO3 cycle using concentrated solar power - thermodynamic analysis. Energy 31:1379–1389
Nikulshina V, Gavez ME, Steinfeld A (2007) Kinetic analysis of the carbonation reactions for the capture of CO2 from air via the Ca(OH)2-CaCO3-CaO solar thermochemical cycle. Chem Eng J 129:75–83
Petit JR, Jouzel J, Raynaud D, Barkov NI, Barnola JM, Basile I, Bender M, Chappellaz J, Davis M, Delaygue G, Delmotte M, Kotlyakov VM, Legrand M, Lipenkov VY, Lorius C, Pepin L, Ritz C, Saltzman E, Stievenard M (1999) Climate and atmospheric history of the past 420,000 years from the Vostok ice core, Antarctica. Nature 399:429–436
Pontiga F, Valverde JM, Moreno H, Duran-Olivencia FJ (2013) Dry gas-solid carbonation in fluidized beds of Ca(OH)2 and nanosilica/Ca(OH)2 at ambient temperature and low CO2 pressure. Chem Eng J 222546-552
Ranade VV, Chaudhari R, Gunjal PR (2011) Trickle bed reactors: reactor engineering & applications, chapter 4
Ranjan M, Herzog HJ (2011) Feasibility of air capture. Energy Procedia 4:2869–2876
Sculley JP, Zhou HC (2012) Enhancing amine-supported materials for ambient air capture. Angewandte Chemie International Edition 12660–12661
Stephenson JL, Stewart WE (1986) Optical measurement of porosity and fluid motion in packed beds. Chem Eng Sci 42:2161–2170
Tian S, Jiang J, Zhang Z, Manovic V (2018) Inherent potential of steelmaking to contribute to decarbonisation targets via industrial carbon capture and storage. Nat Commun 9:1–8
Yang RY, Zou RP, Yu AB (2003) Effect of material properties on the packing of fine particles. J Appl Phys 94:3025–3034
Zheng L, Hills TP, Fennell P (2016) Phase evolution, characterisation, and performance of cement prepared in an oxy-fuel atmosphere. Faraday Discuss 192:113–124
Funding
The authors would like to thank CanmetENERGY (Natural Resources Canada), the Natural Sciences and Engineering Research Council of Canada, and the Carbon Management Canada for their financial support for this research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 17 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Samari, M., Ridha, F., Manovic, V. et al. Direct capture of carbon dioxide from air via lime-based sorbents. Mitig Adapt Strateg Glob Change 25, 25–41 (2020). https://doi.org/10.1007/s11027-019-9845-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11027-019-9845-0