
Abstract
Alzheimer's disease (AD) is a neurodegenerative disorder with early autophagy deficits. Our study probed the role of lysosomal-related genes (LRGs) in AD. Using the Gene Expression Omnibus (GEO) database, we analyzed differentially expressed genes (DEGs) in AD. AD-related genes and lysosomal-related genes (LRGs) were extracted from public databases. Leveraging the UpSetR package, we identified differentially expressed LRGs (DE-LRGs). Subsequently, consensus cluster analysis was used to stratify AD patients into distinct molecular subtypes based on DE-LRGs. Immune cell patterns were studied via Single-Sample Gene Set Enrichment Analysis (ssGSEA). Molecular pathways were assessed through Gene Set Variation Analysis (GSVA), while Mendelian Randomization (MR) discerned potential gene-AD causations. To reinforce our bioinformatics findings, we conducted in vitro experiments. In total, 52 DE-LRGs were identified, with LAMP1, VAMP2, and CTSB as standout hub genes. Leveraging the 52 DE-LRGs, AD patients were categorized into three distinct molecular subtypes. Interestingly, the three aforementioned hub genes exhibited significant predictive accuracy for AD differentiation across the subtypes. The ssGSEA further illuminated correlations between LAMP1, VAMP2, and CTSB with plasma cells, fibroblasts, eosinophils, and endothelial cells. GSVA analysis underscored significant associations of LAMP1, VAMP2, and CTSB with NOTCH, TGFβ, and P53 pathways. Compellingly, MR findings indicated a potential causative relationship between LAMP1, CTSB, and AD. Augmenting our bioinformatics conclusions, in vitro tests revealed that LAMP1 potentially alleviates AD progression by amplifying autophagic processes. LAMP1 and CTSB emerge as potential AD biomarkers, paving the way for innovative therapeutic interventions
Similar content being viewed by others
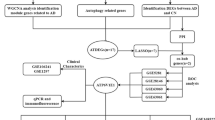

Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a profoundly distressing neurodegenerative disorder predominantly afflicting the elderly, leading to significant cognitive decline (Rostagno 2022). Current estimations highlight the grave magnitude of this ailment, affecting approximately 50 million individuals globally while indirectly impacting the lives of countless others (2021). The characteristic pathological hallmarks of AD encompass the formation of amyloid-beta (Aβ) plaques in distinct brain regions, the presence of neurofibrillary tangles within neurons, and the insidious loss of synapses and neurons (Li et al. 2017). Despite extensive research, the precise etiology of AD remains enigmatic. Nonetheless, a complex interplay of genetic predispositions, environmental exposures, and lifestyle choices is posited to underlie its pathogenesis (2021). Current pharmacological interventions primarily provide symptomatic relief, aiming to retard the progression of cognitive, behavioral, and psychological symptoms associated with dementia (Passeri et al. 2022). While these treatments may temporarily stabilize or improve cognitive functioning and manage behavioral manifestations, their effects are largely palliative. They alleviate AD’s symptoms without addressing its root cause (Cummings et al. 2019; Fish et al. 2019; Passeri et al. 2022). The unmet need for a comprehensive understanding of AD’s underlying mechanisms and novel therapeutic targets remains a pressing concern.
Central to our cellular machinery, autophagy, an evolutionarily conserved lysosome-dependent process in eukaryotes, plays an indispensable role in modulating protein metabolism. By overseeing the degradation and subsequent recycling of damaged organelles and misfolded proteins, autophagy ensures protein homeostasis (Zhang et al. 2021). Impaired autophagy is postulated to aggravate the accumulation of toxic amyloid-beta (Aβ) aggregates, signature pathological features of AD (Zhang et al. 2021). Lysosomes, specialized cellular organelles laden with hydrolytic enzymes, are integral to autophagy (Yu et al. 2018). As our understanding of neurodegenerative diseases evolves, the interconnection between lysosomal function and neurodegeneration gains prominence. Specifically, lysosomal dysfunctions in glial and neuronal cells are implicated in the propagation of Aβ and tau proteins in AD, as well as α-synuclein in Parkinson’s disease (Udayar et al. 2022). Based on transgenic dual-fluorescence probe, Lee et al. identified unique autophagy dysregulation in five different APP-AD mouse models, and such failure was established to originate from an early decrease of autolysosomes/lysosomes acidification (Lee et al. 2022), highlighting the potential of autophagy/lysosome regulators as therapeutic agents in AD. For example, as a lysosomal cysteine protease, Cathepsin B has been recognized as candidate drug target for AD (Hook et al. 2020). Lysosomal-related genes (LRGs) have been observed to exhibit altered expression profiles in an array of diseases, including Parkinson’s disease (Chen et al. 2021), hepatocellular carcinoma (Tian et al. 2023), and gastric cancer (Wang et al. 2023). While direct correlations between LRGs and AD are still in their formative stages, the evident dysregulation of LRGs in other conditions sparks a compelling hypothesis regarding their potential involvement in AD.
In light of these observations, our study endeavors to elucidate the role of DE-LRGs in AD through a combination of bioinformatics and rigorous experimental validations. We earnestly hope that our contributions not only enhance the current knowledge paradigm about this debilitating condition but also guide the development of innovative therapeutic strategies, offering respite to millions affected by this devastating ailment.
Materials and methods
Data collection and preprocessing
Gene chip datasets specific to AD, GSE132903 (97 disease cases, 98 controls) and GSE138260 (17 disease cases, 19 controls), were sourced from the Gene Expression Omnibus (GEO) database. Probes were annotated according to the gene chip platforms GPL10558 and GPL27556, respectively. Pertinent genes for AD were retrieved from the GeneCards (Safran et al. 2010) and DisGeNET (Piñero et al. 2017) databases. After eliminating duplicates and computing the intersection, the relevant AD-associated genes were selected. Lysosomal-related genes (LRGs) and pathways were acquired from the Human Lysosome Gene Database (hLGDB) (Brozzi et al. 2013) and the Molecular Signatures Database (MSigDB) (Liberzon et al. 2015). Duplication among the LRGs extracted from these databases was further pruned.
Differential gene expression analysis
For the GSE132903 dataset, the “limma” package (Ritchie et al. 2015) was employed to identify differentially expressed genes (DEGs) by comparing AD and normal samples, applying criteria of P < 0.05 and |logFC| > 0.1. The overlap between DEGs and LRGs was computed using the UpSetR package (Conway et al. 2017), leading to the identification of DE-LRGs.
Construction of protein-protein interaction (PPI) network
The STRING database (von Mering et al. 2003), a well-established resource for studying protein-protein interactions including both physical interactions and functional associations, was utilized to construct the PPI network. Interactions with a confidence score between 0.4 and 10 were considered valid. Hub genes within the network were pinpointed using the CytoHubba plugin and its MCC, MNC, and Degree algorithms.
Single-sample gene set enrichment analysis (ssGSEA)
Marker genes for 23 common human immune cells were derived from the CellMarker database (Zhang et al. 2019), a comprehensive resource providing cell markers for various cell types in both human and mouse tissues. Using the GSE132903 dataset, the ssGSEA method within the “GSVA” package of R 4.2.2 was employed to infer the scores of immune cells across samples. Subsequently, the t-test was used to compare the immune cell scores between the AD and normal samples.
Gene set variation analysis (GSVA)
The Hallmark gene set (h.all.v2023.1.Hs.symbols.gmt) was acquired from the MSigDB database and used as the background gene set. With GSE132903 dataset as the foundation, the “gsva” method in the GSVA package was used to perform GSVA enrichment analysis on this background gene set. Significant differences in pathways between the disease and control groups were subsequently pinpointed using the t-test.
Identification of AD subtypes through consensus clustering
Based on the DE-LRGs, unsupervised consensus clustering analysis was conducted to identify AD molecular subtypes and define the number of clusters via the “ConsensusClusterPlus” R package in the GSE132903 dataset. Clustering was determined where the cumulative distribution function value showed stability. Post this, differences in immune cell infiltration scores between groups were analyzed using a t-test, identifying immune cells with differential infiltration scores. Further, the DrugBank database was utilized to search for AD-related drugs and their target genes. A total of two drugs (memantine and ginkgo biloba) and their six target genes (NMDA1, HTR3A, GRIN1, GLRA1, NOS2, and MAOA) were identified. Differential expression of these six target genes among the subtypes was validated using the t-test.
Mendelian randomization (MR) study
MR is a causal inference method grounded in genetic variation. Essentially, it leverages the naturally randomized allocation of genotypes and their impact on phenotypes to infer the effects of biological factors on diseases. Using the R 4.2.2 and the TwoSampleMR package, SNP data that can affect core genes was searched from the GWAS database (https://gwas.mrcieu.ac.uk/) to act as exposure factors. With AD as the outcome variable, MR analysis was carried out. The relationship between the core gene levels and AD risk was evaluated using the Inverse Variance Method (IVM). Further sensitivity analysis was executed using the MR-Egger method.
Cell culture and treatment
Human neuroblastoma SH-SY5Y cells were procured from Pricella (Wuhan, China). The cells were maintained in Dulbecco’s Modified Eagle’s Medium (Gibco, NY, USA) enriched with 10% fetal bovine serum (Gibco). Cultures were kept at 37 °C under a 5% CO2 humidified atmosphere. Cells were exposed to 25 µM of Aβ25-35 (MedChemExpress, Shanghai, China) for 24 h to establish an Aβ-induced neurotoxicity model as previously described (Li et al. 2020).
Cell transfections
The lentiviral vector system was utilized to overexpress LAMP1, and the used vectors were obtained from VectorBuilder Inc (Guangzhou, China). The sequence of mature LAMP1 was obtained from the NCBI database. Vectors that carry LAMP1 (oe-LAMP) and corresponding negative control (oe-NC) sequences were prepared, and together with lentiviral packaging plasmids (pMDLg/pRRE: pVSV-G: pRSV-Rev = 5 : 3 : 2) were transfected into 293T cells utilizing HighGene transfection reagent. Lentivirus particles were collected and concentrated following 48 h for transfecting. For lentivirus infections, SH-SY5Y cells were seeded to a 6-well plate at a density of 2 × 105/mL, and lentivirus (1 × 108 TU/mL) was added to the medium at 70–90% fusion degree. Following 48 h for infections, stable transfected cells were obtained.
Total RNA isolation and quantitative real-time PCR (qRT-PCR) analysis
Total RNA was harvested utilizing Trizol reagent (Invitrogen, CA, USA). The isolated RNAs were then reverse transcribed to cDNA using the PrimeScript™ RT-PCR Kit (Takara, Beijing, China). qRT-PCR was conducted on a CFX Connect instrument (Bio-Rad, CA, USA). The reaction mixture, with a total volume of 20 µL, contained 10 µL of SYBR Green PCR Master (Lifeint, Xiamen, China). The thermal cycler conditions were set as follows: an initial denaturation at 95 °C for 3 min, followed by 40 cycles of 95 °C for 12 s, 62 °C for 40 s. The relative expression of the genes was determined by the 2−ΔΔCT method, taking GAPDH as the reference gene for normalization. The primer sequences employed for amplifying the genes of interest were as follows: LAMP1: Forward: 5ʹ-GGT AAC GCC GCT GTC TCT A-3ʹ, Reverse: 5ʹ-TGT TCA CAG CGT GTC TCT CC-3ʹ. GAPDH: Forward: 5ʹ-GAA GGT CGG AGT CAA CGG AT-3ʹ, Reverse: 5ʹ-CTT CCC GTT CTC AGC CAT GT-3ʹ.
Cell viability assessment
For the purpose of analyzing cell viability, SH-SY5Y cells (5 × 103 cells per well) were seeded in 96-well plates. The cell viability was examined at 24 h post-culture using the CCK-8 kit (Beyotime, Shanghai, China). Absorbance measurements were taken using the microplate reader (Wuxi Hiwell Diatek, China).
Transmission electron microscopy (TEM)
SH-SY5Y cells were initially fixed using chilled 2.5% glutaraldehyde and subsequently post-fixed employing 1% osmium tetraoxide. Following a graded dehydration in escalating ethanol concentrations, cells were encapsulated in Epon 812 resin and sectioned. The obtained ultra-thin slices were treated with uranyl acetate and lead citrate for staining. Microscopic observations were conducted using a transmission electron microscope (FEI, OR, USA).
Western blotting
Cells were lysed using RIPA buffer (Beyotime). Equal protein amounts were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was blocked with 5% nonfat milk in Tris-buffered saline 0.1% Tween 20 (TBST) for 1 h and then incubated overnight at 4 °C with the primary antibodies (anti-LAMP1, 1:1000, ab278043; anti-LC3, 1:2000, ab192890; anti-p62, 1:1000, ab207305; and anti-GAPDH, 1:2500, ab181602; Abcam, Cambridge, UK). After washing, the membrane was treated with the secondary antibody (1:10000, Abcam, ab205718) for 1 h at room temperature. Post-wash, bands were visualized using ECL (Merck Millipore, MA, USA).
Statistical analysis
Results are represented as means ± standard deviation, derived from a minimum of three independent trials. For multiple comparisons, one-way ANOVA followed by Tukey’s post hoc test was employed. A p-value below 0.05 was deemed statistically significant. All statistical evaluations were carried out using GraphPad software (version 7.0, CA, USA).
Results
Identification of DE-LRGs
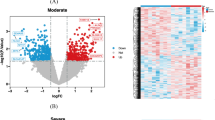
By mining the GeneCards and DisGeNET databases, we extracted 2458 AD-associated genes after duplicate elimination (Fig. 1A). Within the gene chip dataset GSE132903, 5828 DEGs (3004 upregulated and 2824 downregulated) were pinpointed (Fig. 1B) and the expression of top 30 DEGs have obvious distinction between AD and normal groups (Fig. 1C). From the hLGDB and MSigDB databases, 567 LRGs were retrieved. Cross-referencing AD-related genes, DEGs, and LRGs yielded 52 DE-LRGs (Fig. 1D).

Identification of differential expressed lysosomal-related genes (DE-LRGs). A Venn diagram of 2458 Alzheimer's disease (AD)-related genes from two public databases. B Volcano plot of differential expressed genes (DEGs) in GSE132903 dataset between AD patients and normal. C Expression heatmap of the top 30 DEGs. D Venn diagram of 52 DE-LRGs obtained based on the AD-related genes, DEGs, and lysosomal-related genes (LRGs)
PPI construction and hub genes identification
Utilizing STRING, a PPI network comprising 52 DE-LRGs was structured and subsequently visualized via Cytoscape (Fig. 2A). The CytoHubba plugin, employing MCC, MNC, and Degree as topological criteria, designated the top 10 hub genes (Fig. 2B). An intersection of these methods highlighted seven pivotal genes: LAMP1, VPS35, HSPA8, VAMP2, CHMP2B, IGF2R, and CTSB (Fig. 2C). Notably, LAMP1 and VAMP2 displayed significantly reduced expression in AD, contrary to CTSB which was upregulated (P < 0.05; Fig. 2D).

Construction of the Protein-Protein Interaction (PPI) network and identification of hub genes. A PPI network of the 52 DE-LRGs, visualized using the STRING database and Cytoscape. B Identification of the top 10 hub genes using three topological analysis methods: MCC, MNC, and Degree. C Venn diagram illustrating the intersection of the three methods to finalize seven critical hub genes. D The expression of LAMP1, VAMP2, and CTSB in GSE138260 dataset was analyzed. *P < 0.05, **P < 0.01, ***P < 0.001
Assessment of immune cell infiltration
Using ssGSEA function of GSEA to analyze the scores of 23 immune cells, and the boxplot showed that AD patients manifested increased ssGSEA scores of eight immune cells (plasma cells, T cells CD4 memory, NK cells, eosinophils, stromal cells, fibroblasts, endothelial cells, and T cells), while exhibiting diminished B cells naive scores (Fig. 3A). The association between 23 categories of immune cells demonstrated that the nine immune cells predominantly exhibited a positive correlation with each other (Fig. 3B).

Immune cell infiltration analysis in AD patients. A Boxplot displaying Single-Sample Gene Set Enrichment Analysis (ssGSEA) scores of the 23 immune cells in AD patients versus controls. B Correlation matrix of all 23 types of immune cell subtype compositions; blue represents positive correlation; orange represents negative correlation. ns, no significance; *P< 0.05, **P < 0.01, ***P < 0.001
Pathway enrichment analysis via GSVA
Utilizing a rigorous t-test examination paired with exhaustive literature screening, three pathways (NOTCH, TGFβ, and P53), were pinpointed as significantly associated with AD. As shown in Fig. 4, the scores of three identified pathways were markedly elevated in the AD group compared to the normal group.

Pathway Enrichment Analysis using Gene Set Variation Analysis (GSVA). Illustration of the identified pathways (NOTCH, TGFβ, and P53) and their associated expression levels in AD versus control groups. ****P< 0.0001
Consensus clustering analysis of AD subtypes
Consensus clustering analysis was performed using the “ConsensusClusterPlus” R package. The 52 DE-LRGs expressions were used to determine that k = 3 provided the most stable grouping (Fig. 5A). Since then, the AD samples in GSE132903 dataset have been split into three distinct categories in consensus clustering analysis (Fig. 5B). As shown by the PCA plot, gene expression patterns were distinct between the clusters (Fig. 5C). Then, the scores of 12 immune cells (T cells CD4 naive, plasma cells, B cells memory, monocytes, macrophages, dendritic cells, mast cells, eosinophils, neutrophils, fibroblasts, endothelial cells, and B cells) and in the expression levels of three drug-targeted genes (GRIN1, GLRA1, and MAOA) were significant differences in these three AD subtypes (P < 0.05; Fig. 5D, E), suggesting that the three subtypes of patients with AD are of good clinical relevance.

Consensus clustering of AD subtypes based on 52 DE-LRG expressions. A Determination of the optimal number of clusters (k = 3) using the “ConsensusClusterPlus” R package. B Representation of the three distinct AD subcategories in the gene chip data GSE132903. C Principal Component Analysis (PCA) plot, showcasing distinct gene expression patterns across the clusters. D, E Comparative analysis indicating variations in immune cell scores and expression levels of drug-targeted genes among the three AD subtypes. *P< 0.05, **P < 0.01, ****P < 0.0001
Hub genes correlation analysis
Through an intersectional analysis between the nine significantly differentiated immune cells across AD and normal groups and the 12 immune cells across three AD subtypes, a commonality of four immune cells (plasma cells, fibroblasts, eosinophils, and endothelial cells) was discerned (Fig. 6A). Subsequently, Pearson correlation analysis was conducted between the expression levels of three hub genes, the scores of the four immune cells, and the three pathways. As delineated in Fig. 6B, LAMP1 and CTSB expression was positively correlation with four immune cells, whereas VAMP2 expression was negatively correlation with them. Moreover, the scatter plots showcasing the significantly positive correlation between LAMP1 and three pathways, CTSB and TGFβ signaling; negative correlation between VAMP2 and NOTCH signaling or P53 pathway (Fig. 6C).

Analysis of hub gene correlations with immune cells and pathways. A Venn diagram representing the intersection between differentially expressed immune cells across AD versus control groups and among the three AD subtypes. B Scatter diagram detailing Pearson correlation between hub gene and four immune cells. C Scatter plots highlighting significant positive and negative correlations between hub genes and three pathways
MR results
MR analysis have indicated a causal relationship between genes LAMP1 and CTSB with AD, while no such link was identified for the gene VAMP2. As exhibited in Fig. 7A, B, using the IVW method, our analysis revealed that LAMP1 and CTSB was significantly associated with the risk of developing AD, with an odds ratio (OR) of 1.164 (95% CI = 1.021–1.326, P = 0.023) and 1.721 (95% CI = 1.043–2.840, P = 0.034). Conversely, the MR-Egger method did not yield any significant statistical association [LAMP1: OR = 0.980, 95% CI = 0.642–1.496, P = 0.925; CTSB: OR = 1.132, 95% CI = 0.330–3.884, P = 0.844]. The funnel plot displaying causal effects was approximately symmetrical (Fig. 7C), further suggesting that pleiotropy is unlikely to bias the causal effect.

Mendelian Randomization (MR) results for determining causal relationships between hub genes and AD risk. A Scatter plot showing the causal effect of LAMP1 and CTSB on the risk of AD. B Forest plot showing the causal effect of each SNP on the risk of AD. C Funnel plots to visualize overall heterogeneity of MR estimates for the effect of LAMP1 and CTSB on AD
LAMP1 augments autophagy in Aβ25-35-treated SH-SY5Y cells
To validate our bioinformatics findings, we detected the effect of LAMP1 on Aβ25-35-induced AD cellular model. Utilizing qRT-PCR, we observed a pronounced decreasing in the expression level of both LAMP1 mRNA and protein in SH-SY5Y cells treated with Aβ25−35 compared to controls, which was reversed by LAMP1 overexpression (P < 0.01; Fig. 8A, B). oe-LAMP1 transfection significantly reversed the decreased viability induced by Aβ25−35 (P < 0.01; Fig. 8C). TEM showing a significant increase in autolysosomes in cells overexpressing LAMP1 when exposed to Aβ25–35 (Fig. 8D). Western blotting corroborated these findings, showing LAMP1 overexpression reversed LC3-II/I downregulation and p62 upregulation in Aβ25−35-treated SH-SY5Y cells (P < 0.01; Fig. 8E).

LAMP1Augments Autophagy in Aβ25‐35-treated SH-SY5Y Cells. A, B Quantitative real-time PCR and western blot depicting expression levels of LAMP1 mRNA and protein in Aβ25-35 treated SH-SY5Y cells with and without LAMP1 overexpression. C The cell viability of SH-SY5Y cells across treatments over 24 h is illustrated. D, E Representative images (D) and quantification (E) of transmission electron micrographs showing the counts of autolysosomes across treatment groups, scale bar = 500 nm. F Western blot evaluations present the expression patterns of LC3-II/I and p62 among the various treatment groups. **P < 0.01 versus the control group, #P < 0.05, ##P< 0.01 versus the Aβ25‐35 group
Discussion
The intricate molecular architecture of AD is characterized by complex interactions that are crucial in determining its inception and progression. In our quest to better understand AD, our investigation centered on the role of DE-LRGs, revealing associations that not only enhance our understanding but also pinpoint potential therapeutic interventions.
Initially, we discerned 52 DE-LRGs by meticulously cross-referencing AD-specific genes, DEGs, and LRGs from diverse databases. The integration of gene sets derived from three distinct methodologies (MCC, MNC, and Degree), anchored in the PPI network, steered us towards three seminal DE-LRGs: LAMP1, VAMP2, and CTSB. Strikingly, LAMP1 and VAMP2 presented with a marked decrease in their expression within AD. Echoing our findings, research of Yamamoto et al. elucidated that an elevated transcription factor EB leads to an upsurge in LAMP1, influencing the autophagy-lysosomal pathway, thereby playing a pivotal role in Aβ generation within neurons (Yamamoto et al. 2019). The narrative surrounding direct linkage of VAMP2 to AD remains sparse in extant literature. However, modulation of CALM on autophagy, by orchestrating the endocytosis of soluble NSF attachment protein receptors (notably VAMP2), affects various facets of the autophagy trajectory, encompassing autophagosome inception to its degradation (Moreau et al. 2014). In tandem, the decline or absence of CTSB-CTSL, known regulators of lysosomal function and facilitators of the fusion between autophagosomes and lysosomes, culminates in lysosomal anomalies and subsequent Aβ accrual (Cermak et al. 2016). Bolstering this perspective, our MR outcomes reinforced the causative nexus between LAMP1 and CTSB in relation to AD. This inferred causality, especially when juxtaposed with empirical validations on SH-SY5Y cellular models, underscores the pivotal role of LAMP1 in autophagy modulation in AD scenarios.
Role of the immunological framework in pathology of AD is gaining increasing cognizance. Our analyses divulged a nuanced picture of immune cell infiltration within AD sufferers, echoing findings underscoring neuroinflammation as a distinctive feature of AD (Rajesh and Kanneganti 2022). This neuroinflammatory response could either influence or be impacted by dysregulated expression of LRGs. Our pathway enrichment exploration highlighted an indisputable connection of the NOTCH, TGFβ, and P53 pathways with AD. This liaison holds immense relevance considering the cardinal roles of these pathways in cellular dynamics like proliferation, differentiation, and apoptosis. Historical data correlates NOTCH signaling with neuronal genesis and sustenance, with its aberration potentially exacerbating AD (A Kapoor and Nation 2021). Similarly, the TGFβ and P53 pathways are intrinsically intertwined with neuronal vitality and programmed cell death in AD contexts (Jazvinšćak Jembrek et al. 2018; Kapoor and Chinnathambi 2023).
Furthermore, our consensus clustering accentuated inherent molecular diversity in the context of AD, insinuating the existence of distinct molecular subtypes. Such heterogeneity could elucidate disparities in manifestation, trajectory, and therapeutic responsiveness of AD — a paradigm gradually crystallizing within AD research circles (Badhwar et al. 2020).
Conclusion
In sum, our work elucidates the cardinal roles that DE-LRGs, particularly LAMP1, VAMP2, and CTSB, play in AD’s narrative. While these revelations present a formidable foundation for subsequent inquiry, a holistic comprehension demands rigorous in vivo experimentation, translational studies, and clinical assessments. By broadening our comprehension of LRGs, we advance incrementally towards demystifying AD, potentially heralding groundbreaking therapeutic avenues.
Data availability
No datasets were generated or analysed during the current study.
References
(2021) 2021 Alzheimer’s disease facts and figures. Alzheimers Dement 17:327–406. https://doi.org/10.1002/alz.12328
Badhwar A, McFall GP, Sapkota S, Black SE, Chertkow H, Duchesne S, Masellis M, Li L, Dixon RA, Bellec P (2020) A multiomics approach to heterogeneity in Alzheimer’s disease: focused review and roadmap. Brain 143:1315–1331. https://doi.org/10.1093/brain/awz384
Brozzi A, Urbanelli L, Germain PL, Magini A, Emiliani C (2013) hLGDB: a database of human lysosomal genes and their regulation. Database (Oxford) 2013:bat024. https://doi.org/10.1093/database/bat024
Cermak S, Kosicek M, Mladenovic-Djordjevic A, Smiljanic K, Kanazir S, Hecimovic S (2016) Loss of cathepsin B and L leads to lysosomal dysfunction, NPC-Like cholesterol sequestration and accumulation of the key Alzheimer’s proteins. PLoS ONE 11:e0167428. https://doi.org/10.1371/journal.pone.0167428
Chen YP, Gu XJ, Song W, Hou YB, Ou RW, Zhang LY, Liu KC, Su WM, Cao B, Wei QQ, Zhao B, Wu Y, Shang HF (2021) Rare variants analysis of lysosomal related genes in early-onset and familial Parkinson’s disease in a Chinese cohort. J Parkinsons Dis 11:1845–1855. https://doi.org/10.3233/jpd-212658
Conway JR, Lex A, Gehlenborg N (2017) UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33:2938–2940. https://doi.org/10.1093/bioinformatics/btx364
Cummings JL, Tong G, Ballard C (2019) Treatment combinations for Alzheimer’s disease: current and future pharmacotherapy options. J Alzheimers Dis 67 779– 94. https://doi.org/10.3233/jad-180766
Fish PV, Steadman D, Bayle ED, Whiting P (2019) New approaches for the treatment of Alzheimer’s disease. Bioorg Med Chem Lett 29 125– 33. https://doi.org/10.1016/j.bmcl.2018.11.034
Hook V, Yoon M, Mosier C, Ito G, Podvin S, Head BP, Rissman R, O’Donoghue AJ, Hook G (2020) Cathepsin B in neurodegeneration of Alzheimer’s disease, traumatic brain injury, and related brain disorders. Biochim Biophys Acta Proteins Proteom 1868:140428. https://doi.org/10.1016/j.bbapap.2020.140428
Jazvinšćak Jembrek M, Slade N, Hof PR, Šimić G (2018) The interactions of p53 with tau and Aß as potential therapeutic targets for Alzheimer’s disease. Prog Neurobiol 168 104– 27. https://doi.org/10.1016/j.pneurobio.2018.05.001
Kapoor M, Chinnathambi S (2023) TGF-β1 signalling in Alzheimer’s pathology and cytoskeletal reorganization: a specialized tau perspective. J Neuroinflammation 20:72. https://doi.org/10.1186/s12974-023-02751-8
Kapoor A, Nation DA (2021) Role of Notch signaling in neurovascular aging and Alzheimer’s disease. Semin Cell Dev Biol 116 90– 7. https://doi.org/10.1016/j.semcdb.2020.12.011
Lee JH, Yang DS, Goulbourne CN, Im E, Stavrides P, Pensalfini A, Chan H, Bouchet-Marquis C, Bleiwas C, Berg MJ, Huo C, Peddy J, Pawlik M, Levy E, Rao M, Staufenbiel M, Nixon RA (2022) Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nat Neurosci 25:688–701. https://doi.org/10.1038/s41593-022-01084-8
Li Q, Liu Y, Sun M (2017) Autophagy and Alzheimer’s disease. Cell Mol Neurobiol 37 377– 88. https://doi.org/10.1007/s10571-016-0386-8
Li LH, Peng WN, Deng Y, Li JJ, Tian XR (2020) Action of trichostatin A on Alzheimer’s disease-like pathological changes in SH-SY5Y neuroblastoma cells. Neural Regen Res 15:293–301. https://doi.org/10.4103/1673-5374.265564
Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P (2015) The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1:417−25. https://doi.org/10.1016/j.cels.2015.12.004
Moreau K, Fleming A, Imarisio S, Lopez Ramirez A, Mercer JL, Jimenez-Sanchez M, Bento CF, Puri C, Zavodszky E, Siddiqi F, Lavau CP, Betton M, O’Kane CJ, Wechsler DS, Rubinsztein DC (2014) PICALM modulates autophagy activity and tau accumulation. Nat Commun 5:4998. https://doi.org/10.1038/ncomms5998
Passeri E, Elkhoury K, Morsink M, Broersen K, Linder M, Tamayol A, Malaplate C, Yen FT, Arab-Tehrany E (2022) Alzheimer’s disease: treatment strategies and their limitations. Int J Mol Sci 23. https://doi.org/10.3390/ijms232213954
Piñero J, Bravo À, Queralt-Rosinach N, Gutiérrez-Sacristán A, Deu-Pons J, Centeno E, García-García J, Sanz F, Furlong LI (2017) DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res 45:D833–d9. https://doi.org/10.1093/nar/gkw943
Rajesh Y, Kanneganti TD (2022) Innate immune cell death in neuroinflammation and Alzheimer’s disease. Cells 11. https://doi.org/10.3390/cells11121885
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43:e47. https://doi.org/10.1093/nar/gkv007
Rostagno AA (2022) Pathogenesis of Alzheimer’s disease. Int J Mol Sci 24. https://doi.org/10.3390/ijms24010107
Safran M, Dalah I, Alexander J, Rosen N, Iny Stein T, Shmoish M, Nativ N, Bahir I, Doniger T, Krug H, Sirota-Madi A, Olender T, Golan Y, Stelzer G, Harel A, Lancet D (2010) GeneCards Version 3: the human gene integrator. Database (Oxford).2010, baq020. https://doi.org/10.1093/database/baq020
Tian W, Li C, Ren J, Li P, Zhao J, Li S, Dong D (2023) Identification of PPT1 as a lysosomal core gene with prognostic value in hepatocellular carcinoma. Biosci Rep 43. https://doi.org/10.1042/bsr20230067
Udayar V, Chen Y, Sidransky E, Jagasia R (2022) Lysosomal dysfunction in neurodegeneration: emerging concepts and methods. Trends Neurosci 45 184– 99. https://doi.org/10.1016/j.tins.2021.12.004
von Mering C, Huynen M, Jaeggi D, Schmidt S, Bork P, Snel B (2003) STRING: a database of predicted functional associations between proteins. Nucleic Acids Res 31:258–261. https://doi.org/10.1093/nar/gkg034
Wang Q, Liu Y, Li Z, Tang Y, Long W, Xin H, Huang X, Zhou S, Wang L, Liang B, Li Z, Xu M (2023) Establishment of a novel lysosomal signature for the diagnosis of gastric cancer with in-vitro and in-situ validation. Front Immunol 14:1182277. https://doi.org/10.3389/fimmu.2023.1182277
Yamamoto F, Taniguchi K, Mamada N, Tamaoka A, Kametani F, Lakshmana MK, Araki W (2019) TFEB-mediated enhancement of the autophagy-lysosomal pathway dually modulates the process of amyloid β-Protein generation in neurons. Neuroscience 402:11–22. https://doi.org/10.1016/j.neuroscience.2019.01.010
Yu L, Chen Y, Tooze SA (2018) Autophagy pathway: cellular and molecular mechanisms. Autophagy 14:207–15. https://doi.org/10.1080/15548627.2017.1378838
Zhang X, Lan Y, Xu J, Quan F, Zhao E, Deng C, Luo T, Xu L, Liao G, Yan M, Ping Y, Li F, Shi A, Bai J, Zhao T, Li X, Xiao Y (2019) CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res 47:D721–d8. https://doi.org/10.1093/nar/gky900
Zhang Z, Yang X, Song YQ, Tu J (2021) Autophagy in Alzheimer’s disease pathogenesis: therapeutic potential and future perspectives. Ageing Res Rev 72:101464. https://doi.org/10.1016/j.arr.2021.101464
Acknowledgements
The authors have no acknowledgments to report.
Funding
This work was supported by [Zhejiang Traditional Chinese Medicine Administration] (Grant numbers [2023ZF104]).
Author information
Authors and Affiliations
Contributions
Xiangzhen Zhu: conceptualization, writing–original draft, and formal analysis. Jingfang Gao: project administration, visualization. Chao Qiu: conceptualization, funding acquisition and writing–review & editing. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, X., Gao, J. & Qiu, C. Integrative analysis reveals key lysosomal genes as potential therapeutic targets in Alzheimer’s disease. Metab Brain Dis (2024). https://doi.org/10.1007/s11011-024-01409-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11011-024-01409-5