
Abstract
Recently, de novo MS/MS peptide sequencing has enabled the application of affinity selections to synthetic peptide mixtures that approach the diversity of phage libraries (> 108 random peptides). In conjunction with ‘split-mix’ solid phase synthesis to access equimolar peptide mixtures, this approach provides a straightforward means to examine synthetic peptide libraries of considerably higher diversity than has been feasible historically. Here, we offer a critical perspective on this work, report emerging data, and highlight opportunities for further methods refinement. With continued development, ‘affinity selection–mass spectrometry’ may become a complimentary approach to phage display, in vitro selection, and DNA-encoded libraries for the discovery of synthetic ligands that modulate protein function.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Synthetic peptide libraries arguably spurred the development of combinatorial chemistry (Lowe 1995), and developed in parallel with phage libraries (Scott 1992)—a powerful tool for discovering binding molecules from libraries of random peptides (Smith and Petrenko 1997; Sidhu et al. 2000; Kehoe and Kay 2005). As discussed in a classic review, central to the phage display technique are affinity selections, which separate binding molecules from non-binders in a single physical step, and yield mixtures of reduced complexity that are enriched for binding molecules (Clackson and Wells 1994). In contrast to screens, which measure the activity of many compounds individually, affinity selections require a method to sequence individual peptides from complex mixtures (Clackson and Wells 1994). For phage display, this task was facilitated by (1) use of sequential rounds of selection and propagation of selected phage, which serve to reduce mixture complexities by amplifying high-affinity clones; and (2) the ease with which individual clones can be isolated for Sanger sequencing, by plating at dilution. More recently, next-generation sequencing can be used to sequence many clones in parallel, after a single affinity selection step (Dias-Neto 2009; Matochko et al. 2012).
Historically, the challenge of sequencing individual peptides from mixtures precluded the use of affinity selections with libraries of synthetic peptides. Therefore, although many early peptide libraries were accessed by chemical synthesis, their use was more appropriate for screening—in arrayed formats (Fodor, et al. 1991; Frank 1993), while bound to solid support beads (the ‘one-bead one-compound’ approach) (Lam et al. 1991), or as a series of mixtures, each with a known fixed position (the ‘positional scanning’ approach) (Houghten et al. 1991). A number of early uses of synthetic peptide libraries in affinity selections are suggestive of interest in the concept (Flynn et al. 1991; Zuckermann et al. 1992; Zhou et al. 1993). However, analysis of selected peptides was limited to either pooled sequencing (yielding positional frequencies only), or to sequence inference by unique molecular mass (applicable to small libraries only).
A solution to the challenge of sequencing individual peptides from complex mixtures—such as those generated by affinity selection from synthetic libraries—is provided by liquid chromatography/tandem mass spectrometry. In the mid 1990s, this approach was used by at least two groups to identify individual peptides post-affinity selection, by manual interpretation of tandem MS spectra (Chu et al. 1996; Kelly et al. 1996). However, despite early interest in the general use of mass spectrometry to characterize synthetic libraries (Metzger et al. 1993; Till et al. 1994), peptide sequencing by tandem MS was not routine. Moreover, when mass spectrometry has been applied to peptide libraries, it has generally been through ‘ladder sequencing’ (not applicable to peptide mixtures) (Chait et al. 1994; Youngquist et al. 1995), or by tandem MS-based sequencing of individual peptides (Paulick et al. 2006).
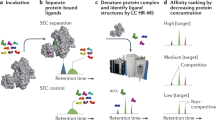
Here, we discuss how automated de novo peptide sequencing using nano-liquid chromatography/tandem MS has become a practical approach for sequencing synthetic peptide mixtures (Vinogradov et al. 2017), and enables the application of affinity selections to synthetic libraries of high diversity (up to 108–109 random peptides) (Touti et al. 2019; Quartararo et al. 2020) (Fig. 1). These numbers are comparable to early phage libraries, and motivate a re-examination of synthetic peptide libraries as sources of novel binding molecules. In our view, success hinges on two key issues: (1) whether de novo sequencing provides sufficient coverage of selected mixtures with high accuracy, such that sequencing data is meaningful; and (2) whether affinity selections can recover rare binding molecules efficiently with high enrichment, such that determinants of binding affinity are discernable amongst sequenced peptides. We discuss both recently published results and new data that speak to these questions, provide the interdisciplinary background necessary to place the work in context, and highlight open questions. Emphasis is placed on the p53/MDM2 interaction as a system for benchmarking the performance of synthetic libraries.

De novo sequencing enables affinity selections from synthetic peptide libraries
‘Split-Mix’ Synthesis Enables Access to Synthetic Peptide Libraries of Defined Diversity
Straightforward access to equimolar peptide mixtures for use in affinity selection–mass spectrometry is enabled by ‘split-mix’ synthesis (Lam et al. 1991; Houghten et al. 1991; Furka et al. 1991) on mono-sized resin beads (Rapp et al. 1992; Bayer 1991) (Fig. 2). Following chain assembly, the resulting peptide mixture is cleaved from the solid support, isolated, and ready for input to affinity selections (Fig. 1). A strength of this approach is its flexibility: both the number of unique peptides (the library diversity) and the quantity of individual peptides accessed can be controlled by a combination of the synthetic program, the quantity of resin beads employed, and the resin bead particle size and loading capacity. How these variables interact to determine the outcome of ‘split-mix’ synthesis is essential background, as they define the range of libraries accessible to the approach.

‘Split-mix’ synthesis enables access to equimolar peptide mixtures. a Iterative cycles of ‘divide-couple-recombine’ yield peptide libraries of increasing length. b Theoretical diversities obtained for libraries of increasing length are shown, using 18 distinct amino acids in each cycle. c Mono-size Tentagel resin beads are available commercially, and enable access to billion-member libraries in sufficient quantity for mass spectrometry. The quantity of peptide/bead assumes a bulk loading of 0.28 mmol/g (http://www.rapp-polymere.com/)
In general, the number of peptides accessed by ‘split-mix’ synthesis is determined by either of two factors: (1) the theoretical diversity of the library, when small; or (2) the number of beads employed, when theoretical diversity is high (‘theoretical diversity’ is the number of unique compounds that would be present, if all possible combinations of amino acids were sampled, and is determined by the synthetic program—the number of varied positions, and the number of monomers used at each). For ‘low diversity’ libraries, each compound is represented on many individual beads, and the library is said to be ‘over-sampled’; using more beads increases the quantities of individual peptides generated, but not their total number. For ‘high diversity’ libraries, in contrast, only a fraction of possible sequence combinations are sampled, and each peptide is represented on a single bead only. The resulting libraries are said to be ‘under-sampled’ or ‘sparsely-sampled’Footnote 1; using more beads increases the number of unique peptides accessed, but not their individual quantities. Both library types will be discussed: low diversity libraries for benchmarking de novo sequencing (“Automated De Novo MS/MS Sequencing Provides Acceptable Sequencing Coverage of Peptide Mixtures, with Good Accuracy” section), and high diversity libraries for both benchmarking de novo sequencing and discovering binding molecules by affinity selection (“Single-Pass Affinity Selections Recover Binders from Random Peptide Libraries” section).
Originally, ‘split-mix’ synthesis was devised to avoid an issue with a simpler alternative for combinatorial synthesis, wherein a single portion of resin beads is treated with activated amino acid mixtures (Flynn et al. 1991; Ivanetich and Santi 1996). In this approach (originally used to prepare degenerate oligonucleotides) (Ike et al. 1983), every possible sequence combination is sampled, but in unequal proportion, due to differences in amino acid coupling rates. ‘Split-mix’ synthesis avoids this issue, since couplings occur in distinct vessels, such that competition between amino acids is avoided. Additionally, ‘split-mix’ synthesis provides a second important advantage: the ability to access libraries of high theoretical diversity by ‘sparse’ sampling, while still generating a sufficient quantity of each peptide for downstream use. In contrast, libraries prepared by coupling of reagent mixtures sample all sequence combinations, but in diminishingly useful quantities.
Figure 2c illustrates the number of beads per gram of resin, at different particle sizes, and the quantities of peptide contained in single beads. For context, low femtomole sample quantities have long been accessible to commercial mass spectrometers (Wilm et al. 1996; McCormack et al. 1997), suggesting that even the smallest commercial beads generate sufficient quantities of individual peptides for affinity selection and tandem MS sequencing. Using 10 micron beads at a reasonable lab scale (e.g. 1–10 g of beads), 109–1010 unique peptides could be accessed, each in sufficient quantity for at least 10 affinity selections (based on a selection scale of 10 fmol/peptide).
Automated De Novo MS/MS Sequencing Provides Acceptable Sequencing Coverage of Peptide Mixtures, with Good Accuracy
As described elsewhere (McCormack et al. 1997; Eng et al. 1994; Steen and Mann 2004; Coon et al. 2005), mass spectrometry is ideally-suited for analysis of peptide mixtures, because of the ease with which individual ions can be isolated based on unique mass to charge ratio. When coupled to liquid chromatography, which is conveniently interfaced with electrospray ionization, an additional separation dimension is achieved, enabling individual ions to be isolated from mixtures containing many thousands of unique peptides. Once isolated, individual peptide ions are fragmented into ‘daughter ions’, which are analyzed as composite spectra in MS/MS. The differences between peaks in the resulting MS/MS spectra correspond to amino acid residue masses, characteristic of the peptide sequence.
In proteomics, peptide sequences are typically inferred from MS/MS spectra by the method of database searching (Eng et al. 1994; Domon and Aebersold 2006; Zhang et al. 2013; Sinitcyn et al. 2018). In this approach, experimental spectra are matched to predicted spectra generated from sequence databases (Eng et al. 1994), or to sequence-annotated spectral libraries (Lam et al. 2007). Database searching routinely matches a high proportion of MS/MS spectra (50% or more) (Cox and Mann 2008; Michalski et al. 2011; Senko et al. 2013; Hebert et al. 2014) with high accuracy (e.g., such that ~ 99% of peptide-spectrum matches are correct, at a false discovery rate of 1%) (Elias and Gygi 2007; Nesvizhskii et al. 2007; Marx et al. 2013). The approach is suitable when relevant databases are available, and contain a reasonable number of largely unrelated peptides. However, it is less suitable for random peptide libraries, which comprise trillions of possible sequences with significant overlap in local sequence. De novo sequencing (Taylor and Johnson 2001; Seidler et al. 2010; Hunt et al. 1992) is an alternative approach to interpreting MS/MS spectra that is better-suited to peptide libraries, since it is applicable in principle to any peptide sequence, provided complete series of fragment ions can be assigned (with the exception that Ile and Leu are not routinely distinguished). But compared to database searching, the reliability of automated de novo sequencing continues to be viewed with skepticism (Muth et al. 2018; O’Bryon et al. 2020).
In this section, we discuss evidence regarding the performance of automated de novo sequencing, and its suitability for analyzing peptide mixtures of unknown complexity. Broadly, performance evaluation involves performing de novo sequencing on a set of MS2 spectra (often from a data repository), and comparing the resulting sequence assignments to reference peptides associated with the spectra (‘ground truth’ peptides). Typically, ground truth peptides have been taken as peptide-spectrum matches from database searching. As a complimentary approach, we focus on the use of synthetic libraries as a source of ground truth peptides. For both approaches, emphasis is placed on the value of precision-recall analysis (Davis and Goadrich 2006)—a concept borrowed from information science—for evaluating sequencing performance, and on the use of confidence score thresholds for obtaining sequencing results of improved accuracy.
Precision and Recall Applied to De Novo Sequencing
In general terms, precision and recall report on the quality and quantity, respectively, of retrieved data. For de novo sequencing, recall is the fraction of ground truth peptides that are correctly sequenced, and precision is the fraction of de novo sequence assignments that are accurate (in other words, the fraction of total de novo assignments that correspond to ground truth peptides) (Fig. 3a). In some scenarios, precision and recall are equivalent—for example, when just a single spectrum/de novo assignment are associated with each ground truth peptide (Fig. 3b), and when all de novo assignments are considered regardless of confidence score. But in other scenarios—such as when the number of spectra/de novo assignments exceeds the number of ground truth peptides (Fig. 3c), or when some de novo assignments are excluded based on a confidence score threshold—precision and recall will differ.

The performance of de novo sequencing can be evaluated in terms of precision and recall (a). In (b) and (c), hypothetical de novo assignments and ground truth peptides are illustrated, alongside confidence scores for the de novo assignments (in parentheses). Mis-sequenced portions of the assignments are underlined. In (c), ground truth peptides are derived from a combinatorial Ala scan library, for illustration (with Ala variants in bold)
By considering de novo sequence assignments with highest confidence scores only, sequencing precision may be improved, at the expense of recall. This tradeoff can be visualized in a precision-recall curve, where precision and recall are plotted as a function of confidence score. Precision-recall analysis has been applied to de novo sequencing, but generally at the level of amino acids only (Ma 2015; Tran et al. 2017; Yang et al. 2019). Recently, the analysis was extended to the level of peptide sequences (Vinogradov et al. 2017; Devabhaktuni and Elias 2016; Miller et al. 2018). This type of analysis is of considerable practical importance, since it provides guidance on appropriate confidence score thresholds to use when sequencing unknown samples, and the precision/recall to be expected at those thresholds. As discussed below, continued work along these lines will be important.
Evaluating De Novo Sequencing: Ground Truth from Database Searching
The use of test spectra with high-confidence sequence annotations is perhaps the best way to evaluate the performance of de novo sequencing algorithms, absent other factors (Fig. 3b). When evaluated in this fashion using proteome-scale datasets, and with ground truth peptides taken as peptide-spectrum matches from database searching, the best de novo algorithms have produced sequence assignments identical to database peptides for ~ 10–50% of matched spectra (Tran et al. 2017; Pevtsov et al. 2006; Muth and Renard 2018). This type of studies suggest that de novo sequencing performs significantly worse than database searching in terms of both precision and recall. However, they do not generally speak to the precision that may be obtained by considering high-confidence assignments only.
A recent study demonstrated that with an appropriate confidence score threshold, the commercial software PEAKS recalled ~ 50% of accurate de novo assignments with a precision of 90% (Devabhaktuni and Elias 2016). As above, ground truth peptides were peptide-spectrum matches from database searching. The results clearly demonstrate the effectiveness of the PEAKS confidence score (‘average local confidence score’; ALC) at differentiating accurate assignments at the level of peptides, and of bringing the precision of de novo sequencing closer to that of database matching (99%). There is a tradeoff, as expected, with respect to recall. Based on a maximum recall of ~ 10–50% (above), and a reduction by ~ half at 90% precision, a recall of ~ 5–25% can be expected, compared to database searching at 99% precision. Further studies of this kind would be helpful in generalizing the results for PEAKS, and in establishing similar guidelines for new software.
Evaluating De Novo Sequencing: Ground Truth from Synthetic Libraries
The performance of de novo sequencing on test spectra with matches from database searching may not reflect the performance on raw datasets, collected from peptide mixtures. Synthetic peptide libraries provide a means to test this situation, which is of direct relevance to affinity selection–mass spectrometry and differs in several respects from evaluation using database matches. Whereas for database matches each ground truth peptide is associated with a single test spectrum, for a library analysis the pairings of individual spectra and associated peptides are unknown, and the number of spectra may exceed the number of input peptides. For these reasons, de novo assignments are considered accurate if they match any ground truth peptide, rather than a specific ground truth peptide associated with each spectrum (Fig. 3c).
Recently, the performance of PEAKS was assessed by analysis of a high-diversity synthetic library (Vinogradov et al. 2017). The exact sequences of analyzed peptides were unknown, but the library design was constrained by alternating varied positions with distinct amino acid subsets, such that ground truth peptides were taken with some confidence as de novo sequence assignments consistent with the library design (Fig. 4a). Using design-compliant assignments as ground truth, precision and recall were then calculated for the partially-processed de novo output (all assignments of correct length and C-terminal identity, to simulate the degree of processing that would be possible for a fully random library with fixed C-terminus). Similar to above (“Evaluating De Novo Sequencing: Ground Truth from Database Searching” section), good recall was achieved at 90% precision (~ 60% here, vs. ~ 50% previously). In this case, 90% precision corresponded to confidence scores > 75, which may provide guidance on the appropriate score threshold to use when analyzing unknown mixtures.

a Adapted with permission from Vinogradov, A. A. et al. Library Design-Facilitated High-Throughput Sequencing of Synthetic Peptide Libraries. ACS Comb. Sci. 19, 694–701 (2017). Vinogradov et al. 2017. Copyright 2017 American Chemical Society. The number of input peptides was controlled by counting beads from split-mix synthesis, prior to cleavage from the solid support (Vinogradov et al. 2017). b This work. Because the library diversity was sufficiently low, the entire library was analyzed
De novo sequencing provides acceptable coverage of synthetic peptide mixtures, with good accuracy. Each point corresponds to the precision/recall values obtained as described, for de novo assignments above a varied score threshold (from 50 to 99). The precision and recall values at score threshold = 80 are indicated (black points). Where applicable, the minimum score thresholds that achieved 90% precision are specified.
Here, we have applied a similar analysis to synthetic libraries of low diversity, where the sequences of analyzed peptides were known exactly. Two libraries were prepared by ‘combinatorial scanning’ of a K-Ras-binding peptide (Niida et al. 2017)—one of which contained a fixed C-term Lys, presumed to facilitate productive fragmentation for MS/MS (Fig. 4b). The two libraries were analyzed by nLC-MS (with cysteines in acetamide-modified form), and the total PEAKS de novo output was processed as above, keeping all assignments with correct length and fixed terminal residues for evaluation. Precision and recall were then calculated, using the known set of library sequences as ground truth. For the library containing C-term Lys, good recall was achieved at 90% precision (69%), in broad agreement with prior work. But for the second, lacking C-term Lys, significant recall was achieved at lower precisions only, and the maximum recall was lower (34%, at 59% precision). Further work is needed to determine the actual cause of this effect, which was reproduced on a second instrument (Fig. S1) and could potentially result from over-fragmentation of the non-Lys library (expected to fragment at lower energy) (Dongré et al. 1996).
Together, the results across three peptide libraries suggest that within individual datasets, PEAKS confidence scores are effective at differentiating correct de novo assignments, since for each library precision increased with increasing score threshold. However, the significance of confidence scores is not absolute, since the precision achieved at a given score threshold varied considerably between libraries (for example, either 0.97, 0.94, or 0.80 at score threshold = 80; Fig. 4). Careful benchmarking will be important when applying de novo sequencing in new contexts (e.g., to peptoids), since a high confidence score alone does not imply assignment accuracy. But for libraries of relatively short canonical peptides (e.g. 10-mers), a sensible approach would be to keep design-compliant candidates above a score of 80, and to anticipate that at least 80% are accurate.
Summary and Outlook—De Novo Sequencing
Existing de novo MS/MS sequencing tools can provide acceptable coverage of synthetic peptide mixtures with good accuracy (~ 50% recall at ~ 90% precision), and with continued refinement, additional performance gains may be expected (Tran et al. 2019; Gessulat et al. 2019). At the same time, the approach is not yet competitive with peptide identification by database searching. At a minimum, closing the performance gap requires a tenfold reduction in the frequency of erroneous sequence assignments, from roughly 1 in 10 at present to 1 in 100. Toward this goal, benchmarking should focus on precision at the peptide level, and consider the effect of assignments to all MS/MS spectra in raw datasets, rather than subsets of spectra only.
Synthetic libraries provide a powerful tool for benchmarking de novo sequencing, but not without limitation. The sequencing precisions obtained for synthetic libraries benefit from removing a priori incorrect assignments (e.g. of incorrect length, or lacking the correct C-terminus) prior to evaluation (Fig. S2). Where this processing is not warranted (e.g., when peptides of varying length are analyzed), precision may be lower. Additionally, precision and recall may be overestimated, if erroneous assignments happen to match ground truth peptides incidentally.
With respect to sequencing coverage, good recall was obtained for the mixture complexities studied here (~ 500 peptides per sample). More work to test the effect of mixture complexity on sequencing coverage would be helpful, over a wider range of complexities. As the recovery of de novo assignments is an indirect measure of the number of peptides present in a mixture, an orthogonal method to determine mixture complexity would be valuable, when analyzing unknown mixtures.
Single-Pass Affinity Selections Recover Binders from Random Peptide Libraries
As discussed in “Automated De Novo MS/MS Sequencing Provides Acceptable Sequencing Coverage of Peptide Mixtures, with Good Accuracy” section, the available data suggest that de novo sequencing provides adequate coverage and accuracy for mixtures containing hundreds to perhaps thousands of synthetic peptides. The next question is whether mixtures of this complexity can be obtained by affinity selection from high-diversity libraries, and whether de novo sequencing can be applied reliably when the complexity of selected mixtures is unknown. Equally important is whether affinity selections provide efficient recovery of binding molecules with high enrichment, such that rare binding motifs can be discerned among sequenced peptides.
Here, we discuss recent evidence that at least in some cases, single-stage affinity selections can provide sufficient enrichment to identify binders from libraries of random synthetic peptides. We focus on selections using magnetic bead affinity reagents (Quartararo et al. 2020; Pomplun et al. 2020; Pomplun et al. 2021), which have identified a number of interesting ligands from synthetic peptide libraries (Fig. 5) and have been used in phage display for many years (Fowlkes et al. 1992). Recent foundational results are highlighted, as well as new data that replicates a key benchmark: recovery of p53-like peptides by selection for MDM2 binding (Quartararo et al. 2020). The new results are encouraging, but differ from the original report in several important aspects. Possible strategies to further improve selection performance are discussed, including proof of concept data for a multi-stage selection that illustrate the feasibility of recovering femtomole quantities of individual peptides over two sequential selection steps. The need for a measure of peptide mixture complexity that is ‘orthogonal’ to de novo sequencing is also discussed, along with proof-of-concept data for a possible solution.

Affinity selections from high-diversity synthetic libraries have identified several interesting ligands. Examples using libraries with a limited number of ‘fixed’ positions (‘library design’ column, blue) are shown. Non-proteogenic side chains and backbone structures are indicated (orange and blue shading, respectively). Note added in proof: Very recently, high-diversity synthetic libraries were used as a source of novel ligands for angiotensin converting enzyme 2 (Zhang et al. 2022) (Color figure online)
Establishing Selection Performance as a Function of Library Diversity
Recently, selections for binding to a monoclonal antibody were used to test the performance of affinity selections, using random 9-mer libraries of increasing diversity (from 2 × 106 to 2 × 109 peptides) (Quartararo et al. 2020). The results are foundational in several respects: (1) in demonstrating further the reliability of de novo sequencing, since the expected binding epitope was identified among selected peptides; (2) in establishing the possibility of achieving very high enrichment by single-pass affinity selection, since the vast majority of selected peptides contained the binding epitope; and (3) in formally testing the benefit of library diversities beyond those accessible to standard synthetic combinatorial approaches.
The recovery of sequenced epitope-containing peptides from libraries of 106, 107, 108, and 109 random 9-mers is reproduced in Fig. 6a. The results show that higher diversity is generally beneficial, since epitope-containing peptides were identified in proportion to diversity over a wide range (2 × 106–2 × 108), with no loss in enrichment. So long as the recovery of binders is maintained, as it was in this range, then additional diversity will help to identify a rare binding motif, since the number of times a motif is sampled will increase with library diversity. Beyond diversity of ~ 108, however, recovery of epitope-containing peptides suffered (Fig. 6a). Similar results were obtained by selection for MDM2 binders from libraries of random 12-mers (Fig. 6b). Therefore, ~ 108 was taken as a practical limit to library diversity amenable to the ‘affinity selection–mass spectrometry’ approach, and subsequent applications focused on libraries of this size (Pomplun et al. 2020, 2021).

Adapted with permission from Quartararo, A. J. et al. Ultra-large chemical libraries for the discovery of high-affinity peptide binders. Nat. Commun. 11, 3183 (2020), licensed under CC BY (https://creativecommons.org/licenses/by/2.0/)
Selection from synthetic libraries recovers motif-containing sequenced peptides, with high enrichment. The selections were performed on 1 mL scale, using a fixed quantity of selection target (130 pmol) and varied number of input peptides (10 fmol/peptide for 106, 107, and 108-member libraries; 2 fmol/peptide for 109-member libraries) (Quartararo et al. 2020). ‘Sequenced peptides’ are library design-compliant de novo assignments with PEAKS software score > 80.
The underlying cause of poor recovery of sequenced peptides from libraries beyond 108 members has not been definitively proven, and should be revisited, since its correction could lead to further gains in accessible diversity, and the chances of identifying rare binding motifs. It was hypothesized that low recovery was caused by ‘peptide interference’, when selections yielded more peptides than are compatible with tandem MS sequencing (Quartararo et al. 2020). This is a plausible hypothesis, but would be strengthened by a direct measurement of the number of retained peptides, independent of de novo sequencing. Absent such a measure, the recovery of de novo assignments cannot distinguish between changes in the recovery of binders by selection, and changes in the coverage of selected binders by de novo sequencing (changes in sequence recall).
Another open question is the absolute recovery of sequenced binders, based on the total number input (which can be estimated based on statistical motif frequencies, but is not known with certainty). The proportionality of sequenced epitope peptides and total input peptides establishes that relative recovery was constant across conditions (Fig. 6a), but does not speak to the fraction of epitope peptides that were recovered and sequenced from the individual libraries. As we will discuss below, there is reason to believe that absolute recoveries could be improved.
Selections for MDM2 Binding: Replicating a Benchmark Experiment
Based on findings from the anti-haemagglutinin system, and to reproduce an important result from phage display, recent work tested libraries of 2 × 108 random 12-mers in selections for MDM2 binding, with the goal of recovering p53-like peptides (Quartararo et al. 2020). Remarkably, these selections yielded just a handful of library design-compliant de novo assignments, with PEAKS software score > 80 (‘sequenced peptides’). About half of sequenced peptides contained the ‘FxxxWxxL’ motif, in essence reproducing the phage display outcome. But as for the anti-haemagglutinin system, the absolute recovery of peptides from these selections was unclear, as was their coverage by de novo sequencing.
We have replicated selections for MDM2 binding from synthetic libraries of similar design (Fig. 7). The results are encouraging, in that recovery of sequenced motif peptides was ~ tenfold higher than reported originally (23 ‘FxxxWxxL/V’ peptides here, vs. 2 previously). Of the motif peptides, many contained a tyrosine residue not present in the p53 protein but that emerges from phage display (‘FxxYWxxL’) (Böttger et al. 1996). Additionally, many peptides containing a partial ‘FxxxW’ motif were identified, which may be legitimate binders (117 total). At the same time, the enrichment achieved by the selection was poorer than reported previously, since p53-like peptides comprised a smaller fraction of total sequenced peptides (1666 here, vs. 5 previously). Potentially, some of the non-motif peptides may be true binders, but these would be without strong precedent from phage display (Böttger et al. 1996; Kay et al. 1998; Hu et al. 2007; Pazgier et al. 2009).

Compared to literature precedent, selection for MDM2 binding recovers more p53-like sequenced peptides, but with poorer enrichment. ‘Sequenced peptides’ are library design-compliant de novo assignments with PEAKS software score > 80. The selection conditions and library preparation were as described (Quartararo et al. 2020), with the exception that Arg was excluded from the libraries here, to avoid interference with peptide fragmentation by collision-induced dissociation (the Orbitrap Fusion Lumos used previously was equipped with electron-transfer dissociation, which mitigates the loss in sequencing coverage that would result from this effect)
We have not identified a single causal variable that explains the differences between these and other recent results. However, the results here demonstrate that absolute recovery of binders in recent work was not optimal, and motivate continued refinement of selection procedures that will increase the odds of success in more challenging applications, where binders are less frequent in a library. The key challenge will be to improve the recovery of binders, ideally from libraries of 109 or higher, while maintaining the high enrichment necessary to distinguish binding motifs from unrelated ‘background’.
Quantifying Peptides Recovered by Selection
As discussed above, the number of sequenced peptides obtained by de novo sequencing may not correspond to the number actually present in a mixture. If the number of peptides becomes sufficiently high—as it may when library diversity is increased, or when a high degree of non-specific binding occurs—then sequencing coverage is expected to drop, as the isolation of individual precursor ions becomes more challenging and ‘composite’ tandem spectra interfere with de novo sequencing (Michalski et al. 2011). Therefore, a means of measuring sample complexity independent of de novo sequencing would be informative.
Primary MS ‘feature extraction’ is a potential solution, which extracts MS signals from raw data and could be used to estimate the number of peptides in a mixture, if features correspond reliably to peptides. We tested this approach using data from the MDM2 selection, and a commercial feature extraction algorithm. By comparing the extracted features to sequenced peptide precursor ions, the correspondence of features with sequenced peptides was evaluated. As shown in Fig. 8, extracted features significantly outnumbered precursors, and the majority were singly-charged. Therefore, when overlaid with precursor ions, little overlap was found, since MS2 spectra were collected on ions of z = 2 and higher only. For higher charge states (especially z = 3 ions), extracted features clustered with sequenced peptide precursors, suggestive of correspondence. However, even for z = 3 ions, the total features still exceeded sequenced precursors by > tenfold. Similar results were found for a sample of known complexity, obtained by cleaving a ~ 2000 bead aliquot from ‘split-mix’ synthesis of a random 12-mer library (‘library QC’, Fig. S8).

Among ions of higher charge state, extracted features overlay with sequenced peptide precursors. Extracted features from Thermo Proteome Discoverer (blue) and sequenced peptide precursors (orange) are shown as kernel density maps (Color figure online)
The significance of extracted features was studied further by determining the proportion of features that could be ‘matched’ to sequenced peptide precursors (z = 3 ions only). To ensure that each feature matched to at most a single unique precursor, each list was filtered for uniqueness, such that precursors were internally unique within 2 ppm, and features within 4 ppm. Then, for each precursor, the feature list was searched for matches (Δm/z < 2 ppm). This analysis was repeated considering the most abundant features only, at each of 5 different peak area thresholds (Fig. 9).

The majority of sequenced peptide precursors are identified by feature extraction, but comprise a minority of total features. Ions of charge state z = 3 were analyzed as described in the main text
At lower area thresholds, most of the sequenced precursors were identified among the features, demonstrating the ability of feature extraction to correctly identify sequenced peptides. However, since total features exceeded sequenced precursors, the vast majority of features remained unmatched. Of the highest-abundance features, a significant fraction matched with sequence peptide precursors (10–25%; Fig. 9), and the significance of these matches was supported by the agreement of retention times for matched feature-precursor pairs (Fig. S9). But even here, the proportion of matched features underestimated sequencing coverage for the ‘library QC’ sample, where the number of input peptides was known approximately. Based on the similar proportion of matched features between the ‘library QC’ and affinity selection samples, we find no evidence of dramatically different sequencing coverage, and conclude that de novo sequencing did not grossly underestimate the total peptides recovered by affinity selection. However, more work is warranted before feature extraction is used as a quantitative measure of sample complexity.
Possible Routes to Improved Enrichment—Parallel Selections with Unrelated Targets
In the anti-haemagglutinin system, it was shown that motif-containing peptides were recovered by the target of interest only, whereas non-motif peptides were also isolated by an unrelated selection target (Quartararo et al. 2020). Therefore, it was concluded that parallel selections might be useful in distinguishing specific vs. non-specific binders, and improving enrichment for binders at the level of data analysis. We re-investigate this topic here, using parallel selections between MDM2 and streptavidin.
Overlap between sequenced peptides from MDM2 and streptavidin selections is shown in Fig. 10a. In contrast to the anti-haemagglutinin system, the majority of total peptides isolated in the MDM2 conditions (> 90% of which lack a binding motif) were unique to MDM2 and not isolated by streptavidin. While some degree of overlap is evident, it is less than the overlap between replicate MDM2 selections. This result suggests that ‘subtracting’ peptides isolated by unrelated targets is unlikely to be a general strategy to improve enrichment for binders.

Parallel selections do not distinguish motif vs. non-motif peptides. a The majority of non-motif peptides from MDM2 selection were not recovered by an unrelated target (streptavidin). b In replicate MDM2 selections, motif-containing peptides are no more likely to be isolated reproducibly, compared to non-motif peptides
The overlap between replicate selections for MDM2 binding was examined further, to test whether motif-containing peptides were more likely to be isolated in replicate (Fig. 10b). Overlap between the total sequenced peptides from each replicate was visualized, alongside overlap between ‘FxxxW’ and ‘FxxxWxxL/V’ peptides. The similar degree of peptide overlap in each case demonstrates that peptides recovered from replicate selections are no more likely to contain a binding motif, compared to peptides isolated from a single replicate only. Therefore, when evaluating a selection outcome, isolation from replicates should not be an absolute criteria for confidence.
Possible Routes to Improved Enrichment—Multi-stage Selections
Two-stage selection procedures are commonly employed with DNA-encoded libraries (Goodnow et al. 2017), which are similar to synthetic libraries in that they cannot be propagated for sequential rounds of selection. Compared to DNA sequencing, however, the feasibility of tandem MS sequencing after two sequential selections (and accompanying material losses) is less clear. As a test of feasibility, we piloted a two-stage selection for MDM2 binding. The quantity of library employed was doubled, such that after the first selection, half of the eluate could be sequenced directly, and the remaining carried through a second selection stage. In this way, the enrichment for ‘FxxxWxxL’ peptides achieved at each stage could be compared.
The results of the two-stage selection are shown in Fig. 11. Encouragingly, ‘FxxxWxxL’ peptides were identified after each stage, proving the approach’s feasibility. However, the enrichment conferred by stage 2 was only ~ 2.5-fold, compared to ~ 2000 for stage 1Footnote 2. For the 108-member library studied here, an optimized procedure would probably use less selection target in the second stage, to maintain stringency as the quantity of input peptides is decreased. Libraries of even higher diversity may be amenable to an optimized two-stage selection, if enrichment of ~ 103 could be achieved in each stage (~ 106 overall).

Two-stage sequential selection for MDM2 binding recovers p53-like sequenced peptides with improved enrichment. As above, ‘sequenced peptides’ are library design-compliant de novo assignments with PEAKS software score > 80
Summary and Outlook—Affinity Selections
Even with confidence in the performance of de novo sequencing under controlled conditions, its use in characterizing unknown mixtures—such as those generated by affinity selection from peptide libraries—remains an analytical challenge. The sophistication of commercial MS instrumentation is considerable, and the quantity of data generated necessitates its automated handling. However, when differences between experimental outcomes arise at the level of sequenced peptides, sound conclusions cannot be made without reference to the underlying data. As discussed, a reliable means of inferring sample complexity from primary MS data would be helpful, and should ideally supplement de novo sequencing. Additionally, analytical standards and metrics tailored specifically for de novo sequencing may help to standardize results across laboratories.
An optimal affinity selection would recover all of the binders present in a library, with high enrichment. The enrichment for MDM2 binders obtained here is in-line with expectation from phage display (~ 104), but lower than a recent application of synthetic libraries (binders comprised 1% or 12% of total sequenced peptides after single stage selection here, compared to 40% previously). At the same time, many more sequenced binders were recovered here (up to 73 from a single stage selection, compared to only 2 previously). Optimizing selection performance in systems where the frequency of binders is measurable will be essential before applying the method in new contexts, especially where binders are less frequent. In our view, an optimized procedure will likely involve two sequential selections.
Conclusion
Affinity selections have long been an attractive means to examine the high-diversity peptide libraries accessible by ‘split-mix’ synthesis. As discussed here, automated de novo sequencing is a solution to the challenge of sequencing individual peptides from selected mixtures, and opens up new possibilities for examining synthetic libraries of diversities not conventionally accessible. Discovery of binding molecules from libraries of synthetic polymers is a topic of longstanding interest (Cho et al. 1993). However, absent a practical approach to examine such libraries for binding molecules with sufficient throughput, the ability of non-proteogenic structures (e.g. beta-amino acids, N-Me-amino acids) to fundamentally alter library ‘fitness’ has remained largely a question in principle. Going forward, ‘affinity selection–mass spectrometry’ is poised to make an impact.
‘Affinity selection–mass spectrometry’ (Muchiri and Breemen 2021; Prudent et al. 2021) accurately conveys the experimental workflow described here, and accordingly, the ‘AS-MS’ designation is not inappropriate. However, we note that discovery of binding molecules by selection from synthetic peptide mixtures is a longstanding goal, which predates the formal designation ‘AS-MS’. Because the enabling aspect of ‘AS-MS’ applied to peptide libraries is reliable de novo sequencing and not MS detection per se, and because a typical ‘AS-MS’ approach does not involve a general sequencing strategy, we suggest this term be used with caution. We look forward to continued progress in the space, driven by applications, unusual libraries, optimized methods, and continued progress in de novo sequencing.
Data Availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Notes
In the early days, it was considered important for synthetic libraries to achieve complete sampling. But given that some of the earliest phage libraries were under-sampled, and the relative success of the phage approach, this point seems less important in retrospect.
By definition, enrichment is the fold-change in motif-peptide frequency that accompanies selection. The enrichment achieved in the second stage was measured directly, since the proportion of motif-peptides was determined both before and after selection (p53-like peptides increased in proportion from 12% of total sequenced peptides after stage 1, to 32% after stage 2). The enrichment achieved in the first stage was estimated based on the statistical frequency of motif-peptides in the library, and the measured proportion of sequenced motif-peptides after stage 1. The statistical frequency of ‘FxxYWxxL’ peptides was used, since most ‘F-W-L’ peptides from each stage contained a tyrosine, and was taken as 6.0 × 10–5 (based on a statistical frequency of 1.2 × 10–5 ‘FxxYWxxL’ 8-mer motifs, using 17 randomized residues, and 5 possible motif registers in a library of 12-mers).
References
Bayer E (1991) Towards the chemical synthesis of proteins. Angew Chem Int Ed Engl 30:113–129
Böttger V et al (1996) Identification of novel mdm2 binding peptides by phage display. Oncogene 7:195–225
Chait BT, Wang R, Beavis R, Kent SBH (1994) Protein ladder sequencing. Science 262:89–92
Cho C et al (1993) An unnatural biopolymer. Science 261:1303–1305
Chu YH, Dunayevskiy YM, Kirby DP, Vouros P, Karger BL (1996) Affinity capillary electrophoresis - Mass spectrometry for screening combinatorial libraries. J Am Chem Soc 118:7827–7835
Clackson T, Wells JA (1994) In vitro selection from protein and peptide libraries. Trends Biotechnol 12:173–184
Coon JJ, Syka JEP, Shabanowitz J, Hunt DF (2005) Tandem mass spectrometry for peptide and protein sequence analysis. Biotechniques 38:519–523
Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26:1367–1372
Davis J, Goadrich M (2006) The relationship between precision-recall and ROC curves. In: Proc. 23rd Int. Conf. Mach. Learn. https://doi.org/10.1145/1143844.1143874
Devabhaktuni A, Elias JE (2016) Application of de novo sequencing to large-scale complex proteomics data sets. J Proteome Res 15:732–742
Dias-Neto E et al (2009) Next-generation phage display: Integrating and comparing available molecular tools to enable costeffective high-throughput analysis. PLoS ONE 4:e8338
Domon B, Aebersold R (2006) Mass spectrometry and protein analysis. Science 312:212–217
Dongré AR, Jones JL, Somogyi Á, Wysocki VH (1996) Influence of peptide composition, gas-phase basicity, and chemical modification on fragmentation efficiency: evidence for the mobile proton model. J Am Chem Soc 118:8365–8374
Elias JE, Gygi SP (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4:207–214
Eng JK, Mccormack AL, Yates JR (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5:976–989
Flynn GC, Pohl J, Flocco MT, Rothman JE (1991) Peptide binding specificity of the molecular chaperon BiP. Nature 353:726–730
Fodor SPA et al (1991) Light-directed, spatially addressable parallel chemical synthesis. Science 251:767–773
Fowlkes DM, Adams MD, Fowler VA, Kay BK (1992) Multipurpose vectors for peptide expression on the M13 viral surface. Biotechniques 13:422–428
Frank R (1993) Strategies and techniques in simultaneous solid phase synthesis based on the segmentation of membrane type supports. Bioorg Med Chem Lett 3:425–430
Furka A, Sebestyén F, Asgedom M, Dibó G (1991) General method for rapid synthesis of multicomponent peptide mixtures. Int J Pept Protein Res 37:487–493
Gessulat S et al (2019) Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat Methods 16:509–518
Goodnow RA, Dumelin CE, Keefe AD (2017) DNA-encoded chemistry: enabling the deeper sampling of chemical space. Nat Rev Drug Discov 16:131–147
Hebert AS et al (2014) The one hour yeast proteome. Mol Cell Proteomics 13:339–347
Houghten RA et al (1991) Generation and use of synthetic peptide combinatorial libraries for basic research and drug discovery. Nature 354:84–86
Hu B, Gilkes DM, Chen J (2007) Efficient p53 activation and apoptosis by simultaneous disruption of binding to MDM2 and MDMX. Cancer Res 67:8810–8817
Hunt DF et al (1992) Peptides presented to the immune system by the murine class II major histocompatibility complex molecule I-Ad. Science 256:1817–1820
Ike Y, Ikuta S, Sato M, Huang T, Itakura K (1983) Solid phase synthesis of polynucleotides. VIII. Synthesis of mixed oligodeoxyribonucleotides by the phosphotriester solid phase method. Nucleic Acids Res 11:477–488
Ivanetich KM, Santi DV (1996) Preparation of equimolar mixtures of peptides by adjustment of activated amino acid concentrations. Methods Enzymol 267:247–260
Kay BK, Kurakin AV, Hyde-Deruyscher R (1998) From peptides to drugs via phage display. Drug Discov Today 3:370–378
Kehoe JW, Kay BK (2005) Filamentous phage display in the new millennium. Chem Rev 105:4056–4072
Kelly MA et al (1996) Characterization of SH2-ligand interactions via library affinity selection with mass spectrometric detection. Biochemistry 35:11747–11755
Lam KS et al (1991) A new type of synthetic peptide library for identifying ligand-binding activity. Nature 354:82–84
Lam H et al (2007) Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics 7:655–667
Lowe G (1995) Combinatorial chemistry. Chem Soc Rev 24:309–317
Ma B (2015) Novor: real-time peptide de novo sequencing software. J Am Soc Mass Spectrom 26:1885–1894
Marx H et al (2013) A large synthetic peptide and phosphopeptide reference library for mass spectrometry-based proteomics. Nat Biotechnol 31:557–564
Matochko WL et al (2012) Deep sequencing analysis of phage libraries using Illumina platform. Methods 58:47–55
McCormack AL et al (1997) Direct analysis and identification of proteins in mixtures by LC/MS/MS and database searching at the low-femtomole level. Anal Chem 69:767–776
Metzger DJW, Wiesmuller DK-H, Gnau V, Brünjes J, Jung G (1993) Ion-spray mass spectrometry and high-performance liquid chromatography-mass spectrometry of synthetic peptide libraries. Angew Chem Int Ed 32:894–896
Michalski A, Cox J, Mann M (2011) More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC-MS/MS. J Proteome Res 10:1785–1793
Miller SE, Rizzo AI, Waldbauer JR (2018) Postnovo: postprocessing enables accurate and FDR-controlled de novo peptide sequencing. J Proteome Res 17:3671–3680
Muchiri RN, van Breemen RB (2021) Affinity selection–mass spectrometry for the discovery of pharmacologically active compounds from combinatorial libraries and natural products. J Mass Spectrom 56:e4647
Muth T, Renard BY (2018) Evaluating de novo sequencing in proteomics: already an accurate alternative to database-driven peptide identification? Brief Bioinform 19:954–970
Muth T, Hartkopf F, Vaudel M, Renard BY (2018) A potential golden age to come—current tools, recent use cases, and future avenues for de novo sequencing in proteomics. Proteomics 18:1700150
Nesvizhskii AI, Vitek O, Aebersold R (2007) Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat Methods 4:787–797
Niida A et al (2017) Investigation of the structural requirements of K-Ras(G12D) selective inhibitory peptide KRpep-2d using alanine scans and cysteine bridging. Bioorg Med Chem Lett 27:2757–2761
O’Bryon I, Jenson SC, Merkley ED (2020) Flying blind, or just flying under the radar? The underappreciated power of de novo methods of mass spectrometric peptide identification. Protein Sci 29:1864–1878
Paulick MG et al (2006) Cleavable hydrophilic linker for one-bead-one-compound sequencing of oligomer libraries by tandem mass spectrometry. J Comb Chem 8:417–426
Pazgier M et al (2009) Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX. Proc Natl Acad Sci USA 106:4665–4670
Pevtsov S, Fedulova I, Mirzaei H, Buck C, Zhang X (2006) Performance evaluation of existing de novo sequencing algorithms. J Proteome Res 5:3018–3028
Pomplun S, Gates ZP, Zhang G, Quartararo AJ, Pentelute BL (2020) Discovery of nucleic acid binding molecules from combinatorial biohybrid nucleobase peptide libraries. J Am Chem Soc 142:19642–19651
Pomplun S et al (2021) De novo discovery of high-affinity peptide binders for the SARS-CoV-2 spike protein. ACS Cent Sci 7:156–163
Prudent R, Annis DA, Dandliker PJ, Ortholand JY, Roche D (2021) Exploring new targets and chemical space with affinity selection-mass spectrometry. Nat Rev Chem 5:62–71
Quartararo AJ et al (2020) Ultra-large chemical libraries for the discovery of high-affinity peptide binders. Nat Commun 11:3183
Rapp W, Fritz H, Bayer E (1992) Monosized 15 micron grafted microspheres for ultra high speed peptide synthesis. Peptides Chemistry and Biology. In: Proceedings of the Twelfth American Peptide Symposium, ESCOM, Leiden, The Netherlands
Scott JK (1992) Discovering peptide ligands using epitope libraries. Trends Biochem Sci 17:241–245
Seidler J, Zinn N, Boehm ME, De Lehmann WD (2010) De novo sequencing of peptides by MS/MS. Proteomics. https://doi.org/10.1002/pmic.200900459
Senko MW et al (2013) Novel parallelized quadrupole/linear ion trap/orbitrap tribrid mass spectrometer improving proteome coverage and peptide identification rates. Anal Chem 85:11710–11714
Sidhu SS, Lowman HB, Cunningham BC, Wells JA (2000) Phage display for selection of novel binding peptides. Methods Enzymol 328:333–363
Sinitcyn P, Rudolph JD, Cox J (2018) Computational methods for understanding mass spectrometry-based shotgun proteomics data. Annu Rev Biomed Data Sci 1:207–234
Smith GP, Petrenko VA (1997) Phage display. Chem Rev 97:391–410
Steen H, Mann M (2004) The ABC’s (and XYZ’s) of peptide sequencing. Nat Rev Mol Cell Biol 5:699–711
Taylor JA, Johnson RS (2001) Implementation and uses of automated de novo peptide sequencing by tandem mass spectrometry. Anal Chem 73:2594–2604
Till JH, Annan RS, Carr SA, Miller WT (1994) Use of synthetic peptide libraries and phosphopeptide-selective mass spectrometry to probe protein kinase substrate specificity. J Biol Chem 269:7423–7428
Touti F, Gates ZP, Bandyopdhyay A, Lautrette G, Pentelute BL (2019) In-solution enrichment identifies peptide inhibitors of protein–protein interactions. Nat Chem Biol 15:410–418
Tran NH, Zhang X, Xin L, Shan B, Li MD (2017) De novo peptide sequencing by deep learning. Proc Natl Acad Sci USA 114:8247–8252
Tran NH et al (2019) Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nat Methods 16:63–66
Vinogradov AA et al (2017) Library design-facilitated high-throughput sequencing of synthetic peptide libraries. ACS Comb Sci 19:694–701
Wilm M et al (1996) Femtomole sequencing of proteins from polyacrylamide gels by nano-electrospray mass spectrometry. Nature 379:466–469
Yang H, Chi H, Zeng WF, Zhou WJ, He SM (2019) PNovo 3: Precise de novo peptide sequencing using a learning-to-rank framework. Bioinformatics 35:i183–i190
Youngquist RS, Fuentes GR, Lacey MP, Keough T (1995) Generation and screening of combinatorial peptide libraries designed for rapid sequencing by mass spectrometry. J Am Chem Soc 117:3900–3906
Zhang G, Brown JS, Quartararo AJ, Li C, Tan X, Hanna S, Antilla S, Cowfer AE, Loas A, Pentelute BL (2022) Rapid de novo discovery of peptidomimetic affinity reagents for human angiotensin converting enzyme 2. Commun Chem. https://doi.org/10.1038/s42004-022-00625-3
Zhang Y, Fonslow BR, Shan B, Baek M, Yates JR (2013) Protein analysis by shotgun proteomics. Chem Rev 113:2343–2394
Zhou S et al (1993) SH2 domains recognize specific phosphopeptide sequences. Cell 72:767–778
Zuckermann RN, Kerr JM, Siani MA, Banville SC, Santi DV (1992) Identification of highest-affinity ligands by affinity selection from equimolar peptide mixtures generated by robotic synthesis. Proc Natl Acad Sci USA 89:4505–4509
Acknowledgements
This work was supported by two A*STAR Industry Alignment Fund Pre-Positioning Programmes: Peptide Engineering Programme (IAF-PP H17/01/a0/010); and Macromolecular Interactions Program (IAF-PP H20H6a0030). We thank Charlie Johannes, Yee Hwee Lim, Chris Brown, Radek Sobota, David Lane, and Ashok Venkitaraman for encouragement and support; Alex Vinogradov, Ethan Evans, and Farid Ghadessy for critical feedback on the manuscript; Yuri Frosi for providing recombinant MDM2; and Loo Chien Wang for assistance with nLC-MS operation and Thermo Proteome Discoverer. We thank Jin Yong Neo and Shu Ting Rachel Chan for many fruitful discussions, and their engagement in related work that will hopefully be published soon.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Koh, L.Q., Lim, Y.W. & Gates, Z.P. Affinity Selection from Synthetic Peptide Libraries Enabled by De Novo MS/MS Sequencing. Int J Pept Res Ther 28, 62 (2022). https://doi.org/10.1007/s10989-022-10370-9
Accepted:
Published:
DOI: https://doi.org/10.1007/s10989-022-10370-9