
Abstract
Chikungunya virus is a major arbovirus of great public health concern in the whole world, but no vaccine is yet available, still advance therapeutic treatment and effective vaccines are in progress. The present multistep screening and structural binding analysis with CHIKV proteome exploration can be crucial in the development phase of CHIKV epitope based vaccine. The approach employed in two phases (i) Sequence based screening of peptides through propred and IEDB Server (ii) Structure based study through autodocking and NAMD VMD simulation analysis. Among all 29 extracted peptides, only two peptides 2LLANTTFPC10 of protein E3 and 98VNSVAIPLL106 of protein nsP3 were observed most prominent over all consider parameters such as peptide conserve nature, supertype population coverage, TAP binding, docking and simulation study. During docking interaction study, the best peptide and allele docked complexes such as 2LLANTTFPC10–B*0702 allele and 98VNSVAIPLL106–A*0301 allele exhibited best binding energy of − 3.13 kcal/mol and − 3.19 kcal/mol, respectively, with stable bonding patterns and their motion during NAMD simulation which confirm conserve peptide and allele stable interaction. The current study also exhibited the good docking interaction of both peptides 2LLANTTFPC10 and 98VNSVAIPLL106 with c TAP1 protein (1jj7 -PDB ID) cavity which confirm as a channel passageway to peptide transport through the cytoplasm to lumen of ER during antigen processing and presentation. Overall, this multistep screening and crosscheck structural binding analysis with an exploration of the complete proteome of CHIKV can be a novel step in the development of CHIKV epitope based vaccine as well as diagnostic development with aspect of time, cost and side effects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chikungunya virus is a major arbovirus of great public health concern in the whole world which belongs to togaviridae family (Sanyaolu et al. 2016). As arbovirus, CHIKV transmission in humans occurs through mosquito vectors Aedes aegypti and Aedes albopictus. CHIKV infection cause generally chikungunya fever but its infection severity result morbidity with chronic stages such as poly-arthralgia and prolong myalgia (Cunha et al. 2020). In the acute-infected individuals other severe complication like gastrointestinal upset, encephalitis, depression, lung, kidney, heart dysfunction, loss of vision, sleep disorder and memory loss being perceived (Sanyaolu et al. 2016; Qamar et al. 2018). The risk of Chikungunya may increase in the older patient more than 65 years and younger ones, posses more severe symptoms like neurological, cardiac and mortality also (Horwood and Buchy 2015). The present time, WHO also recorded high rate of CHIKV infections in India as well as rest of the developing countries due to urbanization activities, poor santization and weak mosquito vector control. Along with this, same vector and similar infection of CHIKV and Dengue virus which misleads treatment plans of both infections, causing major problems for health workers (Dutta et al. 2017; Saxena and Mishra 2021). On other side, the available therapeutic drugs and conventional vaccines lack the potential to address this global problem of CHIKV infection (WHO 2020; Weaver et al 2012) still advance therapeutic treatment and effective vaccines are in progress. CHIKV is a single positive-strand RNA virus with genome size 11.8 kb which translated in to structural capsid protein, envelope protein (E1, E2, 6 K and E3) and enzymatic non structural nsP1, nsP2, nsP3 and nsP4 proteins (Lee & Chu 2015; Qamar et al. 2018).
In the development of vaccine research, a specific approved or therapeutic vaccine for CHIKV is not yet available (WHO 2020). Therefore, the only option to verify the extent of CHIKV is quarantine and vector control with symptomatic therapeutic treatment at this time. In some way, this is effective only in the developed countries because of well develop techniques, but in the developing countries, these methods not working properly because of the large population and lacking of resources (Silva and Dermody 2017). So that's why persistency of this disease has remained very high because of insufficient therapeutic and control of this disease. Consequently, all facts demands specific therapeutic treatment as well as effective and advance vaccines to control the severe CHIKV infections globally. The traditional vaccines, likes- heat-killed and live attenuated vaccines having some safety issues because of whole micro-organism administration as well as time and cost are also major issues. (De Groot et al. 2002; Sharma et al. 2018). In current scenario, the advent of immunoinformatics tools and databases are helping in the development of multi epitope vaccines, which rising as a new hope to resolve all above limitations of conventional vaccines for human pathogenic viruses, bacteria (Joshi and Kaushik 2020), fungi (Akhtar et al. 2021) as well as veterinary pathogens also (Jain et al. 2021).
Vaccines based on epitopes are better than conventional vaccines and can also overcome side effects and safety issues (Krishnan et al. 2020; Srivastava et al. 2020). In the development of epitope based vaccines requires complete knowledge of immunology to initiate immune response. The epitope peptides cannot generate immunity alone; epitopes should attach to Major Histocompatibilty Complex (MHC) membrane proteins of host cell. Generally MHC molecules in human are called Human Leukocyte Antigen (HLA) which show high diversification in human population which raises the issue of population coverage during development of vaccines (Gupta and Kumar 2020a,b). To initiate an immune response towards an antigen; it has to undergo antigen processing and presentation process under which range of epitopes were generated to bind with HLA membrane molecules. Different ethnic population shows variations in binding pattern between HLA alleles and epitope. Therefore, each ethnic populations can be represent by a group of HLA alleles called as Supertypes and those epitopes bind with these supertype alleles group known as superantigens. The idea of supetypes which is representing a particular population has lower the chance of antigen escape from immune response as well as population coverage issues. (Kangueane et al. 2005; Shekhar et al.2012).
During development of epitope based vaccine, screening of highly conserved epitopes along with different supertypes alleles binding analysis strengthened the finding of best superantigen as vaccine reagents which were already shown in several other viruses vaccine development such as H1N1, JEV, DENGUE and WNV etc. ( Kumar et al. 2013; Sharma et al. 2014, 2021). Recently, other than these human pathogens zika virus, fungal pathogen as well as veterinary pathogens also showed potential results in development of epitope vaccine (Sharma et al. 2019; Jain et al. 2021). In several studies, epitope based vaccine immunizations against various infections showed significant results with proven cellular as well as humoral immunity (Hajissa et al. 2019). Approaches using concept of HLA supertype, with conserve epitope nature have been proven boon for mankind against various infectious disease which change the trends in various vaccine development approaches (Saxena and Mishra 2020). In short, several significances of epitopes based vaccines such as complete proteome representation through conserve epitopes, population coverage, less experimental burden, time, cost and doses overcome various issues with conventional vaccines (Krishnan et al. 2021).
Along with this, the process of antigen processing and presentation involved number of proteins such as Transporter Associated Protein (TAP) which embedded in rough endoplasmic reticulum (RER) membrane. The TAP worked as carrier for cytosolic epitopes to transport them through cytosol towards HLA molecule for presentation over membrane. These presented epitopes with HLA alleles make complex with T cell receptors to activate T cell to give immune response. Therefore, cytosolic molecular pathways showed binding of epitopes with TAP and HLA molecules as a major factor to generate immunogenicity against antigen peptide (Procko and Gaudet 2009; Gaudet and Wiley 2001). TAP binding analysis of top screened conserved and superantigenic peptides strengthen the vaccine candidacy of an epitope with more positivity (Sharma et al.2017; Mishra et al. 2020).
The present work is targeted to find most potential vaccine candidates from whole CHIKV proteome with least time as well as least experimental load which is ultimately reducing the burden of CHIKV infection globally. Therefore, this study started with the finding of the range of epitopes from each protein of virus which will be conserved and superantigenic through a sequence based epitope–alleles binder algorithms. After that, these conserved peptides and HLA alleles were generated in their structural models by PEPstrMODE and Swiss-prot for further structural binding analysis by docking. The binding of conserve superantigen epitopes with HLA molecule as well as cTAP substantiate that the epitopes generated during antigen processing were well transported with cTAP through cytosol and presented with HLA molecules over the membrane. The superantigenic epitopes peptides which showed best binding with HLA molecules were further docked with cTAP to confirm whether these peptides are really processed and presented during cytosolic pathway. Finally, the most potential candidates were undergone for molecular simulation study to confirm stable binding between Epitope and HLA molecule by NAMD-VMD. Overall, this multistep screening and crosscheck structural binding analysis with an exploration of the complete proteome of CHIKV can be a novel step in the development of CHIKV epitope based vaccine.
Methodology
In the present study, NCBI database (http: //www.ncbi.nlm.nih.gov/ entrez) was used to access fasta sequences of all required proteins of Indian CHIKV study strain Accession No. EF210157.2 (Santhosh et al.2009) as well as all geographical CHIKV regions strains such as India, Italy, Senegal, Thailand and USA. After retrivation of all required sequences of proteins, this study will be divided in two phases such as sequences based study and structure based study. Sequences based study involve the screening of epitopes from study strain, conservancy analysis, supertype analysis pI analysis and finally cross check with other algorithms to validate them. The structure based study was done to confirm the potential of epitopes to bind with HLA molecule by docking and simulation study, to generate an immune response. Prepared and Propred I was employed to retrieve HLA class II and Class I binding epitopes with recommended parameters. Therefore, the epitopes comparative analysis with all geographical regions strains were done to observe variations and conserve nature of propred screened epitopes with the help of IEDB platform. Along with, mutated or varied all epitopes were tested for their pI value. The above all screened epitopes through propred platform were further tested by using IEDB Artificial Neural Network (ANN) and (SMM) based algorithms to cross check the results.
Extraction of T Cell Binding Epitopes by Sequence Based Study
The sequence based study will extract out large pools of epitopes through Propred and Propred I server. Propred server was working out for finding of HLA class II binding peptide epitopes for 51 HLA alleles (Singh and Raghava 2001) as well as Propred I was employed for HLA class I binding peptide epitopes for 47 HLA alleles with recommended parameters (Singh and Raghava 2003). MHC Pred 2.0 were used to for epitope TAP binding analysis (Guan et al.2006). Above extracted epitopes under gone for validation by various Algorithms such as SMM and ANN (Nielsen et al. 2003) by IEDB server.
Epitope Conserve Nature Analysis, Among Different Geographical Regions
Extracted CHIKV T cell epitopes were employed to epitope conserve nature analysis, among different geographical regions such as India, Italy, Senegal, Thailand and USA. IEDB epitope conservancy study tool has been used to perform this analysis and required protein sequences of all proteins of different geographical strains were extracted from NCBI database. IEDB conservancy analysis was done by applying minimum five sequences of the same protein randomly for all geographical regions (Bui et al. 2007). Epitopes conservancy was done for all above five different geographical regions to get entirely conserved T cell epitopes. Altogether epitopes were kept in in three groups (i) entirely conserved (ii) having only a single mutation with 80% conservancy (iii) more than one mutation with less than 80% conservancy. Among these three groups, the epitopes having more than one mutation with less than 80% conservancy were taken out from further study and rest epitopes means single mutated and entire conserve epitopes were taken in consideration for further cross check and rigid screening procedure.
Finding of Common T Cell and B Cell Epitopic Regions in the Whole CHIKV Proteome
BC pred method algorithm was used to find linear B cell epitopes regions of 20 amino acid from whole CHIKV proteome (EL-Manzalawy et al. 2008; Terry et al. 2015). B cell and T cell epitopes interaction with antibody and MHC molecule, respectively, is must for neutralizing the antigen. This finding will reveal the common peptide region, which includes both T cell and B cell epitopes region and have possibility to initiate both B cell and T cell immune response (Saxena and Mishra 2020).
HLA Supertype Study for CHIKV T Cell Epitopes to Cover Maximum Population by Vaccine Candidates
The concept of HLA supertype in addition to above epitope conservancy analysis will increase more potency to cover maximum population by sorted conserve epitopes. As already known there are five (A3, A2, A24, B15, B7) HLA class I supertypes (23 HLA alleles) and five (Main DP2, DP2, DR3, DR2, DR) HLA class II supertypes (14 HLA alleles) which cover the maximum population (Reche and Reinherz 2005). The extracted promising conserve epitopes will studied for binding analysis to all HLA alleles supertype groups in comparison with already known superantigen positive controls to validate epitopes super-antigenicity. The known + ve controls which have been taken in the study are 141STLPETTVV149 peptide of Hepatitis core protein (Accession No.CAA59535) and 265ILRGSVAHK273 peptide of H1N1 Nucleoprotein (P03466) (Sharma et al. 2021; Ansari et al. 2009). This study was done by using IEDB platform.
Structural Binding Study of Extracted T Cell Epitopes to Relevant HLA Supertype Alleles and cTAP Protein
Before going to binding study between T cell epitopes and relevant alleles, the three dimensional structural models of each peptide and Supertype alleles were generated through PEPstrMOD and Swiss prot, respectively (Singh et al.2015; Saxena and Mishra 2020) which have been required to perform all docking and simulation study. The present docking study was done by Autodock 4.2 (Morris et al. 2009). In all docking experiments all recommended parameters were applied to extract the file.pdbqt. The autodock results were observed and analyzed with autodock tools to confirm epitope and alleles binding.
MD Simulation Analysis of Top Docked HLA Allele and Epitopes Binary Complexes
The molecular dynamic simulation (MD) of allele and epitope complexes and their physical interactions within in time window during docking were performed by NAnoscale Molecular Dynamics (NAMD) (James et al. 2005) and their visual assessment done by Visual MD platform (VMD) (Humphrey et al. 1996). For simulation analysis the three dimensional models of each epitopes and alleles were optimized by employing recommended force field parameters such as potential energy of bonding and nonbonding interactions with possible variations. To run NAMD the PSF building tool was used to generate.PSF structure file through.PDB file by extracting all atomic coordinates, velocity, force and sequence data. The pdb files are most necessary raw data for NAMD due to its high accuracy. The.DCD trajectory file was created by NAMD and Visual MD (VMD) generated RMSD.TCl source file. During the whole simulation process the RMSD values were stored in a RMSD.DAT file with regard to time frame and RMSD graph was obtained. This molecular simulation of protein–protein interaction will show physical stable bonding patterns and their motion during interaction which confirm epitope conserve peptide stable interaction with HLA alleles (Mishra et al. 2020; Peele et al. 2020).
Results
The present hierarchical approach will result finest T cell peptide epitopes as most promising candidates for vaccine as well as diagnosis of CHIKV infection.
Finding of Promising T Cell Peptide Epitopes of CHIKV Through Sequence Based Analysis
The complete proteome of all 8 proteins undergone in propred analysis to give a range of epitopes with sequence based matrix algorithm by keeping 5% threshold value. These large numbers of epitopes further sorted on the basis of HLA allele frequency of allele binding, conservancy, Tap binding nature. Among all, top most 29 identified epitopes, six structural protein epitopes viz. 2LLANTTFPC10, 31KGRVVAIVL39, 33LKIQVSLQI41, 490AALILIVVL498, 274LILIVVLCV279, 418LQISFSTAL426 and three nonstructural protein epitope 98VNSVAIPLL106, 93VTRLGVNSV103, 187VLLPNVHTL195 were exhibited more than 90% conservancy with high coverage of population through supertype analysis (Table 1). Furthermore, the peptide 39IDNADLAKL47 of capsid protein also showed good binding with HLA class I alleles with completely conservancy.
In the Table 1, under Different Geographical region of CHIKV section, the amino acid letter with green color in peptide shows amino acid variation or mutation in the other Geographical region.
Explanation with example for CHIKV Geographical region section: YEKEPEE(S3,K4,K5)TL, here original peptide is YEKEPEETL. Glutamic acid (E) seventh residue is mutated with Serine (S) in 3rd (Senegal) geographical region, Lysine (K) in 4th (Thailand) and 5th (USA) geographical region.
Conserve Nature of Extracted Peptide Epitopes
During IEDB analysis of conserve nature of extracted peptide epitopes, 8 HLA class II binding peptides and 17 HLA class I binding peptides were found conserve completely. The extracted HLA II peptides exhibited highest 98.46% conserved nature with geographical region I and II, 72.30% with geographical region III, 87.69% with geographical region IV and 87.69% with geographical region V. The extracted HLA I peptides exhibited highest 99,09% conserved nature with geographical region I, 98.18% with geographical region II, 82.72% with geographical region III, 93.63% with geographical region IV and 89.09% with geographical region V. MCLLANTTF, LLANTTFPC, VNSVAIPLL and VTRLGVNSV peptides were shown binding with both HLA class I and II alleles. LLANTTFPC, LQISFSTAL, AALILIVVL epitopes of envelope protein and VNSVAIPLL, VTRLGVNSV, VLLPNVHTL epitopes of non structural protein were exhibited complete conservancy with high HLA allele coverage worldwide.
Peptide Fragments Sharing Both B Cell and T Cell Epitope Property
CHIKV six (NCGGSNEGL, RNEATDGTL GTLKIQVSL, LKIQVSLQI, YAVHAPTSL, RSAEDPERL) T cell peptide epitopes were found in fragment of B cell epitopes but LQISFSTAL and YEKEPEETL peptides reveal moderately share with B cell epitope peptide region. Sharing of B cell as well as T cell epitope property in the same fragment of peptides makes epitope dominancy to induce both humeral as well as adaptive immunity (Sette and Fikes 2003).
Peptide Fragments Sharing Both B Cell and T Cell Epitope (Green Text)
Seq1: KCNCGGSNEGLTTTDKVINN protein E2 envelope.
Seq2: ERIRNEATDGTLKIQVSLQI protein E2 envelope.
Seq3: FPCSQPPCTPCCYEKEPEET protein E3 envelope.
Seq4: REAEIEVEGNSQLQISFSTA protein E1 envelope.
Seq5: KYHCVCPMRSAEDPERLANY NsP1 non structural.
Seq6: IYQDVYAVHAPTSLYHQAIK NsP1 non structural.
CHIKV Proteome Extracted Peptides Population Coverage Study
The peptide epitopes LLANTTFPC, LQISFSTAL, AALILIVVL, KGRVVAIVL, VLLPNVHTL, VNSVAIPLL and VTRLGVNSV exhibited binding to all HLA allele members of B7, A24, A3, A2 supertype HLA class I (Table 2). Likewise peptide epitopes LLANTTFPC, LRMLEDNVM, LKIQVSLQI, LILIVVLCV, VTRLGVNSV and VNSVAIPLL were exhibited binding to almost all allele members of Main DP, DR4, DR3, DR2 class II HLA supertype (Table 3). In this analysis, peptide epitopes were selected which showed their percentile value or range less than 50 but in some case value were found more than 50 percentile value as shown in Table 2. According to analysis less percentile value or range means high affinity with HLA allele. See Table 4.
The above tabulated superantigen peptides LLANTTFPC, LQISFSTAL, AALILIVVL, KGRVVAIVL, VLLPNVHTL, LRMLEDNVM, LKIQVSLQI, LILIVVLCV, VNSVAIPLL, VTRLGVNSV and HLA allele members were three dimensionally modelled for docking and simulation experiments. During IEDB population coverage study, the epitopes LLANTTFPC, LQISFSTAL and AALILIVVL, KGRVVAIVL, VNSVAIPLL showed individual 71.54%, 63.11%, 72.83%, 44.04% and 69.41% population coverage with HLA Class I set of alleles with overall 88.91% and LLANTTFPC, LKIQVSLQI, LILIVVLCV, VNSVAIPLL, VTRLGVNSV and LRMLEDNVM epitopes showed 79.83%, 59.93%, 67.02% 76.80%, 78.76%, and 63.18 coverage with HLA class II set of alleles with overall 81.81% respectively.
The above sequenced based study resulted the most promiscuous T cell peptide CHIKV:LLANTTFPC, CHIKV:LQISFSTAL and CHIKV:AALILIVVL, CHIKV:KGRVVAIVL, CHIKV:VNSVAIPLL, CHIKV:LKIQVSLQI, CHIKV:LILIVVLCV, CHIKV:VTRLGVNSV and CHIKV:LRMLEDNVM with high propred allele frequency, population coverage, superantigenicity and conservancy. Furthermore extracted peptides will be move forward for more stringent binding simulation analysis with most prominent and IEDB recommended sets of HLA class I (HLA:A* 0101, HLA:A* 0201, HLA:A* 0301, HLA:B*0702, HLA:B*3501, HLA:B*5101, HLA:B*5102 and HLA:B*5301) and class II (HLA:DRB1*0101, HLA:DRB1*0401, HLA:DRB1*0405 and HLA:DRB1*0301) supertype alleles along with cTap protein to confirm epitope antigen processing and presentation. See Fig. 1

Workflow of computational proteome exploration for finding of HLA and cTAP binding novel epitopes as CHIKV vaccine candidates
Three Dimensional Structure Modelling of all Extracted Peptides
All nanomer extracted potential peptides (LLANTTFPC, LQISFSTAL, AALILIVVL, KGRVVAIVL, VLLPNVHTL, VNSVAIPLL, VTRLGVNSV, LKIQVSLQI, and LILIVVLCV) were structurally modelled through PEPstrMOD server. See Fig. 2

CHIKV three dimensional structural PEPstrMOD model of peptides. a LLANTTFPC, b VNSVAIPLL
Superantigenic CHIKV Epitopes and HLA Alleles Binding Interaction Study
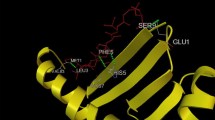
Binding study of superantigen epitopes LLANTTFPC, LQISFSTAL, AALILIVVL, KGRVVAIVL, VLLPNVHTL, VNSVAIPLL, VTRLGVNSV, LKIQVSLQI, and LILIVVLCV with HLA supertype favoured alleles, were done for analyse the interaction of both peptide and allele molecules through Autodock 4.2. During the binding interaction study, the two peptides and allele docked complexes such as 2LLANTTFPC10–B*0702 allele and 98VNSVAIPLL106–A*0301 allele exhibited best binding energy of − 3.13 and − 3.19 kcal/Mol, among all experiments respectively. Peptide LLANTTFPC–B*0702 allele complex appeared with one TRY27:HH Hydrogen bond (Fig. 3). Similarly peptide VNSVAIPLL–A*0301 allele complex appeared with one H bond viz. ARG6:HH12 (Fig. 4).

CHIKV structural envelope protein LLANTTFPC peptide-B*0702 allele docked complex interaction exhibited one TRY27:HH Hydrogen bond through Autodock 4.2

CHIKV non structural protein VNSVAIPLL peptide-A*0301 allele complex interaction exhibited with one H bond viz. ARG6:HH12 through Autodock 4.2
Docking of Top Extracted Class I HLA Binding Peptides with cTap Protein
As the above study revealed extracted several peptides which represent a good binding with HLA class I alleles. These all peptides under gone for antigen processing and presentation confirmation by docking with cTap protein. cTap protein is crucial protein during antigen processing and presentation of class I HLA binding peptides. cTAP channelized and facilitated the peptides passage throughout the transportation from cytoplasm to Endoplasmic reticulam (ER) and provide succesful presentation over the cell membrane to make interaction with T cell receptors. So, the peptides and cTAp interaction also be a major factor to confirm the peptide potential as vaccine candidacy. During this study, these two LLANTTFPC and VNSVAIPLL peptides showed optimum cTAP protein binding pattern with with − 1.00 and − 1.37 kcal/Mol binding energy, respectively (Saxena and Mishra 2020). See Figs. 5 and 6.

A The docking interaction of peptide LLANTTFPC with grey solid phase and cTAP1 (IJJ7 PDB) with white solid phase showed channelling in cTap cavity and facilitated the peptides passage throughout the transportation from cytoplasm to Endoplasmic reticulum (ER). B Peptide LLANTTFPC exhibited two hydrogen bonds with cTAP1 cavity viz. SER545:THR6 and GLU587:CYS9 with − 1. 00 kcal/Mol binding energy

A The docking interaction of peptide VNSVAIPLL with grey solid phase and cTAP1 (IJJ7 PDB) with white solid phase showed channelling in cTap cavity and facilitated the peptides passage throughout the transportation from cytoplasm to Endoplasmic reticulum (ER). B Peptide VNSVAIPLL exhibited three hydrogen bonds with cTAP1 cavity viz. SER545:VAL4, SER545:ILE6 and GLU587:LEU9 with − 1. 37 kcal/Mol binding energy
The Allele and Epitope Complexes, Physical Interaction Study by MD Simulation
Complexes 2LLANTTFPC10–B*0702 allele and 98VNSVAIPLL106–A*0301 allele were studied for their physical interactions within in time window through NAnoscale Molecular Dynamics (NAMD) simulation. During whole simulation process the RMSD values were stored in RMSD.DAT file with respect to time frame and RMSD graph was obtained. During the simulation RMSD graph initially rise up continuously with variations which occurred during interaction between epitope- HLA complex with change in time. Complexes 2LLANTTFPC10–B*0702 allele during simulation observed highest RMSD value 10.4 Å at 6200 picoseconds with stable interaction pattern. Similarly 98VNSVAIPLL106–A*0301 allele complex also observed with stable physical interaction with highest RMSD value 7.7 Å at 8200 picoseconds. This molecular simulation of protein–protein interaction will show physical stable bonding patterns and their motion during interaction which confirm conserve peptide and allele stable interaction.
Discussion
The present hierarchical approach for finding of finest T cell peptide epitopes as most promising candidates for CHIKV vaccine divided in two phase study (i) Sequence based screening of peptides through propred and IEDB Server (ii) Structure based study through docking and simulation analysis. During the study peptides LLANTTFPC of structural envelope protein E3 and VNSVAIPLL of non structural protein nsP3 were observed most prominent over all consider parameters such as peptide conserve nature, supertype population coverage among all extracted peptides from whole proteome. The envelope E1 glycoprotein of virus helps in smooth access of the viral protein into the host cell and the non-structural nsp3 protein of Chikungunya known to function as a viral RNA transcription regulator (Saxena and Mishra 2020). Both the protein have specific prominent role in the virus life cycle and also presents the TAP protein binding efficiency during sequence based analysis. As already known, the peptides and TAP interaction also be a major factor in antigen processing and their presentation over Class I HLA molecule (Mishra et al. 2020). The first phase screening and supertype analysis will result as top two peptides LLANTTFPC of protein E3 and VNSVAIPLL of protein nsP3 as superantigen peptides which observed as a common binder for class I and class II HLA supertypes all allele members (Table 2 and Table 3). Peptide binding with allele members shown good results as IEDB recommended values. The peptide epitopes LLANTTFPC, LQISFSTAL, AALILIVVL, KGRVVAIVL, VLLPNVHTL, VNSVAIPLL and VTRLGVNSV exhibited binding to all HLA allele members of B7, A24, A3, A2 supertype HLA class I (Table 2). Likewise peptide epitopes LLANTTFPC, LRMLEDNVM, LKIQVSLQI, LILIVVLCV, VTRLGVNSV and VNSVAIPLL were exhibited binding to almost all allele members of Main DP, DR4, DR3, DR2 class II HLA supertype (Table 3). Overall the peptide LLANTTFPC and VNSVAIPLL were observed more promising than other as vaccine candidacy. For further confirmation it is necessary to analyze these peptides for their structure based binding simulation study to confirm their physical interaction to concern HLA allele as well as cTap protein.
Binding study of superantigen epitopes 2LLANTTFPC10 and 98VNSVAIPLL 106 with HLA supertype favored alleles, were done through Autodock 4.2. During the binding interaction study, the two peptide and allele docked complexes such as 2LLANTTFPC10–B*0702 allele and 98VNSVAIPLL 106–A*0301 allele exhibited best binding energy of − 3.13 kcal/Mol with one TRY27:HH Hydrogen bond and − 3.19 kcal/Mol with one H bond viz. ARG6:HH12, among all experiments, respectively. Further, these complexes stability confirmation done by using simulation NAMD- VMD platform. Complexes 2LLANTTFPC10–B*0702 allele during simulation observed highest RMSD value 10.4 Å at 6200 picoseconds with stable interaction pattern (Fig. 7). Similarly 98VNSVAIPLL 106–A*0301 allele complex also observed with stable physical interaction with highest RMSD value 7.7 Å at 8200 picoseconds (Fig. 8). This molecular simulation of protein–protein interaction will show physical stable bonding patterns and their motion during interaction which confirm conserve peptide and allele stable interaction.

NAMD-VMD simulation- RMSD graph with time window showed physical interaction of 2LLANTTFPC10–B*0702 allele complex during the simulation which observed highest RMSD value 10.4 Å at 6200 picoseconds with stable interaction pattern

NAMD-VMD simulation- RMSD graph with time window showed physical interaction of 98VNSVAIPLL 106–A*0301 allele complex during the simulation also observed with stable physical interaction with highest RMSD value 7.7 Å at 8200 picoseconds
As both peptides showed binding with class I HLA molecule means these peptides processing and presentation depended on interaction with cTAP protein. Protein cTAP channelized and facilitated the peptides passage throughout the transportation from cytoplasm to Endoplasmic reticulum (ER) and provide successful presentation over the cell membrane to make interaction with T cell receptors. So the peptides and cTap interaction also be a major factor to confirm the peptide potential as vaccine candidacy which will be done by docking of both peptides to cTap protein (Sharma et al. 2018). Peptide LLANTTFPC exhibited two hydrogen bonds with cTAP1 cavity viz. SER545 and GLU587 with − 1.00 kcal/mol binding energy. Peptide VNSVAIPLL exhibited three hydrogen bond 1with cTAP1 cavity viz. SER545 and GLU587 with − 1.37 kcal/Mol binding energy (Sharma et al. 2017; Saxena and Mishra 2020). Both peptides showed optimum binding in cTAP protein cavity, which confirm as a channel passageway to peptide transport through the cytoplasm to lumen of ER. Apart from these two peptides, many potential epitopes shown good results which can employ for diagnostic purpose with use of vaccine candidacy. As reported earlier, that both structural and non-structural proteins presently involved in design and development the peptide-based vaccine for Dengue and Hepatitis (Kaushik et al. 2014; Henriques et al. 2013)., even in fungal and veterinary vaccine development also reported. Recently, several latest researches have been reported, which encourage the Immunoinformatics top down approach to get superantigenic epitopes from whole proteome of viral, bacterial, fungi (Sharma et al. 2019; Joshi and Kaushik 2020; Akhtar et al. 2021) or many more infectious agents to design diagnosis reagents and vaccines against them. Although, computational studies now these days having a major role in research in the way to reduce experimental cost and time, still these computational studies having some limitation to apply them directly to the natural world (Krishnan et al. 2020). To remove this barrier identified products epitopes should be further tested in a wet laboratory as diagnosis purpose as well as vaccine candidacy for Chikungunya. The similar hierarchical approach also done for many infectious viruses such as Dengue, H1N1, JEV, West Nile virus and tuberculosis gave most promising results in vaccine development research (Jardine et al. 2013; De Groot et al. 2013; Feng et al. 2013; Khan et al. 2019; Sharma and Kumar 2010).
Conclusion
As development of tools and techniques in immunoinfomatics top down approach, made easier and feasible to surmount the burden of medical health system by design and development of epitope based vaccine against severe infections like Chikungunya, H1N1, JEV etc. The present analysis, peptides 2LLANTTFPC10 of protein E3 and 98VNSVAIPLL106 of protein nsP3 were observed most superantigenic with consideration of all parameters such as peptide conserve nature, supertype population coverage, tap binding among all extracted peptides from whole proteome. Overall, this multistep screening and crosscheck structural binding analysis with exploration of complete proteome of CHIKV can be a novel step in development of CHIKV epitope based vaccine as well as diagnostic development with aspect of time, cost and side effects.
References
Akhtar N, Joshi A, Kaushik V, Manish K, Mannan AM (2021) In-silico design of a multivalent epitope-based vaccine against Candida auris. Microb Pathog 155:e104879
Ansari HR, Flower DR, Raghava GPS (2009) AntigenDB: an immunoinformatics database of pathogen antigens. Nucleic Acids Res 38:D847–D853
Bui HH, Sidney J, Li W, Fusseder N, Sette A (2007) Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinformatics 8(1):e361
Cunha MS, Costa PAG, Correa IA, de Souza MRM, Calil PT, da Silva GPD, Costa SM, Fonseca VWP, da Costa LJ (2020) Chikungunya virus: an emergent arbovirus to the South American continent and a continuous threat to the world. Front Microbiol 11:1297. https://doi.org/10.3389/fmicb.2020.01297
De Groot AS, Sbai H, Aubin CS, McMurry J, Martin W (2002) Immuno-informatics: mining genomes for vaccine components. Immunol Cell Biol 80:255–269
De Groot AS, Ardito M, Terry F, Levitz L, Ross T, Moise L (2013) Low immunogenicity predicted for emerging avian-origin H7N9. Hum Vaccin Immunother 9(5):950–956
Dutta P, Khan SA, Hazarika NK, Chetry S (2017) Molecular and phylogenetic evidence of chikungunya virus circulating in Assam, India. Indian J Med Microbiol 35:389–393
EL-Manzalawy Y, Dobbs D, Honavar V, (2008) Predicting linear B-cell epitopes using string kernels. J MolRecognit 21:243–255
Feng G, Jiang Q, Xia M, Lu Y, Qiu W, Zhao D, Lu L, Peng G, Wang Y (2013) Enhanced immune response and protective effects of nano-chitosan-based DNA vaccine encoding T cell epitopes of esat-6 and FL against mycobacterium tuberculosis infection. PLoS ONE 8(4):e61135
Gaudet R, Wiley DC (2001) Structure of the ABC ATPase domain of human TAP1, the transporter associated with antigen processing. EMBO J 20:4964–4972
Guan P, Hattotuwagama CK, Doytchinova IA, Flower DR (2006) MHCPred 2.0: an updated quantitative T-cell epitope prediction server. Appl Bioinformatics 5:55–61
Gupta N, Kumar A (2020b) Designing an efficient multi-epitope vaccine against Campylobacter jejuni using immunoinformatics and reverse vaccinology approach. Microb Pathog 147:e104398
Gupta N, Kumar A (2020a) Identification of potent vaccine candidates against Campylobacter jejuni using immunoinformatics approach. Int J Pept Res Ther 26:1303–1312
Hajissa K, Zakaria R, Suppian R, Mohamed Z (2019) Epitope-based vaccine as a universal vaccination strategy against toxoplasma gondii infection: A mini-review. J Adv Vet Anim Res 6(2):174–182
Henriques HR, Rampazo EV, Gonçalves AJS, Vicentin ECM, Amorim JH et al (2013) Targeting the non-structural protein 1 from dengue virus to a dendritic cell population confers protective immunity to lethal virus challenge. PLOS Negl Trop Dis 7(7):e2330
Horwood PF, Buchy P (2015) Chikungunya. Rev Sci Tech off Int Epiz 34(2):479–489
Humphrey W, Dalke A, Schulten K (1996) VMD—visual molecular dynamics. J Mol Graphics 14:33–38
Jain P, Joshi A, Akhtar N et al (2021) An immunoinformatics study: designing multivalent T-cell epitope vaccine against canine circovirus. J Genet Eng Biotechnol 19:121. https://doi.org/10.1186/s43141-021-00220-4
James CP, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802
Jardine et al (2013) Rational HIV immunogen design to target specific germline B cell receptors. Science 340:711–716
Joshi A, Kaushik V (2020) In-silico proteomic exploratory quest: crafting T-cell epitope vaccine against whipple’s disease. Int J Pept Res Ther 27:169–179
Kangueane P, Sakharkar MK, Rajaseger G, Bolisetty S, Sivasekari B, Zhao B, Ravichandran M, Shapshak P, Subbiah S (2005) A framework to sub-type HLA supertypes. Front Biosci 1(10):879–886
Kaushik V, Chauhan G, Singh J (2014) In silico peptide-based vaccine design against non-structural protein 5 of hepatitis c virus. Int J Pharm Pharm Sci 6(9):80–82
Khan F, Srivastava V, Kumar A (2019) Computational identification and characterization of potential T-cell epitope for the utility of vaccine design against Enterotoxigenic Escherichia coli. Int J Pept Res Ther 25:289–302
Krishnan GS, Joshi A, Kaushik V (2020) T cell epitope designing for dengue peptide vaccine using docking and molecular simulation studies. Mol Simul. https://doi.org/10.1080/08927022.2020.1772970
Krishnan GS, Joshi A, Akhtar N, Kaushik V (2021) Immunoinformatics designed T cell multi epitope dengue peptide vaccine derived from non structural proteome. Microb Pathog 150:e104728
Kumar A, Jain A, Shraddha & Verma SK, (2013) Screening and structure based Modeling of T cell epitopes of marburg virus NP, GP and VP40: an ammunoinformatic approach for designing peptide based vaccine. Trends in Bioinformatics 6:10–16. https://doi.org/10.3923/tb.2013.10.16
Lee RCH, Chu JJH (2015) Proteomics profiling of chikungunya-infected Aedes albopictus C6/36 cells reveal important mosquito cell factors in virus replication. PLoS Negl Trop Dis 9(3):e0003544. https://doi.org/10.1371/journal.pntd.0003544
Mishra S, Saxena P, Saxena M (2020) In Silico study on the binding pattern of cTap binding epitopes of S-27 strain with the common HLA alleles for the chikungunya vaccine development. J Biochem Mol Med 2(1):91–96
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) Autodock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 16:2785–2791
Nielsen M, Lundegaard C, Worning P, Lauemøller SL, Lamberth K, Buus S, Brunak S, Lund O (2003) Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci 12:1007–1017
Peele KA, Durthi CP, Srihansa T, Krupanidhi S, Ayyagari VS, Babu DJ, Indira M, Reddy AR, Venkateswarulu TC (2020) Molecular docking and dynamic simulations for antiviral compounds against SARSCoV- 2: a computational study. Inform Med Unlocked 19:e100345
Procko E, Gaudet R (2009) Antigen processing and presentation: TAPping into ABC transporters. Curr Opin Immunol 21:84–91
Qamar MTU, Bari A, Adeel MM, Maryam A, Ashfaq UA, Du X, Muneer I, Ahmad HI, Wang J (2018) Peptide vaccine against chikungunya virus: immuno-informatics combined with molecular docking approach. J Transl Med 16:298
Reche PA, Reinherz EL (2005) PEPVAC: a web server for multi-epitope vaccine development based on the prediction of supertypic MHC ligands. Nucleic Acids Res 33:138–142
Santhosh SR, Dash PK, Parida M, Khan M, Rao PVL (2009) Appearance of EI: A226V mutant chikungunya virus in coastal Karnataka, India during 2008 outbreak. Virology Journal 6:172
Sanyaolu A, Okorie C, Badaru O, Wynveen E, White S, Wallace W, Akl J, Freeze A, Kamel A, Madonna M, Mathur A, Moran R, Perry C (2016) Chikungunya epidemiology: a global perspective. SM J Public Health Epidemiol 2(2):1028
Saxena P, Mishra S (2020) Study of the binding pattern of HLA Class I alleles of Indian frequency and cTAP binding peptide for chikungunya vaccine development. Int J Pept Res Ther 26:2437–2448
Saxena P, Mishra S (2021) Chikungunya outbreak from 1952–2019: a prospective to cope-up with this re-emerging disease. Biointerface Research in Applied Chemistry 11(4):11724–11736
Sette A, Fikes J (2003) Epitope-based vaccines: an update on epitope identification, vaccine design and delivery. Current Opinion in Immunonology 15:461–470
Sharma P, Kumar A (2010) Immunoinformatics: screening of potential T cell antigenic determinants in proteome of H1N1 swine influenza virus for virus epitope vaccine design. J Proteom Bioinform 3:275–278
Sharma P, Saxena K, Mishra S, Kumar A (2014) A comprehensive analysis of predicted HLA binding peptides of JE viral proteins specific to north Indian isolates. Bioinformation 10(6):334–341
Sharma P, Srivastav S, Mishra S, Kumar A (2017) Sequence and structure based binding prediction study of HLA class I and cTAP binding peptides for Japanese encephalitis vaccine development. Int J Pept Res Ther 23:269–279
Sharma P, Sharma P, Mishra S, Kumar A (2018) Analysis of promiscuous T cell epitope for vaccine development against West Nile virus using bioinformatics approaches. Int J Pept Res Ther 24:377–387
Sharma P, Kaur R, Upadhyay AK, Kaushik V (2019) In-Silico prediction of peptide based vaccine against Zika virus. Int J Pept Res Ther 26:85–91
Sharma P, Sharma P, Sheeba KA (2021) Top Down Computational approach: a vaccine development step to find novel superantigenic HLA binding epitopes from dengue virus proteome. Int J Pept Res Ther 27:1469–1480
Shekhar C, Dev K, Verma SK, Kumar A (2012) In silico: screening and modeling of CTL binding epitopes of Crimean Congo hemorrhagic fever virus. Trends in Bioinformatics 5:14–24
Silva LA, Dermody TS (2017) Chikungunya virus: epidemiology, replication, disease mechanisms, and prospective intervention strategies. J Clin Invest 127(3):737–749
Singh H, Raghava GPS (2001) Propred: prediction Of HLA-DR binding sites. Bioinformatics 17(12):1236–1237
Singh H, Raghava GP (2003) ProPred1: prediction of promiscuous MHC class-I binding sites. Bioinformatics 19(8):1009–1014
Singh S, Singh H, Tuknait A, Chaudhary K, Singh B, Kumaran S, Raghava GPS (2015) PEPstrMOD: structure prediction of peptides containing natural, non-natural and modified residues. Biol Direct 10:73. https://doi.org/10.1186/s13062-015-0103-4
Srivastava S, Sharma SK, Srivastava V, Kumar A (2020) Proteomic exploration of Listeria monocytogenes for the purpose of vaccine designing using a reverse vaccinology approach. Int J Pept Res Ther. https://doi.org/10.1007/s10989-020-10128-1
Terry FE, Moise L, Martin RF, Torres M, Pilotte N, Williams SA, Groot ASD (2015) Time for T Immunoinformatics addresses vaccine design for neglected tropical and emerging infectious diseases. Expert Rev Vaccines 14(1):21–35
Weaver SC, Osorio JE, Livengood JA, JA, Chen R, Stinchcomb DT, (2012) Chikungunya virus and prospects for a vaccine. Expert Rev Vaccines 11(9):1087–1101
WHO (2020)- https://www.who.int/news-room/fact-sheets/detail/chikungunya.
Acknowledgements
We are thankful to Prof. Sheeba, Dr. B.R. Ambedkar University, Agra, India for his experimental guidance during the course of experimental work. We are also indebted to Prof. Ajay Kumar, Department of Biotechnology, Faculty of Engineering and Technology, Rama University, Kanpur, India for giving us necessary suggestions and inspiration to successfully accomplish the write-up of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sharma, P., Sharma, P., Ahmad, S. et al. Chikungunya Virus Vaccine Development: Through Computational Proteome Exploration for Finding of HLA and cTAP Binding Novel Epitopes as Vaccine Candidates. Int J Pept Res Ther 28, 50 (2022). https://doi.org/10.1007/s10989-021-10347-0
Accepted:
Published:
DOI: https://doi.org/10.1007/s10989-021-10347-0