
Abstract
Pseudomonas aeruginosa is one of the leading causes of nosocomial infections, characterized by increasing antibiotic resistance, severity and mortality. Therefore, numerous efforts have been made nowadays to identify new therapeutic targets. This study aimed to find potential drug targets and vaccine candidates in drug-resistant strains of P. aeruginosa. Extensive antibiotic-resistant and carbapenem-resistant strains of P. aeruginosa with complete genome were selected and ten common hypothetical proteins (HPs) containing more than 200 amino acids were obtained. The structural, functional and immunological predictions of these HPs were performed with the utility of bioinformatics approaches. Two common HPs (Gene ID: 2877781645 and 2877781936) among other investigated proteins were revealed as potential candidates for pharmaceutical and vaccine purposes based on structural and physicochemical properties, functional domains, subcellular localizations, signal peptides, toxicity, virulence factor, antigenicity, allergenicity and immunoinformatic predictions. The consequence of this predictive study will assist in novel drug and vaccine design through experimental investigations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pseudomonas aeruginosa is a gram-negative bacterium, a gamma proteobacter, belonging to the Pseudomonadaceae family with an excellent adaptability and survival in a variety of different environments and ecological niches (Klockgether and Tümmler 2017; Pang et al. 2019). This bacterium is an opportunistic pathogen and one of the most frequent and severe cause of nosocomial infections (NIs) leading to a wide range of infections, from acute to chronic life-threatening, in humans (Liu et al. 2015; Lund-Palau et al. 2016). The mortality rate due to the infections caused by this bacterium is very high, especially in people with compromised immunity and people with cystic fibrosis (CF) (López-Causapé et al. 2018). Pseudomonas aeruginosa is inherently resistant to a wide range of antibiotics and belongs to the group of ESKAPE pathogens that have potential mechanisms of drug resistance (Santajit and Indrawattana 2016; van Duin and Paterson 2016). The emergence of MDR (multi-drug resistant), XDR (extensively drug-resistant), and PDR (pan-drug resistant) strains of P. aeruginosa and the increasing number of the resistant strains of this bacterium are considered as a major crisis and paints a scary future for current antibiotics and challenges the effectiveness of these antibiotics (Bassetti et al. 2013; Ventola 2015). Despite the efforts made, there is still no vaccine available for this bacterium and the last line of treatment is carbapenem group of antibiotics (meropenem, doripenem, ertapenem and imipenem) but the growing number of carbapenem-resistant strains has made them critical priorities with an urgent need for developing new treatment strategies by the World Health organization (WHO) (Priebe and Goldberg 2014; Haenni et al. 2017; World Health Organization 2019).
The genome of P. aeruginosa is large (5.5–7 million base pairs) and contains many open reading frames predicted as hypothetical proteins (Klockgether et al. 2011; Newman et al. 2017).
Hypothetical proteins (HPs) are predicted proteins with no experimental evidences for their structures and functions but they may be associated with human diseases and may play an important role in understanding biological and functional pathways, finding new structures, functions, domains, motifs and markers, fighting pathogens and early detection or treatment of infectious diseases, and developing new vaccines and drug candidates (Naqvi et al. 2015; Varma et al. 2015; Ijaq et al. 2019). Functional annotation and curation of HPs in several human pathogens have been also reported (Rabbi et al. 2021).
In the present study hypervirulent strains of P. aeruginosa were isolated, and then structural, functional, and immunoinformatics analysis of common hypothetical proteins were performed with the utility of in silico methods to identify potential drug and vaccine candidates against the resistant strains of P. aeruginosa.
Materials and Methods
Sequence Retrieval
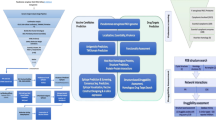
Pseudomonas aeruginosa strains with completed genome sequencing were collected through Integrated Microbial Genome (IMG) site (https://img.jgi.doe.gov/) (Chen et al. 2021). MDR, XDR and strains resistant to carbapenem antibiotics were selected by literatures review. The Pseudomonas database (https://www.pseudomonas.com/) was also used to evaluate the resistance of the strains (Winsor et al. 2016). The most resistant strain was selected as a query and placed in the homologous part in IMG site to find the common hypothetical proteins. In the non-homologous part, Homo sapiens and two human microbiome bacteria (Escherichia coli K-12 MG1655 and E. coli K12-W3110) were placed. Subsequently, 10 common hypothetical proteins with a length of > 200 amino acids were selected for further study (Fig. 1). The sequences of these final proteins were submitted to several servers and predictive databases to characterize the hypothetical proteins.

Schematic workflow of the common hypothetical proteins in silico. Common hypothetical proteins of resistant strains were obtained and then structural, functional and immunological predictions were made for these proteins. Finally, two proteins were introduced as vaccine and drug candidates
Primary Structural Analysis of Hypothetical Proteins
The physicochemical properties of the selected hypothetical proteins were predicted by Protparam tool (https://web.expasy.org/protparam/). The physicochemical properties of a protein provides basic information about its properties (Wilkins et al. 1999).
The subcellular localization of the hypothetical proteins was predicted using 4 servers: PSORTb(V.3) (https://www.psort.org/psortb/) (Yu et al. 2010), CELLO II (http://cello.life.nctu.edu.tw/) (Yu et al. 2006), PSLpred (https://webs.iiitd.edu.in/raghava/pslpred/index.html) (Bhasin et al. 2005) and Gneg-mPLoc (http://www.csbio.sjtu.edu.cn/bioinf/Gneg-multi/) (Shen and Chou 2010). The CELLO II server predicts the position of proteins based on the support vector machine (SVM) method (Yu et al. 2006). The PSLpred server was developed to predict the location of gram-negative bacterial proteins (Bhasin et al. 2005). The Gneg-mPLoc server uses a combination of gene ontology (GO) information and domain function to predict the position of proteins (Shen and Chou 2010). According to various reports, bioinformatics servers cannot accurately detect the location of some proteins to predict the subcellular localization alone. In this study, 4 different servers were used to identify the location of proteins to cover each other's weaknesses (Gardy and Brinkman 2006; Brown et al. 2012).
We used TMHMM, HMMTOP, SignalP and SecretomeP for more precise analysis of HPs. We used TMHMM(V.2) (http://www.cbs.dtu.dk/services/TMHMM/) (Krogh et al. 2001) and HMMTOP (http://www.enzim.hu/hmmtop/) (Tusnády and Simon 2001) to predict the transmembrane helixes for the hypothetical proteins. Both servers are based on hidden Markov model (HMM) (Krogh et al. 2001; Tusnády and Simon 2001).
The SignalP(V.5) server (http://www.cbs.dtu.dk/services/SignalP/) was used to investigate the presence of signal peptides in the hypothetical proteins. This server is based on the deep neural network (Almagro Armenteros et al. 2019). The existence of non-classical secretory pathway in hypothetical proteins was predicted by SecretomeP(V.2) server (http://www.cbs.dtu.dk/services/SecretomeP/) (Bendtsen et al. 2005).
Secondary Structural Analysis of Hypothetical Proteins
Two servers, SOPMA (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html) and PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/), were used to predict the secondary structure of hypothetical proteins. The PSIPRED server uses a two-stage neural network to predict the second structure(Geourjon and Deléage 1995; Buchan and Jones 2019).
Three-Dimensional Structure Analysis of Hypothetical Proteins
The biological functions of a protein are determined by its third structure. The I-TASSER server (https://zhanggroup.org/I-TASSER/) was used to predict the tertiary structure of the putative proteins. The I-TASSER server is an integrated platform for automatically predicting protein structure and its performance is based on sequence-to-structure-to-function pattern (Roy et al. 2010).
Evaluate of the Quality of Three-Dimensional Structures
The quality evaluation of three-dimensional structures of hypothetical proteins was performed using ERRAT tool (https://saves.mbi.ucla.edu/). Types of different atoms due to Energetic and Geometric effects on each other non-randomly proteins are distributed. Errors in model construction lead to a more random distribution of different types of atoms. Statistical methods can be used to distinguish them from correct distributions. Subject that the basis of development is ERRAT (Colovos and Yeates 1993).
Functional Analysis of Hypothetical Proteins
Interpretation of different domains of a protein is a very important topic in protein-related studies. SMART(V.9) (http://smart.embl-heidelberg.de/) (Letunic et al. 2021) and Pfam(V.34) (http://pfam.xfam.org/) databases (Mistry et al. 2021) were used to predict the functional domains in hypothetical proteins. The SMART database is a web resource for identifying and interpreting functional domains (Letunic et al. 2021). The Pfam database is widely used to classify protein sequences in protein families and domains (Mistry et al. 2021).
The CDD database (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) was used to examine the conserved domains of hypothetical proteins. The CDD database is part of the Entrez NCBI search and retrieval system (Marchler-Bauer et al. 2013).
BLASTP. BLASTP tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins) was used to search for similar sequences of hypothetical proteins. Investigating the similarity of the protein sequences studied in the study with other proteins in other organisms can provide useful information about finding different domains. This tool compares the query protein sequences with the protein sequences in the protein database (Altschul et al. 1997).
The toxicity of hypothetical proteins was investigated using the BTXpred server (https://webs.iiitd.edu.in/raghava/btxpred/index.html). This server classifies toxins into two groups: (I) exotoxins and (II) endotoxins (Saha and Raghava 2007).
Function prediction for hypothetical proteins was performed by the VICMpred server (https://webs.iiitd.edu.in/raghava/vicmpred/index.html). This server classifies pathogenic microbial proteins into 4 functional classes: (I) virulence factor, (II) information molecule, (III) cellular process, and (IV) metabolism (Saha and Raghava 2006a, b).
Immunoinformatics Analysis
The antigenicity of hypothetical proteins was predicted with VaxiJen(V.2) server (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html). Predicting conserved antigens is crucial for the development of vaccine candidates. This server predicts conserved antigens using an alignment-independent approach (Doytchinova and Flower 2007). Hypothetical proteins with a threshold above 0.5 were considered as antigens.
AllerTOP(V.2) (https://www.ddg-pharmfac.net/AllerTOP/) (Dimitrov et al. 2014) server was used to predict the allergenicity of hypothetical proteins.
Major histocompatibility complex (MHC) is a basic cell surface protein of the vertebrate immune system (Kaufman 2018). MHC molecules that present antigens are highly polymorphic and are classified into two main classes, MHCI and MHCII (Reche et al. 2004). MHCI is involved in the determination of epitopes for cytotoxic T cells (TCD8+) and MHCII is involved in the determination of epitopes for helper T cells (TCD4+) (Reche et al. 2004). The IEDB database (https://www.iedb.org/) was used to predict MHC-binding epitopes in hypothetical proteins (Vita et al. 2019). For this evaluation, the list of alleles in the default site was used.
Epitopes of hypothetical proteins detected by B lymphocytes were predicted using two servers, ABCpred (https://webs.iiitd.edu.in/raghava/abcpred/index.html) (Saha and Raghava 2006a, b) and Ellipro (http://tools.iedb.org/ellipro/) (Ponomarenko et al. 2008). ABCpred server was used to predict continuous B lymphocyte epitopes with the default threshold. The approach of this server is to use recurrent neural network (Saha and Raghava 2006a, b). The Elllipro server was used to predict discontinuous B lymphocyte epitopes with the default threshold. This server makes predictions based on the geometric properties of the protein structure. Based on the geometrical properties of the protein structure, this server predicts discontinuous epitopes of B lymphocytes (Ponomarenko et al. 2008).
Results
Sequence Retrieval
Thirty five resistant strains with finished genome sequences were detected (Table 1). The 10 common hypothetical proteins are shown in Table 2.
Primary Structural Analysis of Hypothetical Proteins
Physicochemical properties of 10 hypothetical proteins including molecular weight, isoelectric point, instability index, aliphatic index, and hydrophobicity (GRAVY) were evaluated. HP2, HP3, HP4, HP5, and HP6 proteins are stable and others are unstable. Only HP2 protein has pI > 7 and others have pI < 7. The aliphatic index of HP6 protein is the highest. The GRAVY index of all hypothetical proteins is negative, so they are all hydrophilic (Table 3).
Results of prediction of subcellular localization, transmembrane helixes, and prediction of SignalP and SecretomeP are shown in Table 4.
Secondary Structural Analyzes of Hypothetical Proteins
In general, the most common secondary structure in hypothetical proteins was the coil structure. The results of SOPMA and PSIPRED servers for predicting the secondary structure of 10 hypothetical common proteins are given in Supplementary File 1.
Three-Dimensional Structure Analysis of Hypothetical Proteins
The results of I-TASSER server for predicting the tertiary structure of 10 hypothetical proteins are given in Fig. 2.

Models of third structure of 10 hypothetical proteins predicted by the I-TASSER server, a HP1, b HP2, c HP3, d HP4, e HP5, f HP6, g HP7, h HP8, i HP9, and j HP10
Evaluate the Quality of Three-Dimensional Structures of Hypothetical Proteins
The quality of all three-dimensional structure models predicted by the I-TASSER server was evaluated with the ERRAT tool. For the ERRAT tool, a quality tertiary structure model must score above 50. All models obtained from the I-TASSER server had a score above 50.
Functional Analysis of Hypothetical Proteins
Functional and conserved domains of hypothetical proteins were predicted using SMART, Pfam, and CDD databases. No functional and conserved domains were predicted for HP6. The results of three databases are as follows: HP1, HP5, HP7, and HP10 proteins have the DUF domain, HP2, and HP8 proteins have OprD domain, HP4 protein has the MoaF and MoaF-C domains. HP9 protein has the FecR domain in SMART and Pfam database and has the COG4254 domain in CDD database, HP3 protein has the M60-like domain in SMART database and has the peptidase_M60 like super family, IMPa_N_2, and IMPa_helical domains.
BLASTP was used for the prediction of similarity between 10 hypothetical proteins with other proteins in other organisms. In general, the hypothetical proteins were most similar to organisms of Acinetobacter baumannii, Klebsiella pneumonia, Streptococcus dysgalactiae, and Enterobacter cloacae. BLASTP results are given in Supplementary File 2.
The BTXpred server was used to predict the toxicity of 10 hypothetical proteins. According to the results of this server, HP2, HP3, HP4, HP5, HP8, and HP10 are exotoxins and other proteins are not toxins.
The VICMpred server was used for function prediction. Based on the result of this server, HP1, HP4, and HP10 proteins are involved in metabolism, HP2, HP3, and HP9 proteins are involved in virulence factor, and other proteins are involved in cellular process.
Immunoinformatics Analysis
HP2, HP3, HP4, HP5, HP7, HP8, and HP9 are antigens according to the VaxiJen server, while HP1, HP6, and HP10 proteins are not. HP6 protein is an allergen based on AllerTOP server results.
Prediction of T cell epitopes and epitopes identified by B lymphocytes for 10 hypothetical proteins are given in Supplementary File 3 and Supplementary File 4 respectively.
Discussion
Pseudomonas aeruginosa is one of the most important health challenges in the medical community due to its antibiotic resistance. Until now, many efforts have been made to prevent and treat infections caused by this bacterium. In this study, the structure and function of 10 common hypothetical proteins from resistant strains of P. aeruginosa were investigated. The goal of analyzing common hypothetical proteins in resistant strains was to find potential candidates for vaccine and drug design. The structural and functional characterization of hypothetical bacterial proteins can answer many of our questions about bacterial physiology, but experimental approaches are time-consuming and cost a lot of money. In silico techniques along with laboratory techniques can overcome these problems and more accurate results can be achieved (Omeershffudin and Kumar 2019, Uddin et al. 2019).
Immunoinformatics offers new approaches to the design of new vaccines, diagnostic targets, and the study of the pathology of infectious diseases (He et al. 2010). Studies have shown that the best candidates for vaccine design should be able to stimulate both humoral and cellular immunity (Kozakiewicz et al. 2013). Moreover, they should have antigenicity and should be extracellular proteins, not homologous to human proteins and microbiome bacteria and not allergenic to humans (Barat et al. 2012; Sudha et al. 2019). Virulence factors expressed by bacteria are indispensable for the survival and growth of pathogenic bacteria and thus can be a valuable drug targets (Sudha et al. 2019). Vaccines are a safe and cost-effective solution to fight infectious diseases (Doro et al. 2009). Peptide vaccines are suitable, safe, and contain immunogenic epitopes (Rashid et al. 2017). Immunogenic potential is primarily dependent on the affinity of MHC binding (Rashid et al. 2017). Therefore, predicting epitopes with higher binding potential for MHC is essential for designing a peptide vaccine (Rashid et al. 2017).
Considering all the features mentioned in this study, only two proteins, HP2 and HP3, among the 10 common hypothetical proteins are introduced as potential candidates for drugs and vaccines. According to the analysis performed, the location of these two proteins was predicted in an outer membrane. Moreover, for these two proteins, virulence factor function was predicted and they were exotoxins. They had antigenicity and they did not an allergen. Based on the functional and conserved domains analysis, it is predicted that the HP2 protein is a member of the OprD family. This family contains bacterial outer membrane porins with serine protease activity (Yoshihara et al. 1998). It was reported that the OprD2 protein of P. aeruginosa had protease activity (Yoshihara et al. 1998). The OprD protein in P. aeruginosa facilitates the passage of small amino acids and peptides (Ochs et al. 2000). Another role of this protein is to cross the antibiotic imipenem (Ochs et al. 2000). Long-term treatment of patients with imipenem results in the development of imipenem-resistant mutants due to mutations in the OprD gene (Ochs et al. 2000). These mutant strains have severely reduced expression levels of OprD (Ochs et al. 2000). Mutations can also lead to a lack of OprD expression in mutant strains (Ochs et al. 2000). For HP3 protein, M60-like, M60 peptidase family, IMPa helical, and IMPa-N-2 domains were predicted. The peptidase family contains a zinc metallopeptidase motif and has mucinase activity (Nakjang et al. 2012). The IMPa (immunomodulating metalloprotease of P. aeruginosa) domain is a host immune-modulatory metalloprotease in P. aeruginosa that protects the bacterium from neutrophil infestation (Bardoel et al. 2012). This is the domain belonging to the M60 peptidase family (Bardoel et al. 2012). Pseudomonas aeruginosa secretes various proteases that destroy proteins essential for host defense (Noach et al. 2017). The PA0572 protein of P. aeruginosa, which is a PSGL1 (Pselectin Glycoprotein Ligand1) inhibitor, and this secretory protease, called the P. aeruginosa immunomodulator metalloprotease or IMPa (Bardoel et al. 2012).
Conclusion
Antibiotic-resistant bacteria are becoming one of the biggest challenges in medicine and healthcare. Pseudomonas aeruginosa is one of the top priorities of WHO for the immediate development of treatment strategies. Identification of protein functions is very important for understanding the biological processes in bacteria. In this study, the structure and function of 10 common hypothetical proteins of resistant strains of P. aeruginosa were investigated. The results revealed that the two hypothetical proteins (Gene ID: 2877781645 and 2877781936) had the potential to be used in drug and vaccine design. However, in vitro and in vivo studies are required to determine the effectiveness of these candidates for drug and vaccine development.
References
Abdellatif S, Trifi A, Daly F, Mahjoub K, Nasri R, Ben Lakhal S (2016) Efficacy and toxicity of aerosolised colistin in ventilator-associated pneumonia: a prospective, randomised trial. Ann Intensive Care 6(1):26
Almagro Armenteros JJ, Tsirigos KD, Sønderby CK, Petersen TN, Winther O, Brunak S, von Heijne G, Nielsen H (2019) SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol 37(4):420–423
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17):3389–3402
Barat S, Willer Y, Rizos K, Claudi B, Mazé A, Schemmer AK, Kirchhoff D, Schmidt A, Burton N, Bumann D (2012) Immunity to intracellular Salmonella depends on surface-associated antigens. PLoS Pathog 8(10):e1002966
Bardoel BW, Hartsink D, Vughs MM, de Haas CJ, van Strijp JA, van Kessel KP (2012) Identification of an immunomodulating metalloprotease of Pseudomonas aeruginosa (IMPa). Cell Microbiol 14(6):902–913
Bassetti M, Merelli M, Temperoni C, Astilean A (2013) New antibiotics for bad bugs: where are we? Ann Clin Microbiol Antimicrob 12:22–22
Bendtsen JD, Kiemer L, Fausbøll A, Brunak S (2005) Non-classical protein secretion in bacteria. BMC Microbiol 5(1):58
Bhasin M, Garg A, Raghava GP (2005) PSLpred: prediction of subcellular localization of bacterial proteins. Bioinformatics 21(10):2522–2524
Borgatta B, Gattarello S, Mazo CA, Imbiscuso AT, Larrosa MN, Lujàn M, Rello J (2017) The clinical significance of pneumonia in patients with respiratory specimens harbouring multidrug-resistant Pseudomonas aeruginosa: a 5-year retrospective study following 5667 patients in four general ICUs. Eur J Clin Microbiol Infect Dis 36(11):2155–2163
Brown RN, Sanford JA, Park JH, Deatherage BL, Champion BL, Smith RD, Heffron F, Adkins JN (2012) A comprehensive subcellular proteomic survey of Salmonella grown under phagosome-mimicking versus standard laboratory conditions. Int J Proteom 2012:123076
Buchan DWA, Jones DT (2019) The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res 47(W1):W402-w407
Chen IA, Chu K, Palaniappan K, Ratner A, Huang J, Huntemann M, Hajek P, Ritter S, Varghese N, Seshadri R, Roux S, Woyke T, Eloe-Fadrosh EA, Ivanova NN, Kyrpides NC (2021) The IMG/M data management and analysis system vol 6.0: new tools and advanced capabilities. Nucleic Acids Res 49(D1):D751-d763
Colovos C, Yeates TO (1993) Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci 2(9):1511–1519
de Oliveira Santos IC, Pereira de Andrade NF, da Conceição Neto OC, da Costa BS, de Andrade Marques E, Rocha-de-Souza CM, Asensi MD, D’Alincourt Carvalho-Assef AP (2019) Epidemiology and antibiotic resistance trends in clinical isolates of Pseudomonas aeruginosa from Rio de janeiro—Brazil: Importance of mutational mechanisms over the years (1995–2015). Infect Genet Evol 73:411–415
Dimitrov I, Bangov I, Flower DR, Doytchinova I (2014) AllerTOP vol 2–a server for in silico prediction of allergens. J Mol Model 20(6):2278
Ding Y, Teo JWP, Drautz-Moses DI, Schuster SC, Givskov M, Yang L (2018) Acquisition of resistance to carbapenem and macrolide-mediated quorum sensing inhibition by Pseudomonas aeruginosa via ICE(Tn4371) 6385. Commun Biol 1:57
do Nascimento APB, Medeiros Filho F, Pauer H, Antunes LCM, Sousa H, Senger H, Albano RM, Trindade Dos Santos M, Carvalho-Assef APD, da Silva FAB (2020) Characterization of a SPM-1 metallo-beta-lactamase-producing Pseudomonas aeruginosa by comparative genomics and phenotypic analysis. Sci Rep 10(1):13192
Doro F, Liberatori S, Rodríguez-Ortega MJ, Rinaudo CD, Rosini R, Mora M, Scarselli M, Altindis E, D’Aurizio R, Stella M, Margarit I, Maione D, Telford JL, Norais N, Grandi G (2009) Surfome analysis as a fast track to vaccine discovery: identification of a novel protective antigen for Group B Streptococcus hypervirulent strain COH1. Mol Cell Proteom 8(7):1728–1737
Doytchinova IA, Flower DR (2007) VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform 8:4
Espinosa-Camacho LF, Delgado G, Soberón-Chávez G, Alcaraz LD, Castañon J, Morales-Espinosa R (2017) Complete genome sequences of four extensively drug-resistant Pseudomonas aeruginosa strains, isolated from adults with ventilator-associated pneumonia at a tertiary referral hospital in Mexico City. Genome Announc. https://doi.org/10.1128/genomeA.00925-17
Fournier D, Richardot C, Müller E, Robert-Nicoud M, Llanes C, Plésiat P, Jeannot K (2013) Complexity of resistance mechanisms to imipenem in intensive care unit strains of Pseudomonas aeruginosa. J Antimicrob Chemother 68(8):1772–1780
Gardy JL, Brinkman FS (2006) Methods for predicting bacterial protein subcellular localization. Nat Rev Microbiol 4(10):741–751
Geourjon C, Deléage G (1995) SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput Appl Biosci 11(6):681–684
Haenni M, Bour M, Châtre P, Madec J-Y, Plésiat P, Jeannot K (2017) Resistance of animal strains of Pseudomonas aeruginosa to carbapenems. Front Microbiol 8:1847–1847
He Y, Rappuoli R, De Groot AS, Chen RT (2010) Emerging vaccine informatics. J Biomed Biotechnol 2010:218590
Huang W, Hamouche JE, Wang G, Smith M, Yin C, Dhand A, Dimitrova N, Fallon JT (2020) Integrated genome-wide analysis of an isogenic pair of Pseudomonas aeruginosa clinical isolates with differential antimicrobial resistance to ceftolozane/tazobactam, ceftazidime/avibactam, and piperacillin/tazobactam. Int J Mol Sci 21(3):1026
Ijaq J, Malik G, Kumar A, Das PS, Meena N, Bethi N, Sundararajan VS, Suravajhala P (2019) A model to predict the function of hypothetical proteins through a nine-point classification scoring schema. BMC Bioinform 20(1):14
Kaufman J (2018) Unfinished business: evolution of the MHC and the adaptive immune system of jawed vertebrates. Annu Rev Immunol 36:383–409
Klockgether J, Tümmler B (2017) Recent advances in understanding Pseudomonas aeruginosa as a pathogen. F1000Research 6:1261–1261
Klockgether J, Cramer N, Wiehlmann L, Davenport CF, Tümmler B (2011) Pseudomonas aeruginosa genomic structure and diversity. Front Microbiol 2:150–150
Kos VN, Déraspe M, McLaughlin RE, Whiteaker JD, Roy PH, Alm RA, Corbeil J, Gardner H (2015) The resistome of Pseudomonas aeruginosa in relationship to phenotypic susceptibility. Antimicrob Agents Chemother 59(1):427–436
Kozakiewicz L, Phuah J, Flynn J, Chan J (2013) The role of B cells and humoral immunity in Mycobacterium tuberculosis infection. Adv Exp Med Biol 783:225–250
Krogh A, Larsson B, von Heijne G, Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305(3):567–580
Letunic I, Khedkar S, Bork P (2021) SMART: recent updates, new developments and status in 2020. Nucleic Acids Res 49(D1):D458–D460
Liu Q, Li X, Li W, Du X, He J-Q, Tao C, Feng Y (2015) Influence of carbapenem resistance on mortality of patients with Pseudomonas aeruginosa infection: a meta-analysis. Sci Rep 5(1):11715
López-Causapé C, Cabot G, Del Barrio-Tofiño E, Oliver A (2018) The versatile mutational resistome of Pseudomonas aeruginosa. Front Microbiol 9:685
Lund-Palau H, Turnbull AR, Bush A, Bardin E, Cameron L, Soren O, Wierre-Gore N, Alton EW, Bundy JG, Connett G, Faust SN, Filloux A, Freemont P, Jones A, Khoo V, Morales S, Murphy R, Pabary R, Simbo A, Schelenz S, Takats Z, Webb J, Williams HD, Davies JC (2016) Pseudomonas aeruginosa infection in cystic fibrosis: pathophysiological mechanisms and therapeutic approaches. Expert Rev Respir Med 10(6):685–697
Marchler-Bauer A, Zheng C, Chitsaz F, Derbyshire MK, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Lu S, Marchler GH, Song JS, Thanki N, Yamashita RA, Zhang D, Bryant SH (2013) CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res 41(Database issue):D348-352
Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer ELL, Tosatto SCE, Paladin L, Raj S, Richardson LJ, Finn RD, Bateman A (2021) Pfam: the protein families database in 2021. Nucleic Acids Res 49(D1):D412–D419
Miyoshi-Akiyama T, Kuwahara T, Tada T, Kitao T, Kirikae T (2011) Complete genome sequence of highly multidrug-resistant Pseudomonas aeruginosa NCGM2.S1, a representative strain of a cluster endemic to Japan. J Bacteriol 193(24):7010
Miyoshi-Akiyama T, Tada T, Ohmagari N, Viet Hung N, Tharavichitkul P, Pokhrel BM, Gniadkowski M, Shimojima M, Kirikae T (2017) Emergence and spread of epidemic multidrug-resistant Pseudomonas aeruginosa. Genome Biol Evol 9(12):3238–3245
Nakjang S, Ndeh DA, Wipat A, Bolam DN, Hirt RP (2012) A novel extracellular metallopeptidase domain shared by animal host-associated mutualistic and pathogenic microbes. PLoS ONE 7(1):e30287
Naqvi AA, Shahbaaz M, Ahmad F, Hassan MI (2015) Identification of functional candidates amongst hypothetical proteins of Treponema pallidum ssp. pallidum. PLoS ONE 10(4):e0124177
Nascimento A, Filho F, Sousa H, Senger H, Albano R, Trindade dos Santos M, Carvalho-Assef A, da Silva F (2019) Comparative genome analysis of a multidrug-resistant Pseudomonas aeruginosa sequence type 277 clone that harbours two copies of the bla SPM-1 gene and multiple single nucleotide polymorphisms in other resistance-associated genes. bioRxiv. https://doi.org/10.1101/693440
Newman JW, Floyd RV, Fothergill JL (2017) The contribution of Pseudomonas aeruginosa virulence factors and host factors in the establishment of urinary tract infections. FEMS Microbiol Lett. https://doi.org/10.1093/femsle/fnx124
Ng C, Gu X, Goh SG, Chen H, Haller L, Tan B, Gin KY (2018) Draft genome sequences of four multidrug-resistant pseudomonas aeruginosa isolates from hospital wastewater in Singapore. Microbiol Resour Announc. https://doi.org/10.1128/MRA.01193-18
Noach I, Ficko-Blean E, Pluvinage B, Stuart C, Jenkins ML, Brochu D, Buenbrazo N, Wakarchuk W, Burke JE, Gilbert M, Boraston AB (2017) Recognition of protein-linked glycans as a determinant of peptidase activity. Proc Natl Acad Sci USA 114(5):E679-e688
Ochs MM, Bains M, Hancock RE (2000) Role of putative loops 2 and 3 in imipenem passage through the specific porin OprD of Pseudomonas aeruginosa. Antimicrob Agents Chemother 44(7):1983–1985
Omeershffudin UNM, Kumar S (2019) In silico approach for mining of potential drug targets from hypothetical proteins of bacterial proteome. Int J Mol Biol. https://doi.org/10.15406/ijmboa.2019.04.00111
Pang Z, Raudonis R, Glick BR, Lin TJ, Cheng Z (2019) Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and alternative therapeutic strategies. Biotechnol Adv 37(1):177–192
Persyn E, Sassi M, Aubry M, Broly M, Delanou S, Asehnoune K, Caroff N, Crémet L (2019) Rapid genetic and phenotypic changes in Pseudomonas aeruginosa clinical strains during ventilator-associated pneumonia. Sci Rep 9(1):4720
Ponomarenko J, Bui H-H, Li W, Fusseder N, Bourne PE, Sette A, Peters B (2008) ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinform 9(1):514
Priebe GP, Goldberg JB (2014) Vaccines for Pseudomonas aeruginosa: a long and winding road. Expert Rev Vaccines 13(4):507–519
Rabbi MF, Akter SA, Hasan MJ, Amin A (2021) In silico characterization of a hypothetical protein from shigella DYSENTERIAE ATCC 12039 reveals a pathogenesis-related protein of the type-VI secretion system. Bioinform Biol Insights 15:11779322211011140
Rashid MI, Naz A, Ali A, Andleeb S (2017) Prediction of vaccine candidates against Pseudomonas aeruginosa: an integrated genomics and proteomics approach. Genomics 109(3–4):274–283
Reche PA, Glutting JP, Zhang H, Reinherz EL (2004) Enhancement to the RANKPEP resource for the prediction of peptide binding to MHC molecules using profiles. Immunogenetics 56(6):405–419
Roy A, Kucukural A, Zhang Y (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5(4):725–738
Saha S, Raghava GP (2006a) Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 65(1):40–48
Saha S, Raghava GP (2006b) VICMpred: an SVM-based method for the prediction of functional proteins of Gram-negative bacteria using amino acid patterns and composition. Genom Proteom Bioinform 4(1):42–47
Saha S, Raghava GP (2007) BTXpred: prediction of bacterial toxins. In Silico Biol 7(4–5):405–412
Santajit S, Indrawattana N (2016) Mechanisms of antimicrobial resistance in ESKAPE pathogens. Biomed Res Int 2016:2475067
Shen HB, Chou KC (2010) Gneg-mPLoc: a top-down strategy to enhance the quality of predicting subcellular localization of Gram-negative bacterial proteins. J Theor Biol 264(2):326–333
Sudha R, Katiyar A, Katiyar P, Singh H, Prasad P (2019) Identification of potential drug targets and vaccine candidates in Clostridium botulinum using subtractive genomics approach. Bioinformation 15(1):18–25
Tada T, Miyoshi-Akiyama T, Shimada K, Shiroma A, Nakano K, Teruya K, Satou K, Hirano T, Shimojima M, Kirikae T (2016) A Carbapenem-resistant Pseudomonas aeruginosa isolate harboring two copies of blaIMP-34 encoding a metallo-β-lactamase. PLoS ONE 11(4):e0149385
Taiaroa G, Samuelsen Ø, Kristensen T, Økstad OAL, Heikal A (2018) Complete genome sequence of Pseudomonas aeruginosa K34–7, a carbapenem-resistant isolate of the high-risk sequence type 233. Microbiol Resour Announc. https://doi.org/10.1128/MRA.00886-18
Turano H, Gomes F, Barros-Carvalho GA, Lopes R, Cerdeira L, Netto LES, Gales AC, Lincopan N (2017) Tn6350, a novel transposon carrying pyocin S8 genes encoding a bacteriocin with activity against carbapenemase-producing Pseudomonas aeruginosa. Antimicrob Agents Chemother. https://doi.org/10.1128/AAC.00100-17
Tusnády GE, Simon I (2001) The HMMTOP transmembrane topology prediction server. Bioinformatics 17(9):849–850
Uddin R, Siddiqui QN, Sufian M, Azam SS, Wadood A (2019) Proteome-wide subtractive approach to prioritize a hypothetical protein of XDR-Mycobacterium tuberculosis as potential drug target. Genes Genom 41(11):1281–1292
van Duin D, Paterson DL (2016) Multidrug-resistant bacteria in the community: trends and lessons learned. Infect Dis Clin North Am 30(2):377–390
Varma P, Adimulam Y, Kodukula S (2015) In silico functional annotation of a hypothetical protein from Staphylococcus aureus. J Infect Public Health 8:526–532
Ventola CL (2015) The antibiotic resistance crisis: part 1: causes and threats. Pharm Ther 40(4):277–283
Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, Wheeler DK, Sette A, Peters B (2019) The immune epitope database (IEDB): 2018 update. Nucleic Acids Res 47(D1):D339-d343
Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF (1999) Protein identification and analysis tools in the ExPASy server. Methods Mol Biol 112:531–552
Winsor GL, Griffiths EJ, Lo R, Dhillon BK, Shay JA, Brinkman FS (2016) Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res 44(D1):D646-653
World Health Organization (2019) 2019 antibacterial agents in clinical development: an analysis of the antibacterial clinical development pipeline. World Health Organization, Geneva
Xiong J, Déraspe M, Iqbal N, Krajden S, Chapman W, Dewar K, Roy PH (2017) Complete genome of a panresistant Pseudomonas aeruginosa strain, isolated from a patient with respiratory failure in a Canadian Community Hospital. Genome Announc. https://doi.org/10.1128/genomeA.00458-17
Yoshihara E, Yoneyama H, Ono T, Nakae T (1998) Identification of the catalytic triad of the protein D2 protease in Pseudomonas aeruginosa. Biochem Biophys Res Commun 247(1):142–145
Yu CS, Chen YC, Lu CH, Hwang JK (2006) Prediction of protein subcellular localization. Proteins 64(3):643–651
Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Dao P, Sahinalp SC, Ester M, Foster LJ, Brinkman FS (2010) PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26(13):1608–1615
Acknowledgements
The authors wish to thank Department of biology, Shahed university for their support to conduct the present study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shamsinejad, F.S., Zafari, Z. Prediction of Potential Drug Targets and Vaccine Candidates Against Antibiotic-Resistant Pseudomonas aeruginosa. Int J Pept Res Ther 28, 160 (2022). https://doi.org/10.1007/s10989-022-10463-5
Accepted:
Published:
DOI: https://doi.org/10.1007/s10989-022-10463-5