
Abstract
Prosopis cineraria commonly known as Druce are valuable herb that holds antibacterial role, antifungal properties. We identified different peptides from this plant by deploying CADD (Computer-aided-drug-designing) approaches, these peptide sequences are as follows seq1 (RHDEEEEKAKV),seq3(KSNSTVEISQNVQSVDSSKM),seq4(KQVAEMNKPAVGSKTSDANHDLKS),seq5(KTKSAGNDSIQSTKPVPSALTVDKA),seq6(RELEDSNIHHVAASVVLESKSSRT), and seq8(LYSKVELHPFGLHNLGNSCYANAVFSV), these peptides holds therapeutic properties as shows interaction with chitin, a major constituent of fungal cell wall. Molecular docking was conducted by using AutoDock-Vina tool and the results were found to be promising where all binding energies were found in the range of − 9.1 to − 7.5 kcal/mol, it indicates strong binding of peptide sequences with chitin molecule. Even the toxicity analysis supports the considered peptide sequences to hold therapeutic role against fungus with non-toxic effect on humans. These peptides were successfully predicted as important therapeutic agents of P. cinerariaseed that can initiate chitin breakdown, due to their possible strong interaction with fungal cell wall and it also suggests this medicinal plant holds the key for multiple fungal disease treatments. This study will open new research dimensions and integration of computational biology with microbial pathology that will assist scientific and medical community to develop rapid disease prevention strategies against fungal pathogenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Prosopiscineraria (L.) Druce is a valuable herbal plant that has been described by ancient texts. It belongs to family Fabaceae, and holds antibacterial role, antifungal properties and antioxidants; this plant shows multiple therapeutic roles as also involved in wound healing and antifungal role (Napar et al. 2012; Nagori 2011). It has historically can be used to manage pain such as leprosy, typhoid, diabetes, leucoderma, dyspepsia, including allergic reaction (Garg and Mittal et al. 2013). Tannins (gallic acid), steroids (Stigmasterol, campesterol, sitosterol, etc.), Flavone compounds (Prosogerin A, B, C, D, and E), alkaloids (Spicigerine, Prosophylline), and other metabolites were being extracted from the tree. Such chemicals may cause Nuclear_factor_KappaB from reaching the nucleus, preventing the overproduction of proinflammatory cytokines but instead acting as a cure for Lipopolysaccharide-induced cellular harm (Sharma and Sharma et al. 2020). In our recent studies we identified proteins of this plant can inhibit growth of fungus in culture environment, the MALDI-ToF MS/MS experiments revealed 15 peptides that may act as potential fungal attack agents or holds therapeutic properties (Solanki et al. 2018). In one of the previous studies, it was shown that extract of Prosopis cineraria inhibits DPP-4 (Dipeptidyl peptidase) as well as cholinesterase enzyme, which makes it suitable for treatment of Diabetes and neurological disorders (Ram et al. 2019). In another recent study it was shown that the extracts of this plant hold antibacterial property against many bacterial species like Staphylocuccus aurius (Neghabi-Hajiagha et al. 2016).Our current work is extension of our previous study, and here we conducted successful molecular docking and simulation experiments to reveal interaction of these peptides with chitin, to reveal role of Prosopis cinerariaseed in breakdown of fungal cell wall.Immune—competent people, unlike immunocompromised people, are protected from fungal infections by their functioning innate immune system, which identifies and destroys fungal invaders quickly. In one of the recent study it was found that Prosopis juliflora extract holds ant-microbial properties against food spoiling microbes (Saleh and Abu-Dieyeh 2021). It has been also noted that Prosopis cineraria extracts embedded and transferred with nano-particle delivery system shows greater success in cancer treatments (Jinu et al. 2017). The immune system's recognition of fungal cellular characteristics appears to be a critical component of human antifungal defense. Dectin-1, for example, recognizesβ-glucan on the fungal cell wall as a pathogen-associated molecular pattern (PAMP) and triggers pro- and anti-inflammatory cytokines through a myeloid-differentiation-primary-response-gene-88 (MYD88-) dependent signaling cascade. Humans do not biosynthesize chitin, but they do have chitin-degrading enzymes called chitinases. Chitotriosidase (CHIT-1) and acidic mammalian chitinase (AMCase) are two known human chitinases with chitinolytic activity, as well as a number of noncatalytically active chitinases known as chi-lectins. CHIT-1 and AMCase have unclear roles, although they are considered to help in the defence against chitin-containing infections. Serum chitotriosidase levels in guinea pigs, for example, rise in response to systemic fungal infection. The fact that chitinase levels change in response to fungal infections implies that chitinase reactions in the host might be used as a diagnostic (Vega and Kalkum 2012). Our study suggests targeting chitin cell wall of infectious fungus could be possible in all individuals with the help of therapeutic peptides that were isolated from Prosopis spp. Seeds. Figure 1 indicates general workflow of current in-silico study, that clearly show peptides of Prosopis cineraria interacts with chitin by deploying molecular docking and MD simulations. This study will open new dimensions in developing regimens against fungal infections.

Workflow for determining Prosopis cineraria peptides interaction with chitin
Methodology
Peptide Sequence Data Collection
Sequence based data for all peptides of P. cineraria seedswere obtained from our previous MALDI-TOF MS/MS analysis of its protein extracts (Solanki et al. 2018). Chitin structure was downloaded from PubChem database in sdf format, which was converted to pdb format after using openbabel tool (O'Boyle et al. 2011).
Peptides Structural Designing
Peptide’s structure was designed by using PepFold 3.5 server (Thévenet et al. 2012), which allows ab-initio prediction of structures and follows hidden marlov model probability algorithms to produce refine structures for given sequences. All structures were validated by proSA webserver (Wiederstein and Sippl. 2007), this tool assist in determining Z-scores for all protein structures.
Toxicity, Allergenicity Predictions of Peptides
ToxinPred tool (Gupta et al. 2013) was used to check the toxicity of peptides on the basis of SVM scores; also Allergen-FPtool (Dimitrov et al. 2014) was deployed to predict the allergenicity of these P.cineraria seeds peptides. Other biochemical parameters like GRAVY score, Instability index, pI, and Half-life was calculated by subjecting these peptide sequences to ProtParam (Garg et al. 2016) server, these all filters assisted in finalizing sequences from our 15 sequences.
Molecular Interaction Studies Between Chitin and Peptide Sequences
The study of the interactions between the protein and the ligand is known as docking. Ionic contacts, hydrogen bonds, and van der Waals interactions are some of the many forms of protein–ligand associations (Pantsar and Poso, 2018). The total of all molecular interactions between the ligands and the receptor is the binding free energy. The scoring function used to estimate the binding capacity of both the ligands and the receptor once docked is known as Docking Score (Adibpour et al. 2012; Gilad and Senderowitz. 2014). Docking studies were conducted by using AutoDock vina (Trott and Olson 2010) software, for each considered sequences that were docked against chitin molecule. The resulted complexes binding energies was calculated by using this software, also PyMOL software (DeLano and Bromberg 2004) was deployed for visualization of final docked complexes.
Molecular Dynamics Simulation
Molecular dynamic simulations were conducted by using iMODStool (López-Blanco et al., 2014) to reveal eigen value plots, that clearly indicates stability of docked complexes along the trajectory during simulation analysis for default time span. Here variance plots and eigen value plots were calculated for docked complexes to indicate stable interaction between selected peptide sequences and chitin molecule. Further GROMACS tool (Van Der Spoel et al. 2005) was deployed for molecular dynamics and simulation studies for all the subjected complexes to all atom OPLS force field, trajectory analysis was conducted for 50 ns time span, which assisted in generating RMSD and RMSF plots indicating inference about stability of interaction between peptide sequences and chitin. The NVT and NPT ensembles were used to accomplish equilibration in two phases (constant number of particles, volume, and temperature at 50 ns). Following the equilibration phase, the Particle-Mesh Ewald summation technique (Shan et al. 2005) was used, followed by a 20-ns production phase. GROMACS' g rms and g rmsf tools were used to examine the generated trajectories.
Results
Structural Predictions of Peptides
15 peptides sequences were obtained from seeds of P. cineraria by using of MALDI TOF MS/MS were considered for structural predictions. Structures of P. cineraria peptides was predicted successfully by deploying PepFold3.5 tool, and then all structures were also validated on the basis of overall model quality and knowledge-based energy analysis in proSA server, that resulted z-scores in suitable range of − 5 to + 5. The z- cores for finalized sequences are as follows: Seq1 z score: − 1.44, seq3 z score: 0.48, seq4 z score: − 0.72, seq5 z score: 0.18, seq6 z score: 0.02, and seq8zscore: -2.34. All the structures were visualized in Pymol software and are presented in Fig. 2.

Predicted Structures for selected sequence: A Seq1; B Seq3; C Seq4; D Seq5; E Seq6; F Seq8
Toxicity, Allergenicity Predictions
Peptide analysis report generated for allergenicity (Table1), so that only non-allergen peptides could be selected for revealing therapeutic relevance in human fungal treatment strategy development. ProtParam analysis report for 15 peptides assisted in checking stability parameter, out of these 15 peptides only 8 peptides were found to be stable as well as non-allergen. Toxicity analysis reveals that only 6 peptides were non-toxic out of 8 peptides (Table2), and then these structures were subjected for docking to chitin molecule, PubChem CID: 24,978,517: Molecular Formula: C64H106N8O41.
Docking Analysis of Peptides with Chitin
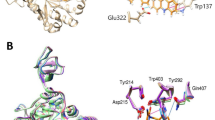
Molecular docking was performed by deploying AutoDockVinatool, which assisted in determining binding energy (Table3) and revealing proper binding pockets (Fig. 3) where enzymatic activity of these peptides occur. These amino residue’s R_groupslike in glutamic acid (Glu) as well as aspartate (Asp) have also been discovered to be essential for these considered peptides' action. Glu functions as a hydrogen donor towards the glycosidic link, slicing the substrate's C-O bond, while Asp functions like a nucleophile to form a glycosyl peptide intermediate. The Glu combines with water to make hydroxyl ion, a greater nucleophile to water, that targets the glycosyl peptide intermediates to produce the breakdown product while keeping overall peptides intact, these peptides behave very similar to lysozyme enzymatic actions. Many of the amino R-groups inside these peptide sequences can behave as nucleophiles and are commonly mentioned in the active site of enzymes. Cysteine, Serine, Threonine, Tyrosine, Glutamic Acid, Aspartic Acid, Lysine, Arginine, and Histidine are some of these amino acids. Therefore, all selected 6 peptide sequences like seq1, seq3, seq4, seq5, seq6, and seq8 follows this mechanism to cleave chitin apart and hence leads to breakdown of chitin cell wall of fungal pathogens.

Docked complexes: A Seq1-chitin B Seq3-chitin C Seq4-chitin D Seq5-chitin E Seq6-chitin F Seq8-chitin
Molecular Dynamics and Simulation analysis
MD simulations were conducted on the basis of normal mode criteria by deploying iMODs tool, here eigen value plot (Fig. 4) indicates deformity associated during simulations of docked complexes; lower the value more deformation in structure, as this value is in direct relation with energy associated for bringing deformity in structure. Also, variance plot was obtained for docked models, as the value of variance is inversely proportional to eigen values (Fig. 5). Here all docked complexes have stable patterns and therefore it indicates perfect docking between peptide sequences and chitin molecule. RMSD (Fig. 6) and RMSF (Fig. 7) plots clear the picture about interaction patterns between all 6 selected peptide sequences with chitin molecule, as here we also used reference molecule (PDB ID: 2D49; Chitinase and chitin complex crystal structure retrieved from RCSB-PDB website) considered for comparative analysis. MD simulation indicates there is less deviation even after 50 ns time span. Interaction snapshots (Fig. 8) after simulation clearly indicates stable binding of peptides to chitin molecule.

Eigen-Value plot for docked complexes: A Seq1-chitin B Seq3-chitin C Seq4-chitin D Seq5-chitin E Seq6-chitin F Seq8-chitin

Variance plot for docked complexes: A Seq1-chitin B Seq3-chitin C Seq4-chitin D Seq5-chitin E Seq6-chitin F Seq8-chitin

Root mean square deviation plot for all 6 complexes and reference chitinase-chitin complex

Root mean square fluctuation plot for all 6 complexes and reference chitinase-chitin complex

Interaction patterns of various peptide sequences with chitin: A Seq1-chitin B Seq3-chitin C Seq4-chitin D Seq5-chitin E Seq6-chitin F Seq8-chitin
Discussion
CADD (Computer-aided-drug-designing) approaches in therapeutic predictions revealed seq1(RHDEEEEKAKV),seq3(KSNSTVEISQNVQSVDSSKM),seq4(KQVAEMNKPAVGSKTSDANHDLKS),seq5(KTKSAGNDSIQSTKPVPSALTVDKA),seq6(RELEDSNIHHVAASVVLESKSSRT), and seq8(LYSKVELHPFGLHNLGNSCYANAVFSV), shows interaction with chitin, a major constituent of fungal cell wall.A CADD study was also found successful in many recent studies, where plant constituents were found to be effective against Sars-Cov2 (Joshi et al. 2020).P. cinerariawas recently found to have one important chemical constituent vitexin that plays major role in inducing apoptosis of leukemia cells (Sarkar et al. 2021). In one of the previous studies, it was shown that extract of Prosopis cineraria inhibits DPP-4 (Dipeptidyl peptidase) as well as cholinesterase enzyme, which makes it suitable for treatment of Diabetes and neurological disorders (Ram et al. 2019). In another recent study it was shown that the extracts of this plant hold antibacterial property against many bacterial species like Staphylocuccusaurius (Neghabi-Hajiagha et al. 2016).In our recent studies we identified proteins of this plant can inhibit growth of fungus in culture environment, the MALDI-Tof experiments revealed 15 peptides that may act as potential fungal attack agents or holds therapeutic properties (Solanki et al. 2018), in current study we extended this work by deploying CADD approach for revealing therapeutic peptides interaction pattern against fungal cell wall.Here we clearly found the best docking scores in more negative to—4.0 kcal/mol range of binding energy that shows strong binding between peptide sequences and chitin (fungal cell wall compound). In one of our recent studies, we also find out that peptides from Dengue virus (Krishnan et al. 2020, 2021), Troperymawhipplei (Joshi and Kaushik 2021), Candida fungus (Akhtar et al. 2021a, b), Nervous Necrosis Virus (Joshi et al. 2021), and Human cytomegalovirus (Akhtar et al. 2021a, b) could be immunogenic for humans, in general by deploying molecular docking and simulation studies. Even the toxicity analysis supports the considered peptide sequences to hold therapeutic role against fungus with non-toxic effect on humans. These peptides were successfully predicted as important therapeutic agents of Prosopis cineraria that can initiate chitin breakdown, due to their possible strong interaction with fungal cell wall and it also suggests this medicinal plant holds the key for multiple fungal disease treatments.
Conclusion
Our research shows peptides of Prosopis cinerariaseed holds good therapeutic potential against fungal pathogens as these peptides are involved in breaking chitin cell wall and leading fungal cells to face oxidative burst. Here we used bioinformatic tools that establish rapid screening of peptides in an efficient manner in terms of money as well as time. As there are large diverse fungi associated diseases, these Antifungal peptides can be used to treat fungal infections, as this study creates new research avenues for future wet-lab analysis of such useful therapeutic peptides. This study will open new research dimensions and integration of computational biology with microbial pathology that will assist scientific and medical community to develop rapid disease prevention strategies against fungal pathogenesis.
References
Adibpour N, Rahim F, Rezaeei S, Khalaj A, Ebrahimi A (2012) In silico designing selective inhibitor of drugs, medicinal plants compounds and experimental ligands for pteridine reductase targeting visceral leishmaniasis. African J Microbiol Res 6(5):917–926
Akhtar N, Joshi A, Kaushik V, Kumar M, Mannan MAU (2021a) In-silico design of a multivalent epitope-based vaccine against Candida auris. Microb Pathog 155:104879
Akhtar N, Joshi A, Singh J, Kaushik V (2021b) Design of a novel and potent multivalent epitope based human cytomegalovirus peptide vaccine: an immunoinformatics approach. J Mol Liquids 335:1165
DeLano, W. L., & Bromberg, S. (2004) PyMOL user’s guide. DeLano Scientific LLC, 629
Dimitrov I, Naneva L, Doytchinova I, Bangov I (2014) AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics 30(6):846–851
Garg A, Mittal SK (2013) Review on Prosopis cineraria: A potential herb of Thar desert. Drug Invention Today 5(1):60–65
Garg VK, Avashthi H, Tiwari A, Jain PA, Ramkete PW, Kayastha AM, Singh VK (2016) MFPPI–multi FASTA ProtParam interface. Bioinformation 12(2):74
Gilad Y, Senderowitz H (2014) Docking studies on DNA intercalators. J Chem Inf Model 54(1):96–107
Gupta, S., Kapoor, P., Chaudhary, K., Gautam, A., Kumar, R., Raghava, G. P., &Open Source Drug Discovery Consortium. (2013). In silico approach for predicting toxicity of peptides and proteins. PloS one, 8(9), e73957.
Jinu U, Gomathi M, Saiqa I, Geetha N, Benelli G, Venkatachalam P (2017) Green engineered biomolecule-capped silver and copper nanohybrids using Prosopis cineraria leaf extract: enhanced antibacterial activity against microbial pathogens of public health relevance and cytotoxicity on human breast cancer cells (MCF-7). Microb Pathog 105:86–95
Joshi A, Kaushik V (2021) In-silico proteomic exploratory quest: crafting T-cell epitope vaccine against Whipple’s disease. Int J Pept Res Ther 27:169–179
Joshi A, Krishnan GS, Kaushik V (2020) Molecular docking and simulation investigation: effect of beta-sesquiphellandrene with ionic integration on SARS-CoV2 and SFTS viruses. J Genetic Engineering and Biotechnol 18(1):1–8
Joshi A, Pathak DC, Mannan MAU, Kaushik V (2021) In-silico designing of epitope-based vaccine against the seven banded grouper nervous necrosis virus affecting fish species. Network Modeling Analysis in Health Informatics and Bioinformatics 10(1):1–12
Krishnan S, Joshi A, Kaushik V (2020) T cell epitope designing for dengue peptide vaccine using docking and molecular simulation studies. Mol Simul 46(10):787–795
Krishnan S, Joshi A, Akhtar N, Kaushik V (2021) Immunoinformatics designed T cell multi epitope dengue peptide vaccine derived from non structural proteome. Microb Pathog 150:104728
López-Blanco JR, Aliaga JI, Quintana-Ortí ES, Chacón P (2014) iMODS: internal coordinates normal mode analysis server. Nucleic Acids Res 42(W1):W271–W276
Nagori BP, Solanki R (2011) Role of medicinal plants in wound healing. Res J Medicinal Plant 5(4):392–405
Napar AA, Bux H, Zia MA, Ahmad MZ, Iqbal A, Roomi S, Shah SH (2012) Antimicrobial and antioxidant activities of Mimosaceae plants; Acacia modesta Wall (Phulai), Prosopis cineraria (Linn) and Prosopis juliflora (Swartz). J Med Plants Res 6(15):2962–2970
Neghabi-Hajiagha M, Aliahmadi A, Taheri MR, Ghassempour A, Irajian G, Rezadoost H, Feizabadi MM (2016) A bioassay-guided fractionation scheme for characterization of new antibacterial compounds from Prosopis cineraria aerial parts. Iranian J Microbiol 8(1):1
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR (2011) Open Babel: an open chemical toolbox. J Cheminformatics 3(1):1–14
Pantsar T, Poso A (2018) Binding affinity via docking: fact and fiction. Molecules 23(8):1899
Ram H, Jaipal N, Kumar P, Deka P, Kumar S, Kashyap P (2019) Dual inhibition of DPP-4 and cholinesterase enzymes by the phytoconstituents of the ethanolic extract of Prosopis cineraria pods: therapeutic implications for the treatment of diabetes-associated neurological impairments. Curr Alzheimer Res 16(13):1230–1244
Saleh I, Abu-Dieyeh MH (2021) Novel Prosopis juliflora leaf ethanolic extract as natural antimicrobial agent against food spoiling microorganisms. Sci Rep 11(1):1–17
Sarkar MK, Kar A, Jayaraman A, Kar Mahapatra S, Vadivel V (2021) Vitexin isolated from Prosopis cineraria leaves induce apoptosis in K-562 leukemia cells via inhibition of the BCR-ABL-Ras-Raf pathway. J Pharmacy and Pharmacol. https://doi.org/10.1093/jpp/rgab085
Shan Y, Klepeis JL, Eastwood MP, Dror RO, Shaw DE (2005) Gaussian split Ewald: A fast Ewald mesh method for molecular simulation. J Chem Phys 122(5):054101
Sharma V, Sharma P (2020) Phyto-therapeutic potential of stem bark of the wonder tree, Prosopis cineraria (L.) Druce in LPS-induced mouse model: An Anti-Inflammatory Study. Clinical Phytoscience 6(1):1–8
Solanki DS, Kumar S, Parihar K, Tak A, Gehlot P, Pathak R, Singh SK (2018) Characterization of a novel seed protein of Prosopis cineraria showing antifungal activity. Int J Biol Macromol 116:16–22
Thévenet P, Shen Y, Maupetit J, Guyon F, Derreumaux P, Tufféry P (2012) PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res 40(W1):W288–W293
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461
Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ (2005) GROMACS: fast, flexible, and free. J Comput Chem 26(16):1701–1718
Vega K, Kalkum M (2012) Chitin, chitinase responses, and invasive fungal infections. Int J Microbiol 2012:1–10
Wiederstein M, Sippl MJ (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 35(2):407–410
Acknowledgements
All the authors are thankful to School of Bioengineering and Biosciences, Lovely Professional University, Punjab, India for conduction of all computational biology support.
Author information
Authors and Affiliations
Contributions
PG and JS have prepared the framework of the study, AJ, VK, DSS, conducted the computational work and manuscript preparation. All authors have proofread manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors have no conflict of interest.
Ethical approval
Not applicable, as it is in-silico work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Joshi, A., Solanki, D.S., Gehlot, P. et al. In-Silico Validation of Prosopis ciniraria Therapeutic Peptides Against Fungal Cell Wall: Better Treatment Strategy for Fungal Diseases. Int J Pept Res Ther 28, 15 (2022). https://doi.org/10.1007/s10989-021-10330-9
Accepted:
Published:
DOI: https://doi.org/10.1007/s10989-021-10330-9