
Abstract
Myostatin, a potent negative regulator of skeletal muscle mass, has garnered significant attention as a therapeutic target for muscle dystrophies. Despite extensive research and promising preclinical results, clinical trials targeting myostatin inhibition in muscle dystrophies have failed to yield substantial improvements in muscle function or fitness in patients. This review details the mechanisms behind myostatin’s function and the various inhibitors that have been tested preclinically and clinically. It also examines the challenges encountered in clinical translation, including issues with drug specificity, differences in serum myostatin concentrations between animal models and humans, and the necessity of neural input for functional improvements. Additionally, we explore promising avenues of research beyond muscle dystrophies, particularly in the treatment of metabolic syndromes and orthopedic disorders. Insights from these alternative applications suggest that myostatin inhibition may hold the potential for addressing a broader range of pathologies, providing new directions for therapeutic development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myostatin (MSTN), also known as growth and differentiation factor 8 (GFD8), is a member of the transforming growth factor β (TGF-β) superfamily of signaling proteins, and functions as a negative regulator of skeletal muscle mass. It was first described by McPherron et al. in 1997, where MSTN-knockout mice displayed a super-muscled phenotype. MSTN is highly conserved among mammalian species, and natural mutations have been observed to cause increased muscle mass in cattle [1,2,3], dogs [4], sheep [5], and humans [6]. Conversely, overexpression of MSTN causes muscle atrophy [7].
MSTN’s relationship with muscle growth has led to the widespread study of its inhibition for the treatment of muscle, bone, and metabolic diseases, as well as enhancing agricultural meat production [8,9,10,11]. Clinical trials utilizing MSTN inhibitors began in the early 2000s, primarily aiming to increase muscular function and survivability in muscular dystrophies. Despite achieving widespread success in preliminary animal trials, the journey to market for MSTN-based drugs has been largely disappointing, as none of the clinically tested inhibitors have been approved for mediating muscle mass [12,13,14]. Although the clinical failures of muscular dystrophy-targeting drugs have branded MSTN inhibitors as a fruitless endeavor, recent advancements in inhibitor application and design offer promising prospects for developing viable MSTN therapeutics. The purpose of this review is to provide an updated overview of current research on MSTN inhibition in the treatment of various pathologies. Specifically, we seek to provide a better understanding of clinical developments and investigate the underlying reasons for the high rate of trial failures. Lastly, we explore potential inhibitor design choices and understudied pathologies that may be better suited for MSTN-inhibition therapeutics.
Mechanism of myostatin actions
MSTN is primarily expressed in skeletal muscle but is also expressed to a lesser extent in adipose tissue [15], heart [16], and kidney [17]. Like most other members of the TGF-β family, MSTN is secreted as an inactive precursor, comprised of an N-terminal signal peptide, N-terminal propeptide, and C-terminal growth factor (GF) domain [18]. In the endoplasmic reticulum, the MSTN precursor undergoes dimerization at the C-terminus, forming a complex known as promyostatin (proMSTN). Initially, the signal peptide is removed by signal peptidase. Furin-like proteases then cleave the MSTN precursor at a conserved tetrabasic site of RXXR amino acid sequence located between the propeptide and GF regions, resulting in an inactive complex consisting of dimerized GF peptides with supporting propeptide regions, which are non-covalently bound to keep MSTN in a latent state (Fig. 1) [19,20,21]. This pro-form of MSTN has higher abundance and longevity than its active mature form, which has a shorter temporal and spatial activity window [20, 22]. MSTN is activated following the cleavage of its propeptides by bone morphogenetic protein-1 (BMP-1)/tolloid (TLD)-like metalloproteinases at an arginine residue. This action releases the propeptides from the dimerized GF region, allowing mature MSTN to interact with non-specific activin receptors (ActRII) on the surface of target cells (i.e., myoblasts) through a paracrine pathway [23, 24]. The MSTN pathway is classically initiated through MSTN binding to ActRIIA/B receptors, with a notably higher affinity for ActRIIB. This binding induces dimerization, which subsequently activates activin-like kinase (ALK4 or ALK5). This receptor activation ultimately leads to the phosphorylation of SMAD2 and SMAD3, which then form a complex with SMAD4. The SMAD complex translocates into the nucleus and negatively regulates myoblast cell activities by modulating gene expressions (Fig. 1). This, in turn, results in the expression of various atrophic E3-Ubiquitin ligases such as Atrogin1 and muscle RING-finger protein-1 (MuRF1) [23, 25, 26]. Additionally, MSTN plays a role in signaling the mitogen-activated protein kinase (MAPK) pathway, specifically the c-Jun N-terminal kinase (JNK), p38, and extracellular signal-regulated kinases (ERK) pathways. These pathways are known to inhibit the transcriptions of a variety of myogenesis-related genes [27,28,29].

Myostatin Activation and SMAD Signaling Pathway, Including Inhibitor Targets. MSTN is secreted as an inactive precursor composed of a signal peptide (S), a 36 kDa propeptide region (PP), and a 12.5 kDa mature, or growth factor (GF) region. In the endoplasmic reticulum, GF regions dimerize, forming promyostatin. Proteolytic processing occurs as detailed in section “Mechanism of myostatin actions”. ProMSTN is cleaved at a dibasic RX site between the S and PP regions, followed by furin-like protease cleavage at an RXXR site between the PP and GF regions, indicated by red arrows. This results in a latent complex of dimerized GF and PP regions held together via non-covalent interactions. MSTN activation occurs through the cleavage of an arginine residue in the supporting PP regions, releasing the mature MSTN dimer. Mature MSTN binds to ActRIIB or ActRIIA, initiating receptor dimerization and recruitment of ALK4/5, triggering various signaling pathways described in section “Mechanism of myostatin actions”. The SMAD pathway is depicted, where phosphorylated Smad2/3 forms a complex with Smad4 and translocates into the nucleus, regulating gene expression associated with muscle homeostasis. The figure also highlights the targets of various MSTN inhibitors discussed in section “Inhibition of Myostatin in animal and human studies”
MSTN plays a pivotal role in skeletal muscle development. During fetal development, muscle fiber formation occurs, and MSTN mRNA is highly expressed in the developing skeletal muscle. In mice, expression begins around 9.5 days post-coitum (dpc) and peaks at approximately 14.5 dpc, indicating its critical role in regulating early muscle growth [30]. Following birth, MSTN levels remain high, though not as elevated as during fetal development [30]. As adulthood sets in, MSTN levels stabilize, maintaining muscle homeostasis [30]. MSTN-knockout animals, lacking MSTN presence during prenatal or neonatal development, exhibit a dramatic 2- to 3-fold increase in muscle mass compared to wild-type animals [30]. Homozygous mutant mice display approximately 30% more body weight, with both larger muscles fibers in cross-sectional area (hypertrophy) and a greater in fiber number (hyperplasia). Furthermore, MSTN-knockout animals demonstrate a higher proportion of type II fibers and a reduced number of type I fibers, along with decreased adipose tissue [30,31,32]. Postnatal suppression of MSTN, achieved through conditional gene targeting or the administration of MSTN inhibitors such as its propeptide, antibody, or follistatin, induces significant but relatively lesser increases in skeletal muscle mass [33,34,35]. In contrast to MSTN-knockout models, muscle growth from postnatal suppression of MSTN results solely from muscle hypertrophy, not hyperplasia, but still predominantly induces type II muscle fibers [36,37,38].
Inhibition of myostatin in animal and human studies
Since its initial description by McPherron et al. in 1997, MSTN inhibition has been widely considered as a potential treatment for muscle wasting diseases. Indeed, the focus of most clinical studies to date has been on diseases such as Duchenne muscular dystrophy (DMD), sporadic inclusion body myositis (sIBM), and limb-girdle muscular dystrophy (LGMD). Muscular dystrophies are genetic disorders characterized by muscle weakness and degeneration resulting from mutations in specific genes. These conditions often lead to mobility issues and premature death [39]. While MSTN inhibition does not address the underlying gene mutations responsible for these diseases, it is anticipated to offer a universal approach to treating muscle wasting that does not rely on correcting individual molecular defects, which can vary between patients and types of muscular dystrophy. Other conditions characterized by muscular wasting, such as sarcopenia and cancer cachexia, are also expected to benefit from improvements in muscular function. Preclinical studies conducted in MSTN-null mdx mice, a model of DMD and Becker muscular dystrophy (BMD) featuring a premature stop codon in the gene for dystrophin, have demonstrated increased muscle size and strength [40]. Furthermore, a three-month treatment regimen with anti-MSTN antibodies in mdx mice resulted in enhancements in body weight, muscle mass, muscle size, and absolute muscle strength, accompanied by a significant reduction in muscle degeneration [41]. Given the consistent success in ameliorating muscle wasting diseases in animal models, various inhibitors have been developed for clinical evaluation (Table 1).
Antibodies
The majority of clinically tested MSTN inhibitors have been MSTN-based antibodies. In 2004, Wyeth Pharmaceuticals (now owned by Pfizer) developed the monoclonal anti-MSTN antibody MYO-029, the first MSTN inhibitor to enter clinical trials. MYO-029 binds to mature MSTN, thereby preventing its interaction with ActRIIA/B receptors. Despite conducting Phase 1 and Phase 2 trials involving patients with BMD, facioscapulohumeral muscular dystrophy (FSHD), and LGMD, administering 2 doses of 1–10 mg/kg MYO-029 every 2 weeks for 6 months found no significant improvements in muscle size, strength, or function, leading to the discontinuation of further development [14].
After MYO-029, the next MSTN inhibitor to reach clinical trials was landogrozumab, an anti-MSTN monoclonal antibody developed by Eli Lilly. Landogrozumab was tested in phase 2 trials targeting sarcopenia, sarcopenia related to hip surgery, and cachexia. Following 5 equally spaced treatments of 315 mg over 20 weeks in patients aged 75 years or older, those who received landogrozumab showed an increase of 0.44 kg in appendicular lean body mass. Additionally, they demonstrated significant improvements in stair climbing (four-step: −0.46 s, 12-step: −1.28 s), chair rising with arms (− 4.15 s), and fast gait speeds (+ 0.05 m/s) [42]. However, subsequent trials failed to show significant increases in appendicular lean mass among patients with osteoarthritis scheduled for elective total hip arthroplasty [43]. Similarly, no improvements in muscle function or survival were observed in pancreatic cancer patients with cachexia [44]. Nonetheless, the former of the two trials did reveal a statistically significant, yet relatively low increase in lean muscle mass [43].
Domagrozumab, developed by Pfizer, is another anti-MSTN/GDF-11 monoclonal antibody, similar in design to its predecessors MYO-029 and landogrozumab. In studies on mdx mice, domagrozumab significantly boosted body weight, muscle weight, and grip strength [45], demonstrating greater enhancements in muscular strength compared to MYO-029 [46]. However, a series of phase 1 and 2 clinical trials targeting DMD and LGMD did not meet their endpoints, with no significant improvement in muscle strength, function, or size among subjects [47, 48]. Further development of domagrozumab was ultimately terminated in 2018 due to the lack of positive results from these studies [49].
Bimagrumab, originally developed by Novartis and later acquired by Lilly, is an antibody targeting MSTN/Activin receptor ActRIIB and ActRIIA, thereby preventing MSTN from binding to the receptor. Initial trials of bimagrumab showed promise, with a 5.7% increase in lean muscle mass and a 14.6% improvement in 6-min walking distance (6MWD) in patients with sIBM given a single dose of 30 mg/kg [50]. However, more extensive studies on sIBM found no improvements in 6MWD and overall mobility [51, 52]. Subsequent studies produced mixed results: administering a single or twice doses of 30 mg/kg bimagrumab to sarcopenia-affected patients resulted in an approximately 6.5% increase in thigh muscle volume (TMV). Notably, improvements in gait speed and 6MWD were explicitly observed in slower walkers, with a mean increase of 0.15 m/s in gait speed and 82 m in 6MWD at Week 16 compared to those on placebo [53]. However, no changes were observed when 700 mg bimagrumab was given monthly to patients following an optimized standard of care for diet and exercise [54]. Additionally, two doses of 30 mg/kg bimagrumab in patients with chronic obstructive pulmonary disease led to an approximate 5% increase in TMV after 24 weeks but did not improve functional capacity [55]. Similarly, bimagrumab administered every 4 weeks for 24 weeks to older adults recovering from hip fracture showed a significant dose-dependent increase in muscle mass up to a mean change of 2.8 kg, but no functional benefit [56]. Clinical trials for bimagrumab are ongoing.
In addition to mature MSTN-targeting antibodies, various other designs exist that target different stages of MSTN expression. One notable example is trevogrumab, developed by Regeneron. It is a monoclonal antibody with an IgG4 Fc domain designed for sarcopenia and inclusion body myositis (IBM) treatment. Trevogrumab targets MSTN in its mature, latent, and pro-forms without cross-reactive binding to GDF11 [57]. Apitegromab, developed by Scholar Rock, specifically targets MSTN in its latent form by stabilizing its conformation, thereby preventing access to prodomain protease cleavage sites [58, 59]. Apitegromab has shown efficacy in increasing muscle mass and function in mouse models of spinal muscular atrophy (SMA) [60]. Similarly, GYM-329 by Roche is designed to treat FSHD by binding to latent MSTN, thus blocking its conversion to its mature form [61]. These antibodies are currently undergoing clinical trials or awaiting the reporting of results.
Fusion proteins
Another class of MSTN inhibitors is fusion proteins, often in the form of soluble activin receptors, which act as ligand traps by binding MSTN and preventing its further interactions. Acceleron, now owned by Merck, first brought ligand traps to clinical trials with ramatercept in 2008. Ramatercept is a fusion protein consisting of human IgG linked to the extracellular domain of ActRIIB, acting as a soluble form of ActRIIB, which binds MSTN and other TGF-β members. Ramatercept showed promise in both phase 1 and phase 2 trials. In a phase 1 trial involving women, total muscle volume (TMV) was improved by approximately 5.1% following a single injection of 3 mg/kg [62]. Additionally, in a phase 2 trial conducted with boys affected by DMD, significant improvements were observed in muscle mass, with the group treated with 1 mg/kg every 2 weeks experiencing an approximate mean increase of 4–5%. Moreover, improvements were noted in 6-min walk distance (6MWD), bone mineral density, and reductions in fat mass [12, 63]. However, non-muscle-related adverse side effects including nosebleeds, gum bleeding, telangiectasia, and erythema led to the discontinuation of further study [12].
Acceleron’s luspatercept is akin to its predecessor, ramatercept, but contains modifications in the extracellular domain of ActRIIB to promote binding of various TGF-β proteins. Luspatercept began clinical trials in 2011 for the treatment of hematologic disorders and demonstrated efficacy in increasing hemoglobin counts in patients with anemia [64, 65], potentially attributed to a reduction in SMAD signaling leading to enhanced erythroid maturation [66]. Luspatercept received FDA approval in 2020 for the treatment of anemia associated with myelodysplastic syndromes, myelofibrosis, and beta-thalassemia [67]. However, the effect of luspatercept on MSTN expression and muscle modulation has not been reported.
In contrast to receptor-based ligand trap designs like ramatercept and luspatercept, PINTA-745, initially developed by biotech company Amgen and later by Atara Biotherapeutics, is a distinctive peptibody fusion protein comprised of a human IgG FC with a MSTN-neutralizing peptide. In a post-stroke muscle loss mouse model, PINTA-745 demonstrated a significant increase in muscle mass, strength, and motor function [68]. However, in a phase 1/2 trial involving patients with prostate cancer undergoing androgen deprivation therapy, weekly injections of 3 mg/kg PINTA-745 for four weeks resulted in a modest 2.2% increase in muscle size, but showed no improvement in physical strength assessed through the Short Physical Performance Battery or the one-repetition maximum knee extension test [13]. These outcomes led to the discontinuation of further development.
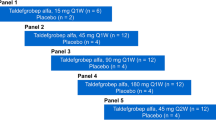
Taldefgrobep alfa, developed by Bristol-Meyers-Squibb and later by Biohaven Pharmaceuticals, is a unique anti-MSTN adnectin. It utilizes an engineered scaffold based on the 10th fibronectin type III domain coupled with a human IgG Fc domain, exhibiting a binding affinity for the C-terminal of mature MSTN and the ActRIIB–MSTN complex, similar to an antibody [69]. Taldefgrobep binding prevents ALK-4/5 recruitment, thereby inhibiting the SMAD pathway. In a phase 1 trial involving healthy adults, a 24-week treatment period with weekly doses ranging from 45 to 150 mg led to a universal decrease in free MSTN by ≥ 90%. Additionally, significant increases in lean body mass, up to a mean increase of 2.69%, and thigh muscle volume, up to a mean increase of 4.75%, were observed [69]. In phase 2 trials with boys affected by DMD, weekly doses of taldefgrobep alfa led to a modest 4.9% increase in lean body mass index in the pooled treatment group compared to placebo, but ultimately found no change in motor function [69]. Further research targeted toward DMD has been terminated, but a phase 3 study evaluating taldefgrobep alfa in SMA is currently underway.
Follistatin
Follistatin (FST) and its related FST-type molecules are naturally antagonists to several TGF-β proteins, and are widely known inhibitors of MSTN [70, 71]. FST binds to mature MSTN with high affinity and inhibits its binding to ActRIIB, but does not interact with proMSTN [20, 72]. The N-terminal α-helical domain of FST interacts directly with a type I receptor binding site of MSTN, causing inactivation [73]. Consequently, FST lacking its C-terminal peptide or fragments of the N-terminal region show similar inhibitory effects [73,74,75]. Transgenic overexpression of FST results in a 2- to 3-fold increase in muscle mass via hypertrophy and hyperplasia [21]. However, the increase in muscle mass is not solely attributed to blocking MSTN, as FST also inhibits the activities of multiple TGF-β family members, some of them play a role in limiting muscle mass [76]. FST-overexpressing MSTN-null mice display an even more extreme fourfold increase in muscle [77]. Contrarily, FST-null mice have reduced muscle mass at birth and perish within a few hours [78]. Delivery of FST-coding mRNA gene therapy and AAV, as well as follistatin peptide derivatives, have all been shown to produce substantial muscle increase in animal models [35, 79, 80].
The number of clinically tested follistatin-based methods is limited. ACE-083, developed by Acceleron/Merck, is a fusion protein consisting of a human IgG2 Fc domain linked with a modified human FST. ACE-083 is designed for intra-muscular injection and causes localized MSTN inhibition. Preclinical studies of ACE-083 demonstrated a dramatic increase in muscle mass and strength in wild-type, Charcot-Marie-Tooth disease (CMT), and DMD disease model mice [81]. In a phase 1 study, ACE-083 injected in to the rectus femoris muscle of healthy women resulted in approximately a 14.5% increase in local muscle mass but no change in muscle strength [82]. Phase 2 studies in patients with FSHD or CMT also revealed significant localized increases in muscle mass but failed to demonstrate any improvement in muscle function [83, 84]. The failure of ACE-083 to meet clinical endpoints ultimately led Acceleron to discontinue its development and shift its focus of TGF-β targeting therapeutics away from muscle-dystrophic diseases.
AAV1.CMV.FS344, developed by Milo Biotechnology/Nationwide Children's Hospital, is a modified human follistatin isoform delivered by adeno-associated virus (AAV) to suppress MSTN. This follistatin AAV has shown promising results in mice, with a single injection significantly increasing long-term muscle mass and strength in wild-type, aged, and mdx models [85]. In humans, AAV.CMV.FS344 has undergone limited testing in a few stage 1 and 2 clinical trials. Administration of the AAV to BMD patients showed improvements to 6MWT, although there was no control group, and only six subjects were tested [86]. AAV injection in a similarly small group of sIBM patients also resulted in increased 6MWT performance [87]. Study of AAV.CMV.FS344 has yet to be terminated, but the continued lack of participants in each study raises questions about the validity of the results.
MSTN propeptide
The MSTN propeptide is a potent inhibitor of MSTN but has seemingly been overlooked in clinical research. MSTN propeptide functions as an inhibitor by binding to the mature MSTN dimer, reverting it to its latent state [88]. Furthermore, two propeptides can bind to the dimerized proMSTN N-terminal region via a critical peptide sequence, preventing the proteolytic processing of proMSTN into its mature form [18]. Transgenic mice overexpressing MSTN propeptide exhibit a dramatic muscling phenotype, with a 17–30% increase in body weight [37]. The inhibitory binding capabilities of MSTN propeptide are due to specific amino acid residues in the propeptide's N-terminus (42–115AA) [18]. Smaller minimum inhibitory peptide sequences (< 25AA) of MSTN propeptide have been identified and proven to inhibit MSTN in vivo [89]. Mutations of MSTN propeptide at the arginine-residue cleavage site can enhance the effectiveness of MSTN inhibition by rendering the propeptide resistant to cleavage by BMP-1/tolloid proteinases, thereby preventing activation of the latent complex [24]. Mice injected with the mutant form of propeptide exhibited more pronounced muscling results than those given wild-type MSTN propeptide [24]. Despite its potent inhibition of MSTN in animals, no attempts have been made to utilize MSTN propeptide in clinical trials.
Challenges in myostatin inhibition
Specificity in inhibitor designs
Despite yielding positive results in various animal studies, MSTN inhibition has not improved human muscular function. A lack of specificity in many MSTN inhibitors could account for unsatisfactory clinical trials. MSTN shares significant structural similarities with other members of the TGF-β superfamily, particularly GDF11, showing nearly 90% sequence identity in their mature domains [90]. Consequently, many anti-MSTN antibodies inadvertently cross-react with GDF11 [91, 92], leading to cross-reaction effects or reduced efficacy. Receptor-based ligand traps encounter similar problems due to ActRIIA and ActRIIB receptors binding to GDF11, activins A, B, and AB, and BMPs 9 and 10 [93]. Inhibiting ActRIIA/B receptors will also affect the signaling of these proteins, potentially causing unintended off-target effects. Likewise, FST has also been shown to bind to GDF11, activins A, B, AB, and E, inhibins A and B, BMPs 2, 4, 6, 7, and 15 [21, 94]. Muramatsu et al. demonstrated the importance of specificity in design by utilizing GYM-329, an antibody which specifically targets the latent form of MSTN. In mice, GYM-329 was shown to increase muscle mass in 3 different models of muscle dystrophy, demonstrating a larger increase in muscle mass and grip strength compared to landogrozumab and domagrozumab, two unspecific-antibodies [61]. GYM-329 treatment also resulted in greater grip strength increases over bimagrumab, the anti-ActRIIA/B antibody [61]. Targeting the latent MSTN complex is likely more efficient not only due to specificity but also due to the increased temporal availability of the latent complex compared to the active mature dimer [20]. In addition to efficacy issues, cross-reactivity poses a serious risk of side effects. As previously mentioned, clinical studies of the soluble receptor ramatercept were prematurely halted due to significant adverse effects, such as nosebleeds, gum bleeding, telangiectasia, and erythema. These adverse events were attributed to the unintended cross-inhibition of BMP9 and BMP10, critical ligands involved in endothelial cell function [12]. Future research and development efforts for MSTN inhibitors should prioritize specificity to mitigate off-target effects and enhance efficacy.
Availability of circulating myostatin
A difference in serum MSTN concentration between healthy and diseased individuals presents another obstacle to developing MSTN inhibitors. Most muscle atrophy and dystrophy diseases are characterized by lower concentrations of circulating myostatin [95]. Patients affected with DMD, for instance, exhibit approximately 65% lower concentrations of serum MSTN compared to healthy adults [96]. Despite a 90% reduction in MSTN compared to pre-treatment levels in DMD patients treated with domagrozumab, muscle mass did not increase significantly [96]. This suggests that the already low MSTN levels in DMD patients may reduce the effectiveness of MSTN inhibitors, as further lowering MSTN might not significantly increase muscle mass, as discussed by Mariot et al. (2017) [95]. Additionally, Mariot et al. (2017) found that in muscle wasting and atrophying diseases, not only is myostatin downregulated, but the activin receptor is also downregulated, along with an increase in the MSTN antagonist follistatin [95]. These factors further complicate the therapeutic potential of MSTN inhibitors in muscle wasting diseases.
MSTN concentrations are also affected by other conditions. Many studies generally suggest that serum MSTN is highest in young individuals and decreases with age [97, 98], which could pose challenges for using MSTN inhibitors to treat sarcopenia in older adults. Furthermore, patients suffering from cancer cachexia also show decreased MSTN concentrations compared to non-cachectic individuals [99, 100]. In patients experiencing severe muscle wasting, the decline in circulating MSTN levels may be attributed to the diminished capacity of muscles to produce myokines, including MSTN.
Much of the data about circulating MSTN levels may be questioned due to potential methodological limitations. Binding reagent assays (e.g., immuno-assays and aptamer-based methods), the most popular method for determining MSTN concentrations, have been shown to cross-react with GDF-11 [101, 102]. However, as GDF-11 is less abundant than GDF-8, its impact on overall MSTN measurements may be inconsequential [102]. Regardless, comprehensive research utilizing refined methodologies to accurately measure serum MSTN concentrations is essential to best determine if reduced MSTN impacts the effectiveness of inhibition therapies [103, 104].
The failure of MSTN inhibitors to effectively treat muscle wasting diseases in humans despite promising results in preclinical studies may stem from species-specific differences in serum MSTN levels. On average, human serum MSTN levels are around 5–10 ng/ml, whereas mice exhibit concentrations exceeding 100 ng/ml, up to a 20-fold difference [105, 106]. This disparity in MSTN availability may contribute to a shift in potency between species. A pharmacokinetic study with MYO-029 found that the concentration of MYO-029 required to elicit a 50% improvement in muscle mass in monkeys was 18 times higher compared to the same improvement in mice [107]. This difference is even more pronounced in diseased models. While mdx mice have lower serum MSTN levels (~ 50 ng/ml) than their wild-type counterparts, their concentration is much higher and proportionally less diminished compared to humans affected by DMD (~ 1 ng/ml), potentially providing a greater scope for the effects of MSTN inhibition [105]. This discrepancy in pharmacokinetics and MSTN serum concentration between diseased human and mouse models may significantly contribute to the difference in results between clinical and animal trials.
Mediating functional strength
Even if MSTN inhibition increases muscle mass, it does not necessarily translate into improved functional strength in muscle wasting disorders. While MSTN inhibition may stimulate muscle hypertrophy, its effectiveness in improving functional strength relies heavily on synergistic motor neuron activation and mechanical signaling induced by exercise. Without adequate fusion of newly formed myotubes with existing muscle fibers, facilitated by neural input, increased muscle mass may not lead to meaningful functional improvements [108]. This limitation is especially relevant in conditions like DMD, where neuromuscular junction vulnerability and reduced neural input contribute to impaired translation of neurological signals to skeletal muscles [109]. In contrast, mdx mice typically exhibit robust contractile function and maintain ambulation throughout their lifespan, which may elucidate why MSTN inhibition in these mice can result in gains in both muscle mass and function [110]. While most clinical trials in muscular wasting disorders prioritize functional improvements as a primary endpoint, if this information holds true, the limited translation of increased muscle mass into functional strength due to neural deficits may impede the achievement of these desired functional improvements.
It is possible that the challenge of improving muscular function could be bypassed by integrating MSTN inhibition therapy with exercise. Studies in mice demonstrate that combining MSTN inhibition therapy with exercise, be it aerobic or resistance training, results in significantly enhanced muscle quality compared to either intervention alone [111, 112]. However, trials involving the combination of bimagrumab with an exercise program in sarcopenia patients, as previously mentioned, did not yield any discernible difference between groups receiving combined therapy or exercise alone [54]. Further clinical research is needed to determine if the combination of MSTN inhibition therapy with exercise could be effective in increasing muscle function in humans.
Expanding therapeutic potential
Myostatin inhibition in obesity, diabetes, and metabolic syndromes
While treating muscle wasting disorders has presented numerous difficulties and demonstrated limited success, inhibition of MSTN may offer a more promising approach to address other pathologies effectively. Unlike conditions like muscular dystrophy, sarcopenia, and cancer cachexia, obesity and diabetes correlate with elevated serum levels of MSTN [113]. A study surveying MSTN serum concentration in human adults found a positive correlation between obesity and increased MSTN, a positive association with insulin resistance, and a negative correlation with insulin sensitivity [114]. This cause-and-effect relationship with insulin resistance is supported by the observed increase in insulin resistance after injection of MSTN in mice [115]. Furthermore, in high-fat diet-induced obesity-susceptible C57BL/6 mice, consumption of high-fat feed led to an increase in MSTN expression, indicating that MSTN may play an essential role in mediating obesity [116]. Moreover, both type 1 and type 2 diabetes patients were shown to have higher serum MSTN concentrations when compared to healthy counterparts [117, 118]. The increased MSTN levels in obesity, insulin resistance, and diabetes suggest that MSTN-targeted inhibitors can improve metabolic function and promote weight loss in obese individuals. By blocking MSTN action, these inhibitors could enhance muscle growth, increase energy expenditure, and improve insulin sensitivity, offering a promising approach to combating diabetes-related health conditions.
Inhibition of MSTN is beneficial for metabolic syndrome and diabetes in various ways. Loss of MSTN function has long been known to reduce total body fat mass. In obesity and T2DM mouse models, loss of MSTN mitigates fat accumulation and deviation from healthy glucose metabolism [119]. Local inhibition by AAV delivery of MSTN propeptide increased glucose uptake and skeletal muscle mass in obese, high-fat-fed mice [120]. Transgenic mouse models with overexpression of MSTN propeptide also resist increases in adipose tissue mass and insulin resistance, which are associated with a high-fat diet, while wild-type counterparts develop prediabetes [121]. Furthermore, crossbreeds between MSTN-knockout and AKITA mice (a model for T1DM) exhibit significantly higher glucose uptake capacity, lower resting blood glucose levels, and reduced diabetic symptoms compared to normal AKITA mice [122]. Additionally, increasing functional muscle mass (assuming it is possible) would be beneficial for treating diabetes, as a loss of skeletal muscle mass is frequently associated with both T1DM and T2DM due to catabolism caused by malfunction of insulin-related issues [123]. MSTN inhibition for the treatment of metabolic disorders in humans has already shown moderate success in clinical trials with bimagrumab. Dosing of bimagrumab in obese adults shows significant increases in lean body mass and reductions in body fat [124, 125]. In obese adults with type 2 diabetes, bimagrumab treatment every four weeks for 48 weeks resulted in a ~ 20% decrease in fat mass, ~ 4% increase in muscle mass, ~ 6.5% decrease in body weight, and ~ 0.8 percentage point decrease in HbA1c level [126]. Ongoing clinical trials with bimagrumab aim to explore its potential in treating obesity; however, no MSTN inhibitor has received approval for use in metabolic syndromes. Expanding the scope of clinical trials targeting obesity, diabetes, and metabolic syndrome is critical for determining MSTN inhibition’s effectiveness in treating these disorders (Fig. 2).

Mechanistic pathways of myostatin inhibition in insulin sensitivity and adipose tissue regulation. Myostatin (MSTN) blockade increases muscle hypertrophy, leading to heightened caloric expenditure and glucose transporter type 4 (GLUT4) expression; it also promotes the activation of adenosine monophosphate-activated protein kinase (AMPK), which facilitates GLUT4 translocation; enhances the expression of adiponectin, further mediating AMPK activation; and leads to the expression of insulin receptor substrates (IRS), activating the phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) pathway. Akt enhances GLUT4 expression by increasing its translocation to the cell membrane and inhibiting the forkhead box O1 (FOXO1) transcription factor, which in turn increases the transcription of GLUT4. Additionally, MSTN blockade induces brown adipose tissue (BAT) formation in white adipose tissue (WAT) via the expression of BAT markers and thermogenic genes; interferon regulatory factor 4 (IRF4), expressed in BAT, inhibits myostatin expression, and BAT promotes thermogenesis. Increased caloric expenditure, adiponectin, and BAT thermogenesis lead to increased energy expenditure and fat utilization, while GLUT4 expression, AMPK activation, IRS, PI3K, and Akt contribute to improved insulin sensitivity and glucose metabolism; these effects collectively result in an improvement in diabetes and obesity symptoms
Mechanistically, MSTN inhibition interacts with insulin sensitivity and obesity through both skeletal muscle-dependent and independent mediation. The primary phenotypic change accompanying MSTN suppression is an increase in skeletal muscle, which partitions nutrients away from adipose tissue to support energy requirements for muscle growth. Skeletal muscle is the primary site for insulin-mediated glucose uptake via glucose transporter type 4 (GLUT4) protein [127]. Indeed, MSTN-knockout mice exhibit upregulated GLUT1 (insulin-independent) and GLUT4 (insulin-dependent) proteins, leading to increased glucose uptake [122]. Conversely, active MSTN reduces GLUT4 expression and glucose uptake through muscle atrophy, inhibition of various insulin-related pathways, and downregulation of gene expression [128]. MSTN inhibits the phosphorylation of insulin receptor substrate (IRS) proteins, which reduces the activation of phosphoinositide 3-kinase (PI3K) and downstream protein kinase B (Akt) [129]. Akt promotes the translocation of GLUT4-containing vesicles to the plasma membrane of muscle cells in response to insulin [130]. It also phosphorylates and inhibits Forkhead box O1 (FoxO1), which is a transcription factor that represses GLUT4 gene transcription [131]. MSTN inhibition upregulates the PI3K/Akt pathway, leading to an increase expression of GLUT4 [132, 133]. Additionally, MSTN inhibits the activation of adenosine monophosphate-activated protein kinase (AMPK), a crucial regulator of mitochondrial biogenesis and energy metabolism, which also promotes GLUT4 translocation in response to insulin-independent energy stress [129, 134]. Furthermore, MSTN knockout has been reported to upregulate adiponectin, a regulator of adipocyte energy metabolism that improves insulin sensitivity and stimulates AMPK [134,135,136]. In our study, transgenic mice overexpressing MSTN propeptide exhibited a significant increase in serum adiponectin levels when fed a high-fat diet, while maintaining normal levels of blood insulin, resistin, and leptin [121].
Although MSTN is not highly expressed in adipose tissue, it plays a significant role in mediating adipose tissue function. Metabolically, MSTN-null mice show increased energy expenditure and leptin sensitivity [137]. Inhibition of MSTN upregulates enzymes involved in lipolysis and mitochondrial fatty acid oxidation, increasing fat breakdown in peripheral tissues, and reducing lipid accumulation [138]. Furthermore, MSTN suppression induces the emergence of brown adipose tissue (BAT) within white adipose tissue (WAT), a fat type known for its thermogenic characteristics, abundance of fat-burning mitochondria, and role in maintaining insulin and glucose homeostasis [138, 139]. MSTN has been shown to mediate the expression of BAT markers and thermogenic genes in WAT, including Ucp1, Prdm16, Pgc-1a, Bmp7, Cidea, Cd137, and Tmem26 [134, 140, 141]. Another possible route of MSTN-mediated BAT formation is the skeletal muscle-derived myokine irisin, which facilitates crosstalk between skeletal muscle and adipose tissue to drive thermogenesis and browning and is increased with inhibition of MSTN [142, 143]. Additionally, MSTN is secreted in BAT and acts as an adipokine, reducing local insulin sensitivity [144]. Furthermore, MSTN is involved in tissue crosstalk between BAT and skeletal muscle through transcription factor interferon regulatory factor 4 (IRF4), which regulates adipogenesis by inhibiting MSTN expression [15]. Expression of IRF4 in BAT is strongly correlated with serum MSTN levels, with loss of IRF4 causing obesity, decreased exercise capacity, and increased serum MSTN [15, 145]. These findings indicate a complex interplay between MSTN and adipose tissue, highlighting distinct effects beyond those mediated by skeletal muscle. Further research is required to fully elucidate the interactions between MSTN inhibition and metabolic disorders.
Myostatin inhibition in orthopedic disorders
MSTN inhibition may also hold therapeutic benefits for orthopedic diseases. Current literature suggests that MSTN acts as a mediator between muscle and bone metabolism, influencing bone formation and remodeling through paracrine and endocrine mechanisms [8]. MSTN negatively impacts bone formation by inhibiting osteogenic differentiation of mesenchymal stem cells and osteoblasts [146, 147]. It also suppresses chondrogenesis, delaying the transition from cartilage to bone during fracture healing, thereby affecting callus formation and bone regeneration [148]. MSTN is shown to be a positive regulator of osteoclast differentiation, which is responsible for the resorption of aged bone and plays a role in bone degradation in arthritis and osteoporosis [149]. Inhibiting MSTN may have therapeutic applications in promoting bone regeneration and healing in bone fractures, osteoporosis, rheumatoid arthritis, and osteoarthritis. In mice, administration of recombinant MSTN propeptide improved fracture healing in a fibula osteotomy model [150]. Additionally, treatment of young mice with ActRIIB-Fc led to increased bone mass [151]. In a mouse model for rheumatoid arthritis, MSTN is highly expressed in synovial tissues, and transgenic or antibody inhibition of MSTN ameliorates joint destruction and arthritis severity [149]. Although MSTN inhibition has shown promise in animal models for increasing bone mass and improving bone strength, no clinical trials targeting orthopedic diseases have been conducted.
Summary and future perspectives
Current attempts at clinical application of MSTN inhibitors have encountered challenges with drug design and disease applications. While preclinical studies indicate that inhibiting MSTN can treat muscle wasting diseases, transitioning into human trials remains challenging due to the differences in MSTN concentration between animals and humans, and the necessity of neural input to improve skeletal muscle functionality. Muscular dystrophies, the primary focus of MSTN-inhibition therapeutics to date, stem from genetic-based pathways that may not be easily remedied solely by inhibiting MSTN or augmenting skeletal muscle mass. Future applications of MSTN inhibition must consider the limitations of targeting skeletal muscle mass and explore more suitable disease applications. Obesity, diabetes, metabolic syndromes, and orthopedic diseases are all potential targets for MSTN inhibitors because they do not face the same challenges with serum MSTN concentration and muscle contraction functions, and their benefits are not solely dependent on boosting muscle function. However, these pathological conditions remain understudied in clinical trials. There may still be potential for MSTN inhibitors in managing muscular dystrophy through combination with exercise, dystrophin, or other gene replacement therapies [95, 152,153,154], or in less severe forms of the condition where elevated levels of residual MSTN are observed, like myotonic dystrophy [105]. These approaches could mitigate the challenges posed by the low MSTN levels in patients with severe muscular dystrophy, which may otherwise reduce the effectiveness of MSTN inhibitors. There are also future application possibilities in non-diseased patients, like targeting MSTN to protect against muscle and bone mass loss during space flight [155]. Regardless of application, future research on MSTN inhibitors should prioritize the development of specific inhibitor designs to mitigate side effects caused by cross-reactivity. Targeting the latent or pro-form of MSTN could offer superior efficacy and reduced cross-reactivity. Most inhibitors tested to date have targeted the mature form or employed broadly reactive receptor-based approaches. Numerous inhibition methods with the potential for greater effectiveness remain unexplored in clinical settings. For instance, MSTN propeptide has demonstrated efficacy as a specific inhibitor but has yet to receive clinical attention. Although MSTN inhibition has yet to fully realize its promise as a muscle-enhancing drug, there is still ample potential for refinement in its therapeutic applications and drug designs.
Data availability
No datasets were generated or analysed during the current study.
References
Grobet L et al (1997) A deletion in the bovine myostatin gene causes the double–muscled phenotype in cattle. Nat Genet 17(1):1. https://doi.org/10.1038/ng0997-71
Kambadur R, Sharma M, Smith TP, Bass JJ (1997) Mutations in myostatin (GDF8) in double-muscled Belgian Blue and Piedmontese cattle. Genome Res 7(9):910–916. https://doi.org/10.1101/gr.7.9.910
McPherron AC, Lee S-J (1997) Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci USA 94(23):12457–12461
Mosher DS et al (2007) A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet 3(5):e79. https://doi.org/10.1371/journal.pgen.0030079
Clop A et al (2006) A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet 38(7):7. https://doi.org/10.1038/ng1810
Schuelke M et al (2004) Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med 350(26):2682–2688. https://doi.org/10.1056/NEJMoa040933
Zimmers TA et al (2002) Induction of cachexia in mice by systemically administered myostatin. Science 296(5572):1486–1488. https://doi.org/10.1126/science.1069525
Cui Y et al (2023) Molecular basis and therapeutic potential of myostatin on bone formation and metabolism in orthopedic disease. BioFactors 49(1):21–31. https://doi.org/10.1002/biof.1675
Kalds P, Zhou S, Huang S, Gao Y, Wang X, Chen Y (2023) When less is more: targeting the myostatin gene in livestock for augmenting meat production. J Agric Food Chem 71(10):4216–4227. https://doi.org/10.1021/acs.jafc.2c08583
White TA, LeBrasseur NK (2014) Myostatin and sarcopenia: opportunities and challenges-a mini-review. GER 60(4):289–293. https://doi.org/10.1159/000356740
Yang M et al (2023) Myostatin: a potential therapeutic target for metabolic syndrome. Front Endocrinol. https://doi.org/10.3389/fendo.2023.1181913
Campbell C et al (2017) Myostatin inhibitor ACE-031 treatment of ambulatory boys with Duchenne muscular dystrophy: results of a randomized, placebo-controlled clinical trial. Muscle Nerve 55(4):458–464. https://doi.org/10.1002/mus.25268
Padhi D, Higano CS, Shore ND, Sieber P, Rasmussen E, Smith MR (2014) Pharmacological inhibition of myostatin and changes in lean body mass and lower extremity muscle size in patients receiving androgen deprivation therapy for prostate cancer. J Clin Endocrinol Metab 99(10):E1967–E1975. https://doi.org/10.1210/jc.2014-1271
Wagner KR et al (2008) A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol 63(5):561–571. https://doi.org/10.1002/ana.21338
Kong X et al (2018) Brown adipose tissue controls skeletal muscle function via the secretion of myostatin. Cell Metab 28(4):631-643.e3. https://doi.org/10.1016/j.cmet.2018.07.004
Breitbart A, Auger-Messier M, Molkentin JD, Heineke J (2011) Myostatin from the heart: local and systemic actions in cardiac failure and muscle wasting. Am J Physiol Heart Circ Physiol 300(6):H1973–H1982. https://doi.org/10.1152/ajpheart.00200.2011
Verzola D, Barisione C, Picciotto D, Garibotto G, Koppe L (2019) Emerging role of myostatin and its inhibition in the setting of chronic kidney disease. Kidney Int 95(3):506–517. https://doi.org/10.1016/j.kint.2018.10.010
Jiang M-S et al (2004) Characterization and identification of the inhibitory domain of GDF-8 propeptide. Biochem Biophys Res Commun 315(3):525–531. https://doi.org/10.1016/j.bbrc.2004.01.085
Chen PR, Lee K (2016) INVITED REVIEW: Inhibitors of myostatin as methods of enhancing muscle growth and development1. J Anim Sci 94(8):3125–3134. https://doi.org/10.2527/jas.2016-0532
Cotton TR et al (2018) Structure of the human myostatin precursor and determinants of growth factor latency. EMBO J 37(3):367–383. https://doi.org/10.15252/embj.201797883
Lee S-J, McPherron AC (2001) Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci 98(16):9306–9311. https://doi.org/10.1073/pnas.151270098
Hill JJ et al (2002) The myostatin propeptide and the follistatin-related gene are inhibitory binding proteins of myostatin in normal serum*. J Biol Chem 277(43):40735–40741. https://doi.org/10.1074/jbc.M206379200
Rebbapragada A, Benchabane H, Wrana JL, Celeste AJ, Attisano L (2003) Myostatin signals through a transforming growth factor β-like signaling pathway to block adipogenesis. Mol Cell Biol 23(20):7230–7242. https://doi.org/10.1128/MCB.23.20.7230-7242.2003
Wolfman NM et al (2003) Activation of latent myostatin by the BMP-1/tolloid family of metalloproteinases. Proc Natl Acad Sci 100(26):15842–15846. https://doi.org/10.1073/pnas.2534946100
McFarlane C et al (2011) Human myostatin negatively regulates human myoblast growth and differentiation. Am J Physiol Cell Physiol 301(1):C195–C203. https://doi.org/10.1152/ajpcell.00012.2011
Rodriguez J et al (2014) Myostatin and the skeletal muscle atrophy and hypertrophy signaling pathways. Cell Mol Life Sci 71(22):4361–4371. https://doi.org/10.1007/s00018-014-1689-x
Huang Z, Chen D, Zhang K, Yu B, Chen X, Meng J (2007) Regulation of myostatin signaling by c-Jun N-terminal kinase in C2C12 cells. Cell Signal 19(11):2286–2295. https://doi.org/10.1016/j.cellsig.2007.07.002
Mitra A, Qaisar R, Bose B, Sudheer SP (2023) The elusive role of myostatin signaling for muscle regeneration and maintenance of muscle and bone homeostasis. Osteoporosis Sarcopenia 9(1):1–7. https://doi.org/10.1016/j.afos.2023.03.008
Philip B, Lu Z, Gao Y (2005) Regulation of GDF-8 signaling by the p38 MAPK. Cell Signal 17(3):365–375. https://doi.org/10.1016/j.cellsig.2004.08.003
McPherron AC, Lawler AM, Lee S-J (1997) Regulation of skeletal muscle mass in mice by a new TGF-p superfamily member. Nature 387:6628. https://doi.org/10.1038/387083a0
Girgenrath S, Song K, Whittemore L-A (2005) Loss of myostatin expression alters fiber-type distribution and expression of myosin heavy chain isoforms in slow- and fast-type skeletal muscle. Muscle Nerve 31(1):34–40. https://doi.org/10.1002/mus.20175
Guo T, Jou W, Chanturiya T, Portas J, Gavrilova O, McPherron AC (2009) Myostatin inhibition in muscle, but not adipose tissue, decreases fat mass and improves insulin sensitivity. PLoS ONE 4(3):e4937. https://doi.org/10.1371/journal.pone.0004937
Camporez J-PG et al (2016) Anti-myostatin antibody increases muscle mass and strength and improves insulin sensitivity in old mice. Proc Natl Acad Sci 113(8):2212–2217. https://doi.org/10.1073/pnas.1525795113
Li Z, Zhao B, Kim YS, Hu CY, Yang J (2010) Administration of a mutated myostatin propeptide to neonatal mice significantly enhances skeletal muscle growth. Mol Reprod Dev 77(1):76–82. https://doi.org/10.1002/mrd.21111
Tsuchida K (2008) Myostatin inhibition by a follistatin-derived peptide ameliorates the pathophysiology of muscular dystrophy model mice. Acta Myol 27(1):14–18
Grobet L et al (2003) Modulating skeletal muscle mass by postnatal, muscle-specific inactivation of the myostatin gene. Genesis 35(4):227–238. https://doi.org/10.1002/gene.10188
Yang J, Ratovitski T, Brady JP, Solomon MB, Wells KD, Wall RJ (2001) Expression of myostatin pro domain results in muscular transgenic mice. Mol Reprod Dev 60(3):351–361. https://doi.org/10.1002/mrd.1097
Zhu X, Hadhazy M, Wehling M, Tidball JG, McNally EM (2000) Dominant negative myostatin produces hypertrophy without hyperplasia in muscle. FEBS Lett 474(1):71–75. https://doi.org/10.1016/S0014-5793(00)01570-2
Dalkilic I, Kunkel LM (2003) Muscular dystrophies: genes to pathogenesis. Curr Opin Genet Dev 13(3):231–238. https://doi.org/10.1016/S0959-437X(03)00048-0
Wagner KR, McPherron AC, Winik N, Lee S-J (2002) Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann Neurol 52(6):832–836. https://doi.org/10.1002/ana.10385
Bogdanovich S et al (2002) Functional improvement of dystrophic muscle by myostatin blockade. Nature 420(6914):418–421. https://doi.org/10.1038/nature01154
Becker C et al (2015) Myostatin antibody (LY2495655) in older weak fallers: a proof-of-concept, randomised, phase 2 trial. Lancet Diabetes Endocrinol 3(12):948–957. https://doi.org/10.1016/S2213-8587(15)00298-3
Woodhouse L et al (2016) A phase 2 randomized study investigating the efficacy and safety of myostatin antibody LY2495655 versus placebo in patients undergoing elective total HIP arthroplasty. J Frailty Aging 5(1):62–70. https://doi.org/10.14283/jfa.2016.81
Golan T et al (2018) LY2495655, an antimyostatin antibody, in pancreatic cancer: a randomized, phase 2 trial. J Cachexia Sarcopenia Muscle 9(5):871–879. https://doi.org/10.1002/jcsm.12331
StAndre M et al (2017) A mouse anti-myostatin antibody increases muscle mass and improves muscle strength and contractility in the mdx mouse model of Duchenne muscular dystrophy and its humanized equivalent, domagrozumab (PF-06252616), increases muscle volume in cynomolgus monkeys. Skelet Muscle 7:25. https://doi.org/10.1186/s13395-017-0141-y
Apgar JR et al (2016) Beyond CDR-grafting: structure-guided humanization of framework and CDR regions of an anti-myostatin antibody. MAbs 8(7):1302–1318. https://doi.org/10.1080/19420862.2016.1215786
Leung DG et al (2021) A phase Ib/IIa, open-label, multiple ascending-dose trial of domagrozumab in fukutin-related protein limb-girdle muscular dystrophy. Muscle Nerve 64(2):172–179. https://doi.org/10.1002/mus.27259
Wagner KR et al (2020) Randomized phase 2 trial and open-label extension of domagrozumab in Duchenne muscular dystrophy. Neuromuscul Disord 30(6):492–502. https://doi.org/10.1016/j.nmd.2020.05.002
“Pfizer Terminates Domagrozumab (PF-06252616) Clinical Studies for the Treatment of Duchenne Muscular Dystrophy | Pfizer.” https://www.pfizer.com/und/news/press-release/press-release-detail/pfizer_terminates_domagrozumab_pf_06252616_clinical_studies_for_the_treatment_of_duchenne_muscular_dystrophy Accessed 03 May 2024.
Amato AA et al (2014) Treatment of sporadic inclusion body myositis with bimagrumab. Neurology 83(24):2239–2246. https://doi.org/10.1212/WNL.0000000000001070
Amato AA et al (2021) Efficacy and safety of bimagrumab in sporadic inclusion body myositis: long-term extension of resilient. Neurology. https://doi.org/10.1212/WNL.0000000000011626
Hanna MG et al (2019) Safety and efficacy of intravenous bimagrumab in inclusion body myositis (RESILIENT): a randomised, double-blind, placebo-controlled phase 2b trial. Lancet Neurol 18(9):834–844. https://doi.org/10.1016/S1474-4422(19)30200-5
Rooks D et al (2017) Treatment of sarcopenia with bimagrumab: results from a phase II, randomized, controlled, proof-of-concept study. J Am Geriatr Soc 65(9):1988–1995. https://doi.org/10.1111/jgs.14927
Rooks D et al (2020) Bimagrumab vs optimized standard of care for treatment of sarcopenia in community-dwelling older adults: a randomized clinical trial. JAMA Netw Open 3(10):e2020836. https://doi.org/10.1001/jamanetworkopen.2020.20836
Polkey MI et al (2019) Activin type II receptor blockade for treatment of muscle depletion in chronic obstructive pulmonary disease. A randomized trial. Am J Respir Crit Care Med 199(3):313–320. https://doi.org/10.1164/rccm.201802-0286OC
Hofbauer LC et al (2021) Bimagrumab to improve recovery after hip fracture in older adults: a multicentre, double-blind, randomised, parallel-group, placebo-controlled, phase 2a/b trial. Lancet Healthy Longev 2(5):e263–e274. https://doi.org/10.1016/S2666-7568(21)00084-2
Latres E et al (2015) Myostatin blockade with a fully human monoclonal antibody induces muscle hypertrophy and reverses muscle atrophy in young and aged mice. Skelet Muscle 5:34. https://doi.org/10.1186/s13395-015-0060-8
Dagbay KB et al (2020) Structural basis of specific inhibition of extracellular activation of pro- or latent myostatin by the monoclonal antibody SRK-015. J Biol Chem 295(16):5404–5418. https://doi.org/10.1074/jbc.RA119.012293
Pirruccello-Straub M et al (2018) Blocking extracellular activation of myostatin as a strategy for treating muscle wasting. Sci Rep 8:2292. https://doi.org/10.1038/s41598-018-20524-9
Long KK et al (2019) Specific inhibition of myostatin activation is beneficial in mouse models of SMA therapy. Hum Mol Genet 28(7):1076–1089. https://doi.org/10.1093/hmg/ddy382
Muramatsu H et al (2021) Novel myostatin-specific antibody enhances muscle strength in muscle disease models. Sci Rep 11:1. https://doi.org/10.1038/s41598-021-81669-8
Attie KM et al (2013) A single ascending-dose study of muscle regulator ace-031 in healthy volunteers. Muscle Nerve 47(3):416–423. https://doi.org/10.1002/mus.23539
Campbell C et al (2012) A phase 2, randomized, placebo-controlled, multiple ascending-dose study of ACE-031, a soluble activin receptor type IIB, in boys with Duchenne muscular dystrophy (DMD) (P04.088). Neurology. https://doi.org/10.1212/WNL.78.1_supplement.P04.088
Cappellini MD et al (2020) A phase 3 trial of luspatercept in patients with transfusion-dependent β-thalassemia. N Engl J Med. https://doi.org/10.1056/NEJMoa1910182
Piga A et al (2019) Luspatercept improves hemoglobin levels and blood transfusion requirements in a study of patients with β-thalassemia. Blood 133(12):1279–1289. https://doi.org/10.1182/blood-2018-10-879247
Taher AT, Musallam KM, Cappellini MD (2021) β-Thalassemias. N Engl J Med 384(8):727–743. https://doi.org/10.1056/NEJMra2021838
Markham A (2020) Luspatercept: first approval. Drugs 80(1):85–90. https://doi.org/10.1007/s40265-019-01251-5
Desgeorges MM et al (2017) Pharmacological inhibition of myostatin improves skeletal muscle mass and function in a mouse model of stroke. Sci Rep 7:14000. https://doi.org/10.1038/s41598-017-13912-0
Muntoni F et al (2024) The clinical development of taldefgrobep alfa: an anti-myostatin adnectin for the treatment of Duchenne muscular dystrophy. Neurol Ther 13(1):183–219. https://doi.org/10.1007/s40120-023-00570-w
Robertson DM et al (1987) The isolation of polypeptides with FSH suppressing activity from bovine follicular fluid which are structurally different to inhibin. Biochem Biophys Res Commun 149(2):744–749. https://doi.org/10.1016/0006-291X(87)90430-X
Ueno N, Ling N, Ying SY, Esch F, Shimasaki S, Guillemin R (1987) Isolation and partial characterization of follistatin: a single-chain Mr 35,000 monomeric protein that inhibits the release of follicle-stimulating hormone. Proc Natl Acad Sci 84(23):8282–8286. https://doi.org/10.1073/pnas.84.23.8282
Amthor H et al (2004) Follistatin complexes myostatin and antagonises myostatin-mediated inhibition of myogenesis. Dev Biol 270(1):19–30. https://doi.org/10.1016/j.ydbio.2004.01.046
Cash JN, Rejon CA, McPherron AC, Bernard DJ, Thompson TB (2009) The structure of myostatin:follistatin 288: insights into receptor utilization and heparin binding. EMBO J 28(17):2662–2676. https://doi.org/10.1038/emboj.2009.205
Nakatani M et al (2008) Transgenic expression of a myostatin inhibitor derived from follistatin increases skeletal muscle mass and ameliorates dystrophic pathology in mdx mice. FASEB J 22(2):477–487. https://doi.org/10.1096/fj.07-8673com
Saitoh M et al (2020) Discovery of a follistatin-derived myostatin inhibitory peptide. Bioorg Med Chem Lett 30(3):126892. https://doi.org/10.1016/j.bmcl.2019.126892
Lee S-J et al (2010) Regulation of muscle mass by follistatin and activins. Mol Endocrinol 24(10):1998–2008. https://doi.org/10.1210/me.2010-0127
Lee S-J (2007) Quadrupling muscle mass in mice by targeting TGF-ß signaling pathways. PLoS ONE 2(8):e789. https://doi.org/10.1371/journal.pone.0000789
Matzuk MM, Lu N, Vogel H, Sellheyer K, Roop DR, Bradley A (1995) Multiple defects and perinatal death in mice deficient in follistatin. Nature 374(6520):360–363. https://doi.org/10.1038/374360a0
Giesige CR et al (2018) AAV-mediated follistatin gene therapy improves functional outcomes in the TIC-DUX4 mouse model of FSHD. JCI Insight 3(22):e123538. https://doi.org/10.1172/jci.insight.123538
Schumann C et al (2018) Increasing lean muscle mass in mice via nanoparticle-mediated hepatic delivery of follistatin mRNA. Theranostics 8(19):5276–5288. https://doi.org/10.7150/thno.27847
Pearsall RS et al (2019) Follistatin-based ligand trap ACE-083 induces localized hypertrophy of skeletal muscle with functional improvement in models of neuromuscular disease. Sci Rep 9:11392. https://doi.org/10.1038/s41598-019-47818-w
Glasser CE, Gartner MR, Wilson D, Miller B, Sherman ML, Attie KM (2018) Locally acting ACE-083 increases muscle volume in healthy volunteers. Muscle Nerve 57(6):921–926. https://doi.org/10.1002/mus.26113
Statland JM et al (2022) Randomized phase 2 study of ACE-083, a muscle-promoting agent, in facioscapulohumeral muscular dystrophy. Muscle Nerve 66(1):50–62. https://doi.org/10.1002/mus.27558
Thomas FP et al (2022) Randomized phase 2 study of ACE-083 in patients with charcot-marie-tooth disease. Neurology 98(23):e2356–e2367. https://doi.org/10.1212/WNL.0000000000200325
Haidet AM et al (2008) Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc Natl Acad Sci USA 105(11):4318–4322. https://doi.org/10.1073/pnas.0709144105
Mendell JR et al (2015) A phase 1/2a follistatin gene therapy trial for becker muscular dystrophy. Mol Ther 23(1):192–201. https://doi.org/10.1038/mt.2014.200
Mendell JR et al (2017) Follistatin gene therapy for sporadic inclusion body myositis improves functional outcomes. Mol Ther 25(4):870–879. https://doi.org/10.1016/j.ymthe.2017.02.015
Thies RS et al (2001) GDF-8 propeptide binds to GDF-8 and antagonizes biological activity by inhibiting GDF-8 receptor binding. Growth Factors 18(4):251–259. https://doi.org/10.3109/08977190109029114
Takayama K et al (2015) Identification of the minimum peptide from mouse myostatin prodomain for human myostatin inhibition. J Med Chem 58(3):1544–1549. https://doi.org/10.1021/jm501170d
Walker RG et al (2016) Biochemistry and biology of GDF11 and myostatin. Circ Res 118(7):1125–1142. https://doi.org/10.1161/CIRCRESAHA.116.308391
Ochsner UA, Green LS, Rice TP, Olivas E, Janjic N, Katilius E (2019) Targeting unique epitopes on highly similar proteins GDF-11 and GDF-8 with modified DNA aptamers. Biochemistry 58(46):4632–4640. https://doi.org/10.1021/acs.biochem.9b00760
St. Andre M et al (2017) A mouse anti-myostatin antibody increases muscle mass and improves muscle strength and contractility in the mdx mouse model of Duchenne muscular dystrophy and its humanized equivalent, domagrozumab (PF-06252616), increases muscle volume in cynomolgus monkeys. Skeletal Muscle 7:25. https://doi.org/10.1186/s13395-017-0141-y
Souza TA et al (2008) Proteomic identification and functional validation of activins and bone morphogenetic protein 11 as candidate novel muscle mass regulators. Mol Endocrinol 22(12):2689–2702. https://doi.org/10.1210/me.2008-0290
Phillips DJ, de Kretser DM (1998) Follistatin: a multifunctional regulatory protein. Front Neuroendocrinol 19(4):287–322. https://doi.org/10.1006/frne.1998.0169
Mariot V et al (2017) Downregulation of myostatin pathway in neuromuscular diseases may explain challenges of anti-myostatin therapeutic approaches. Nat Commun 8:1. https://doi.org/10.1038/s41467-017-01486-4
Wojciechowski J, Purohit VS, Harnisch LO, Dua P, Tan B, Nicholas T (2022) Population PK and PD analysis of domagrozumab in pediatric patients with Duchenne muscular dystrophy. Clin Pharmacol Ther 112(6):1291–1302. https://doi.org/10.1002/cpt.2747
Baczek J, Silkiewicz M, Wojszel ZB (2020) Myostatin as a biomarker of muscle wasting and other pathologies-state of the art and knowledge gaps. Nutrients 12(8):2401. https://doi.org/10.3390/nu12082401
Lehallier B et al (2019) Undulating changes in human plasma proteome profiles across the lifespan. Nat Med 25:12. https://doi.org/10.1038/s41591-019-0673-2
Hedayati M, Nozhat Z, Hannani M (2016) Can the serum level of myostatin be considered as an informative factor for cachexia prevention in patients with medullary thyroid cancer? Asian Pac J Cancer Prev 17(S3):119–123. https://doi.org/10.7314/apjcp.2016.17.s3.119
Loumaye A et al (2015) Role of activin A and myostatin in human cancer cachexia. J Clin Endocrinol Metab 100(5):2030–2038. https://doi.org/10.1210/jc.2014-4318
Egerman MA et al (2015) GDF11 increases with age and inhibits skeletal muscle regeneration. Cell Metab 22(1):164–174. https://doi.org/10.1016/j.cmet.2015.05.010
Rodgers BD, Eldridge JA (2015) Reduced circulating GDF11 Is unlikely responsible for age-dependent changes in mouse heart, muscle, and brain. Endocrinology 156(11):3885–3888. https://doi.org/10.1210/en.2015-1628
Cawthon PM et al (2023) Evaluation of associations of growth differentiation factor-11, growth differentiation factor-8, and their binding proteins, follistatin and follistatin-like protein-3, with measures of skeletal muscle mass, muscle strength, and physical function in older adults. J Gerontol A 78(11):2051–2059. https://doi.org/10.1093/gerona/glad045
Semba RD et al (2019) Relationship of circulating growth and differentiation factors 8 and 11 and their antagonists as measured using liquid chromatography-tandem mass spectrometry with age and skeletal muscle strength in healthy adults. J Gerontol A 74(1):129–136. https://doi.org/10.1093/gerona/gly255
Burch PM et al (2017) Reduced serum myostatin concentrations associated with genetic muscle disease progression. J Neurol 264(3):541–553. https://doi.org/10.1007/s00415-016-8379-6
Lakshman KM et al (2009) Measurement of myostatin concentrations in human serum: circulating concentrations in young and older men and effects of testosterone administration. Mol Cell Endocrinol 302(1):26–32. https://doi.org/10.1016/j.mce.2008.12.019
Singh P, Rong H, Gordi T, Bosley J, Bhattacharya I (2016) Translational pharmacokinetic/pharmacodynamic analysis of MYO-029 antibody for muscular dystrophy. Clin Transl Sci 9(6):302–310. https://doi.org/10.1111/cts.12420
Wu H, Xiong WC, Mei L (2010) To build a synapse: signaling pathways in neuromuscular junction assembly. Development 137(7):1017–1033. https://doi.org/10.1242/dev.038711
Lovering RM, Iyer SR, Edwards B, Davies KE (2020) Alterations of neuromuscular junctions in Duchenne muscular dystrophy. Neurosci Lett 737:135304. https://doi.org/10.1016/j.neulet.2020.135304
Willmann R, Possekel S, Dubach-Powell J, Meier T, Ruegg MA (2009) Mammalian animal models for Duchenne muscular dystrophy. Neuromuscul Disord 19(4):241–249. https://doi.org/10.1016/j.nmd.2008.11.015
Jang J et al (2021) Myostatin inhibition-induced increase in muscle mass and strength was amplified by resistance exercise training, and dietary essential amino acids improved muscle quality in mice. Nutrients 13(5):1508. https://doi.org/10.3390/nu13051508
LeBrasseur NK, Schelhorn TM, Bernardo BL, Cosgrove PG, Loria PM, Brown TA (2009) Myostatin inhibition enhances the effects of exercise on performance and metabolic outcomes in aged mice. J Gerontol A 64A(9):940–948. https://doi.org/10.1093/gerona/glp068
Allen DL et al (2008) Myostatin, activin receptor IIb, and follistatin-like-3 gene expression are altered in adipose tissue and skeletal muscle of obese mice. Am J Physiol-Endocrinol Metab 294(5):E918–E927. https://doi.org/10.1152/ajpendo.00798.2007
Amor M et al (2019) Serum myostatin is upregulated in obesity and correlates with insulin resistance in humans. Exp Clin Endocrinol Diabetes 127(8):550–556. https://doi.org/10.1055/a-0641-5546
Hittel DS, Axelson M, Sarna N, Shearer J, Huffman KM, Kraus WE (2010) Myostatin decreases with aerobic exercise and associates with insulin resistance. Med Sci Sports Exerc 42(11):2023–2029. https://doi.org/10.1249/MSS.0b013e3181e0b9a8
Lyons J-A, Haring JS, Biga PR (2010) Myostatin expression, lymphocyte population, and potential cytokine production correlate with predisposition to high-fat diet induced obesity in mice. PLoS ONE 5(9):e12928. https://doi.org/10.1371/journal.pone.0012928
Dial AG et al (2020) Muscle and serum myostatin expression in type 1 diabetes. Physiol Rep 8(13):e14500. https://doi.org/10.14814/phy2.14500
Wang F, Liao Y, Li X, Ren C, Cheng C, Ren Y (2012) Increased circulating myostatin in patients with type 2 diabetes mellitus. J Huazhong Univ Sci Technol 32(4):534–539. https://doi.org/10.1007/s11596-012-0092-9
McPherron AC, Lee S-J (2002) Suppression of body fat accumulation in myostatin-deficient mice. J Clin Invest 109(5):595–601. https://doi.org/10.1172/JCI13562
Eilers W, Chambers D, Cleasby M, Foster K (2020) Local myostatin inhibition improves skeletal muscle glucose uptake in insulin-resistant high-fat diet-fed mice. Am J Physiol-Endocrinol Metab 319(1):E163–E174. https://doi.org/10.1152/ajpendo.00185.2019
Zhao B, Wall RJ, Yang J (2005) Transgenic expression of myostatin propeptide prevents diet-induced obesity and insulin resistance. Biochem Biophys Res Commun 337(1):248–255. https://doi.org/10.1016/j.bbrc.2005.09.044
Coleman SK, Rebalka IA, D’Souza DM, Deodhare N, Desjardins EM, Hawke TJ (2016) Myostatin inhibition therapy for insulin-deficient type 1 diabetes. Sci Rep 6:1. https://doi.org/10.1038/srep32495
Park SW et al (2009) Excessive loss of skeletal muscle mass in older adults with type 2 diabetes. Diabetes Care 32(11):1993–1997. https://doi.org/10.2337/dc09-0264
Garito T et al (2018) Bimagrumab improves body composition and insulin sensitivity in insulin-resistant individuals. Diabetes Obes Metab 20(1):94–102. https://doi.org/10.1111/dom.13042
Rooks D, Petricoul O, Praestgaard J, Bartlett M, Laurent D, Roubenoff R (2020) Safety and pharmacokinetics of bimagrumab in healthy older and obese adults with body composition changes in the older cohort. J Cachexia Sarcopenia Muscle 11(6):1525–1534. https://doi.org/10.1002/jcsm.12639
Heymsfield SB et al (2021) Effect of bimagrumab vs placebo on body fat mass among adults with type 2 diabetes and obesity: a phase 2 randomized clinical trial. JAMA Netw Open 4(1):e2033457. https://doi.org/10.1001/jamanetworkopen.2020.33457
Merz KE, Thurmond DC (2020) Role of skeletal muscle in insulin resistance and glucose uptake. Compr Physiol 10(3):785–809. https://doi.org/10.1002/cphy.c190029
Elliott B, Renshaw D, Getting S, Mackenzie R (2012) The central role of myostatin in skeletal muscle and whole body homeostasis. Acta Physiol 205(3):324–340. https://doi.org/10.1111/j.1748-1716.2012.02423.x
Liu X-H, Bauman WA, Cardozo CP (2018) Myostatin inhibits glucose uptake via suppression of insulin-dependent and -independent signaling pathways in myoblasts. Physiol Rep 6(17):e13837. https://doi.org/10.14814/phy2.13837
Cong LN et al (1997) Physiological role of Akt in insulin-stimulated translocation of GLUT4 in transfected rat adipose cells. Mol Endocrinol 11(13):1881–1890. https://doi.org/10.1210/mend.11.13.0027
Armoni M, Harel C, Karnieli E (2007) Transcriptional regulation of the GLUT4 gene: from PPAR-γ and FOXO1 to FFA and inflammation. Trends Endocrinol Metab 18(3):100–107. https://doi.org/10.1016/j.tem.2007.02.001
Huang X, Liu G, Guo J, Su Z (2018) The PI3K/AKT pathway in obesity and type 2 diabetes. Int J Biol Sci 14(11):1483–1496. https://doi.org/10.7150/ijbs.27173
Tang L et al (2014) A prepared anti-MSTN polyclonal antibody reverses insulin resistance of diet-induced obese rats via regulation of PI3K/Akt/mTOR&FoxO1 signal pathways. Biotechnol Lett 36(12):2417–2423. https://doi.org/10.1007/s10529-014-1617-z
Braga M, Pervin S, Norris K, Bhasin S, Singh R (2013) Inhibition of in vitro and in vivo brown fat differentiation program by myostatin. Obesity (Silver Spring) 21(6):1180–1188. https://doi.org/10.1002/oby.20117
Asterholm IW, Scherer PE (2010) Enhanced metabolic flexibility associated with elevated adiponectin levels. Am J Pathol 176(3):1364–1376. https://doi.org/10.2353/ajpath.2010.090647
Kubota N et al (2007) Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab 6(1):55–68. https://doi.org/10.1016/j.cmet.2007.06.003
Choi SJ, Yablonka-Reuveni Z, Kaiyala KJ, Ogimoto K, Schwartz MW, Wisse BE (2011) Increased energy expenditure and leptin sensitivity account for low fat mass in myostatin-deficient mice. Am J Physiol Endocrinol Metab 300(6):E1031–E1037. https://doi.org/10.1152/ajpendo.00656.2010
Zhang C et al (2012) Inhibition of myostatin protects against diet-induced obesity by enhancing fatty acid oxidation and promoting a brown adipose phenotype in mice. Diabetologia 55(1):183–193. https://doi.org/10.1007/s00125-011-2304-4
Maliszewska K, Kretowski A (2021) Brown adipose tissue and its role in insulin and glucose homeostasis. Int J Mol Sci 22(4):1530. https://doi.org/10.3390/ijms22041530
Cai C et al (2017) Loss-of-function myostatin mutation increases insulin sensitivity and browning of white fat in Meishan pigs. Oncotarget 8(21):34911–34922. https://doi.org/10.18632/oncotarget.16822
Kim WK, Choi H-R, Park SG, Ko Y, Bae K-H, Lee SC (2012) Myostatin inhibits brown adipocyte differentiation via regulation of Smad3-mediated β-catenin stabilization. Int J Biochem Cell Biol 44(2):327–334. https://doi.org/10.1016/j.biocel.2011.11.004
Dong J, Dong Y, Dong Y, Chen F, Mitch WE, Zhang L (2016) Inhibition of myostatin in mice improves insulin sensitivity via irisin-mediated cross talk between muscle and adipose tissues. Int J Obes 40:3. https://doi.org/10.1038/ijo.2015.200
Shan T, Liang X, Bi P, Kuang S (2013) Myostatin knockout drives browning of white adipose tissue through activating the AMPK-PGC1α-Fndc5 pathway in muscle. FASEB J 27(5):1981–1989. https://doi.org/10.1096/fj.12-225755
Steculorum SM et al (2016) AgRP neurons control systemic insulin sensitivity via myostatin expression in brown adipose tissue. Cell 165(1):125–138. https://doi.org/10.1016/j.cell.2016.02.044
Kong X et al (2014) IRF4 is a key thermogenic transcriptional partner of PGC-1α. Cell 158(1):69–83. https://doi.org/10.1016/j.cell.2014.04.049
Chen Y-S et al (2017) GDF8 inhibits bone formation and promotes bone resorption in mice. Clin Exp Pharmacol Physiol 44(4):500–508. https://doi.org/10.1111/1440-1681.12728
Hamrick MW et al (2007) Loss of myostatin (GDF8) function increases osteogenic differentiation of bone marrow-derived mesenchymal stem cells but the osteogenic effect is ablated with unloading. Bone 40(6):1544–1553. https://doi.org/10.1016/j.bone.2007.02.012
Elkasrawy M, Fulzele S, Bowser M, Wenger K, Hamrick M (2011) Myostatin (GDF-8) inhibits chondrogenesis and chondrocyte proliferation in vitro by suppressing Sox-9 expression. Growth Factors 29(6):253–262. https://doi.org/10.3109/08977194.2011.599324
Dankbar B et al (2015) Myostatin is a direct regulator of osteoclast differentiation and its inhibition reduces inflammatory joint destruction in mice. Nat Med 21(9):1085–1090. https://doi.org/10.1038/nm.3917
Hamrick MW, Arounleut P, Kellum E, Cain M, Immel D, Liang L (2010) Recombinant myostatin (GDF-8) propeptide enhances the repair and regeneration of both muscle and bone in a model of deep penetrant musculoskeletal injury. J Trauma 69(3):579–583. https://doi.org/10.1097/TA.0b013e3181c451f4
Bialek P et al (2014) A myostatin and activin decoy receptor enhances bone formation in mice. Bone 60:162–171. https://doi.org/10.1016/j.bone.2013.12.002
Dumonceaux J et al (2010) Combination of myostatin pathway interference and dystrophin rescue enhances tetanic and specific force in dystrophic mdx mice. Mol Ther 18(5):881–887. https://doi.org/10.1038/mt.2009.322
Lu-Nguyen N, Malerba A, Popplewell L, Schnell F, Hanson G, Dickson G (2017) Systemic antisense therapeutics for dystrophin and myostatin exon splice modulation improve muscle pathology of adult mdx mice. Mol Therapy Nucleic Acids 6:15. https://doi.org/10.1016/j.omtn.2016.11.009
Lu-Nguyen N et al (2019) Functional muscle recovery following dystrophin and myostatin exon splice modulation in aged mdx mice. Hum Mol Genet 28(18):3091–3100. https://doi.org/10.1093/hmg/ddz125
Lee S-J et al (2020) Targeting myostatin/activin A protects against skeletal muscle and bone loss during spaceflight. Proc Natl Acad Sci 117(38):23942–23951. https://doi.org/10.1073/pnas.2014716117
Acknowledgements
We highly appreciate the funding for this research program from Hawaii Community Foundation—George F. Straub Trust (ID#: MedRes_2024_00006475), USDA multi-state project—Nutrient Bioavailability—Phytonutrients and Beyond (W5002) administrated by College of Tropical Agriculture and Human Resources of the University of Hawaii at Manoa (Honolulu, Hawaii), and the USDA-ARS Daniel K. Inouye U.S. Pacific Basin Agricultural Research Center (Hilo, Hawaii).
Funding
Funding was provided by Agricultural Research Service (Grant No. USDA Multi-State Project W5002).
Author information
Authors and Affiliations
Contributions
Brock Wetzlich writes the manuscript with the assistances from Benard Nyakundi. Benard Nyakundi also contributes writing and revising the manuscript. Jinzeng Yang had the initial idea, provided the guidance and the content, and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wetzlich, B., Nyakundi, B.B. & Yang, J. Therapeutic applications and challenges in myostatin inhibition for enhanced skeletal muscle mass and functions. Mol Cell Biochem (2024). https://doi.org/10.1007/s11010-024-05120-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11010-024-05120-y