
Abstract
Context
Habitat fragmentation is expected to erode genetic diversity, which instead needs to be preserved for promoting species adaptation to a changing climate. As this expectation has found mixed support in forest trees, consistent results on the genetic consequences of fragmentation requires adequately replicated experimental designs, as well as an explicit assessment of which landscape features, if any, could mitigate its detrimental effects.
Objective
Evaluating the role of several landscape attributes in buffering the detrimental effects of fragmentation in two metapopulations of silver fir.
Methods
We genotyped 904 silver fir (Abies alba Mill.) trees from 18 local populations forming two metapopulations comparable for size and extension in the Apennines, a Mediterranean mountain range. We identified the signatures left by the fragmentation process on the genetic features of silver fir local populations. After removing potentially confounding effects due to different evolutionary histories, we used a multivariate approach for testing the relative effect of demographic, geographic, environmental and topographic factors on genetic features of both metapopulations.
Results
We found comparable signals of the habitat fragmentation impact on the genetic diversity and structure of both investigated metapopulations. Fragmentation effects were less pronounced in the largest local populations (but not the least isolated), located on gentler slopes with higher soil water availability and lower heat exposure.
Conclusions
Our results suggest the existence of a set of demographic and environmental factors that could have coherently buffered the detrimental genetic effects of fragmentation in both metapopulations. These findings could be useful to plan landscape restoration for the evolutionary rescue of mixed forests that once characterized Mediterranean mountain ecosystems.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Forest ecosystems are seriously threatened by climate change (Allen et al. 2010; Seidl et al. 2017) as well as by other human-driven disturbances, such as habitat fragmentation (Wilson et al. 2016). The impact of such threats on forests largely depends on the responses of trees, which are forests’ pillars. Tree species can cope with global changes by tracking the shift of their ecological niche through migration, as well as by phenotypic plasticity and adaptation, or by combining these responses (Aitken et al. 2008). Genetic diversity underpins adaptive processes guaranteeing local persistence despite changing environmental conditions (Hampe and Petit 2005). Trees developed several strategies to maintain high levels of genetic diversity, such as flexible mating systems, high outcrossing rates and effective long-distance dispersal (Petit and Hampe 2006). Developing a dynamic approach for the conservation of such genetic diversity is vital to maintain the evolutionary potential of tree populations (Alfaro et al. 2014; Fady et al. 2016a), thus fostering forest resilience (Vinceti et al. 2020). The ultimate objective of genetic conservation and management of forest trees should be the maintenance of evolutionary processes acting across a mosaic of diverse populations and environmental gradients, rather than the mere preservation of alleles and genotypes (Koskela et al. 2013). In this perspective, the conservation of peripheral tree populations at the rear edge of species distribution ranges is particularly relevant as they are located where the most significant environmental and evolutionary changes will likely occur (Fady et al. 2016b). Besides preserving the genetic diversity heritage of glacial refugia, rear edge populations also represent reservoirs of potentially pre-adapted genetic variants for reinforcing the adaptive potential of more northern populations (Aitken and Bemmels 2016) and for producing resistant planting material (Konnert et al. 2015).
Habitat fragmentation is a landscape-scale process whose outcomes are the decrease in number and size of populations and the increase of average inter-population distances (Fahrig 2003). It is considered as one of the most serious threats to the persistence of populations and species (Wilson et al. 2016) as it exposes increasingly small and isolated populations to risks related to demographic, environmental and genetic stochasticity (Ouborg et al. 2006). Although fragmented populations are expected to suffer from genetic depletion and to exhibit a larger degree of genetic divergence due to increased genetic drift, elevated inbreeding and reduced gene flow (Young et al. 1996), such theoretical predictions have found mixed support in empirical studies on forest trees leading to what is known as ‘the paradox of forest fragmentation genetics’ (Aguilar et al. 2008; Kramer et al. 2008; Lowe et al. 2015). Besides paradoxical results, studies on the impact of forest fragmentation—highly heterogeneous in terms of fragmentation levels and dispersal syndromes investigated (Lowe et al. 2005; Ottewell et al. 2009)—are often lacking suitable experimental designs, hindering consistent conclusions at the species level (Jump and Peñuelas 2006; Bacles and Jump 2011). This is partially linked to the intrinsic nature of the fragmentation process which, by determining highly diverse and stochastic outcomes, often forces researchers to apply suboptimal, largely descriptive and idiosyncratic sampling designs (Bacles and Jump 2011). Beyond the basic comparison between fragmented and continuous populations, any attempt to reach consistent conclusions about the genetic consequences of fragmentation on forest trees would benefit both from replicating such investigation in more than one metapopulation (i.e., ‘a set of local populations which interact via individuals moving among populations’, sensu Hanski and Gilpin 1991), as well as from an explicit assessment of which and how landscape attributes impact the genetic diversity and structure of tree populations (Manel et al. 2003; Sork and Smouse 2006; McRae and Beier 2007). The combined analysis of genetic and environmental data could help to detect environmental critical thresholds to guarantee sufficient levels of genetic diversity (Bacles and Jump 2011). Nonetheless, very few studies on the genetic consequences of fragmentation have explored the effects of landscape variables besides the standard metapopulation ecology indices (e.g., size, isolation and density of remnant forest patches) (da Silva Carvalho et al. 2015; DeSilva and Dodd 2020; Delnevo et al. 2021) but none applied a suitably replicated experimental design.
Silver fir (Abies alba Mill.) is one of the most relevant conifer species in the montane belts of Mediterranean Europe. With the current climatic change, this species has gained renewed interest due to its unexpected drought tolerance in central and southern Europe (Carrer et al. 2010; Tinner et al. 2013; Vitali et al. 2017; Vitasse et al. 2019). In the Apennines, at one of the southern margins of the species distribution, silver fir features a complex genetic structure (Piotti et al. 2017) and high levels of fragmentation (Rovelli 1995), determined by a controversial combination of past climatic oscillations and relatively recent anthropogenic impacts, that have reduced its role of dominant forest species over the last thousands of years (Di Pasquale et al. 2014; Magri et al. 2015; Piotti et al. 2017; Morales-Molino et al. 2021).
Within these prolonged and widespread conditions of fragmentation, we investigated the genetic consequences of landscape features in two similarly sized silver fir metapopulations, located respectively in the northern and in the central Apennines. We characterized all the existing natural local populations occurring within each metapopulation with a set of microsatellite genetic markers to identify the genetic footprints left by the fragmentation process on the levels of genetic diversity and differentiation, inbreeding, effective population size and spatial genetic structure. Then, after standardizing all parameters at the metapopulation level to remove potential confounding effects due to different evolutionary histories, we used a multivariate approach to investigate the relationships among genetic features and a set of landscape attributes calculated at the local population-level in a GIS environment. Our main goal was testing the relative effect of demographic (i.e., population size), geographic (i.e., degree of isolation), and a set of environmental and topographic factors on altering the genetic layout of fragmented silver fir local populations. The specific experimental design and data treatment applied in this study maximized the chance to isolate the landscape attributes most likely involved in buffering the negative fragmentation effects. We finally discussed the implications of unravelling these synergic factors for the development of genetic-based strategies for the conservation and appropriate management of fragmented forest populations.
Materials and methods
Study areas and sampling activities
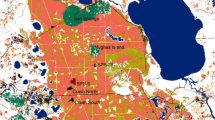
A total of 904 silver fir trees from 18 natural local populations were sampled within two metapopulations (Fig. 1 and Table 1). The first one is located in the northern Apennines, within the Tuscan-Emilian Apennine (TEA) National Park and the second one in the central Apennines, within the Gran Sasso–Monti della Laga (GSL) National Park. In both TEA and GLS metapopulations, silver fir has a highly fragmented distribution, made of small and isolated local populations (Fig. 2) frequently surrounded by plantations of unknown origin (Rovelli 1995; Gallucci and Urbinati 2009; Urbinati 2014; Santini et al. 2018). A previous genetic survey showed that northern and central Apennine silver fir populations belong to different gene pools which separated shortly before the Last Glacial Maximum (Piotti et al. 2017). Whether such separation and more recent demographic dynamics were driven by long-term climatic patterns, neolithic fires and other anthropogenic factors, or a combination of them, it is still debated (Tinner et al. 2013; Magri et al. 2015; Piotti et al. 2017; Morales-Molino et al. 2021). However, the scarcity of palaeobotanical data from the eastern slope of central Apennines prevents developing sound hypotheses on the relative importance of such factors in this geographic area.

Spatial distribution of silver fir local populations sampled (a) within the Tuscan-Emilian Apennine National Park (TEA, blue dots) and within the Gran Sasso–Monti della Laga National Park (GSL, orange dots). See Table 1 for exact local population names and labels. The location of the two metapopulations with respect to the species distribution is reported in box (b). Although highly imprecise regarding natural local populations in these areas, the species distribution according to Caudullo et al. (2017) is overlapped with green lines. Example of the sampling scheme applied (c), where red stars represent the trees sampled for genetic analyses, and the circle the 600-m radius buffer zone around each local population centroid that was used to extract landscape attributes. Dominant Leaf Type data are shown as under-layers, representing non-forested areas (white), broadleaved forests (grey) and coniferous stands (black)

Aerial images of two silver fir local populations in the Tuscan-Emilian Apennine (TEA) National Park at a Lago Ballano (BAL) and b Lago Verde (LAG) sites. Images were taken in winter to enhance the contrast with the broadleaved forest
All natural local populations occurring in both metapopulations were sampled (8 in TEA and 10 in GSL, Table 1 and Fig. 1), collecting approximately 50 trees in each local population, respecting a minimum distance of 20 m among each other. For each sampled tree, fresh needles were collected for genetic analyses and spatial coordinates were recorded with a metric handheld GNSS device (Garmin Ltd., USA).
Genotyping
DNA was extracted from around 50 mg of frozen needles using the DNeasy 96 Plant Kit (Qiagen, Germany) following manufacturer’s instructions. All individuals were genotyped at 16 unlinked nuclear microsatellite markers (nSSRs) (NFF7: Hansen et al. 2005; Aag01, Aat01, Aat02, Aat03, Aat04, Aat05, Aat06, Aat08, Aat09, Aat10, Aat11, Aat13, Aat14, Aat15 and Aat16: Postolache et al. 2014) and three chloroplast microsatellite markers (cpSSRs) (Pt30141 and Pt30249: Liepelt et al. 2001; Pt71936: Vendramin et al. 1996). The procedures for multiplexing and PCR amplification are reported in Postolache et al. (2014) for nSSRs and in Leonarduzzi et al. (2016a) for cpSSRs. All PCR reactions were performed on a GeneAmp PCR System 9700 thermal cycler (Perkin Elmer). PCR products were run on a AB 3500 sequencer (Applied Biosystems, USA), with LIZ-500 as internal size standard. The resulting profiles were sized using GeneMarker (SoftGenetics).
Landscape attribute assessment
Landscape attributes describing local population size, land cover, climate, topography and human pressure data proxies were extracted by public datasets using QGIS software (Table 1). The borders of each local population were drawn using both tree positions collected with a GNSS device during field activities, and Google satellite images for a posteriori check assessment. The resulting convex hull polygons were used to compute the surface area (AREA) of each local population. The attributes available as raster layers were extracted using a 600 m fixed buffer drawn around each local population centroid (Fig. 1c) and computing the median value of all pixels included in each buffer. The size of the adopted buffer was based on the minimum circumscribed circle that includes all sampled trees of the largest local population.
Information on broadleaved (BROAD), coniferous (CONIF) and treeless (TRLES) proportional cover was extracted from the Dominant Leaf Type (DLT) product, available on the Copernicus Land Monitoring Service for the 2018 as reference year (https://land.copernicus.eu/pan-european/high-resolution-layers; last access: March 16, 2023). The main bioclimatic features were extracted from CHELSA database (Karger et al. 2017, 2018), a high resolution (30 arc sec) climate data set currently hosted by the Swiss Federal Institute for Forest, Snow and Landscape Research WSL. In particular, Annual Mean Temperature (AMT), Mean Temperature of Warmest Quarter (MTWQ), Annual Precipitation (AP) and Precipitation of Warmest Quarter (PWQ) were used in the analyses. Topographic features were obtained from the TINITALY DEM (Digital Elevation Model with 10 m resolution, Tarquini et al. 2007): elevation (ELEV), slope (SLO), Heat Load Index (HLI), Compound Topographic Index (CTI) and Roughness Index (ROU). HLI expresses the temperature of a surface by considering the solar radiation, the aspect and slope of that surface (McCune and Keon 2002) and it represents a measure of heat exposure. CTI is a measure of wetness based on the ratio of the catchment area of a given pixel and its slope (Burrough and McDonnell 1998). ROU is a measure of terrain heterogeneity, and it is calculated for each location by summarizing the change in elevation within a 3 × 3 pixel grid (Riley et al. 1999). The mean wind speed (WIND) at 50 m above the ground was obtained by the national grid (1 km-resolution) of the Italian aeolian atlas (http://atlanteeolico.rse-web.it/; last access: October 10, 2022). Isolation value (ISO) was computed averaging all the Euclidean distances between the centroid of single local populations and the others. Finally, proximity of each local population centroid to the closest road (ROAD) was extracted using the national and provincial road network as a proxy of the past and ongoing anthropogenic impact (Dainese et al. 2017; Vitali et al. 2018; Garbarino et al. 2020).
Data analysis
The genetic structure of the 18 local populations was investigated by Principal Component Analysis (PCA) on the matrix of both allele and haplotype counts per population, using the dudi.pca function of the R package adegenet (Jombart and Ahmed 2011).
To explore how different genetic features of silver fir local populations were affected by the fragmentation process, 14 genetic parameters were calculated at the local population level. Within-population genetic diversity was measured by calculating allelic richness (Ar) and expected heterozygosity (HE) on nSSRs, and haplotypic richness (Pb) and diversity (Hk) on cpSSRs. Ar was calculated using hp-rare expected (Kalinowski 2005), HE using GenAlEx v6.5 (Peakall and Smouse 2012) while Pb and Hk using Contrib (Petit et al. 1998). Both Ar and Pb were calculated using a rarefaction method and a minimum sample size of 42 individuals. Within-population genetic differentiation was measured on nSSRs by calculating the average genetic distance among individuals (GD) and its standard deviation (GDsd) using GenAlEx. The average genetic distance among individuals was also calculated on cpSSRs (D2sh), applying the Goldstein et al. (1995)’s distance as implemented in Robledo-Arnuncio et al. (2005). Among-population genetic differentiation was expressed by GST (Nei 1973) and D (Jost 2008), calculated as the average pairwise differentiation of each local population from the others of the same metapopulation. Both GST and D were calculated on nSSRs using the mmod R package (Winter 2012). GST and D range from zero (when two populations are genetically identical) to one (when they are completely fixed for different alleles), but they differ in their sensitivity to the biological processes that drive differentiation and fixation (Jost et al. 2018). The average level of inbreeding characterizing each local population (FIS) was estimated on nSSRs using GenAlEx. The effective population size (Ne), that is the actual number of reproductive trees, was used as a proxy of the intensity of genetic drift, and it was calculated on nSSRs using NeEstimator (Do et al. 2014). Finally, spatial genetic structure (SGS) was assessed with spatial correlograms. Analyses were performed on nSSR data with INEST v2.2, using Nason’s Fij kinship coefficients (Loiselle et al. 1995) and even distance classes (30 m). SGS of each local population was summarized in terms of average kinship coefficient among individuals in the first distance class (F1), slope of the log-linear regression of Fij against spatial distances (b-log) and intensity of SGS (Sp) (Vekemans and Hardy 2004).
The possible occurrence of a bottleneck within each local population was assessed by applying both heterozygosity (H) excess and M-ratio (MR) deficiency tests (Cornuet and Luikart 1996; Garza and Williamson 2001), as implemented in INEST v2.2 (Chybicki and Burczyk 2009). In the calculation, the proportion (pg) and average size (δg) of multi-step mutations were set to 0.22 and 3.1, respectively, as recommended by Peery et al. (2012). The statistical significance of the test was evaluated using the one-tailed Wilcoxon test with 1 × 106 permutations. The bottleneck intensity was assessed by calculating ΔH and ΔMR, which were, respectively, the difference between H and MR values expected at the equilibrium (Heq and MReq) and the observed H and MR values. As H is expected to be higher than Heq, and MR to be lower than MReq under bottleneck, the smaller the ΔH (and the larger the ΔMR), the higher the intensity of the bottleneck. The heterogeneity in the bottleneck intensity among local populations was evaluated by Friedman tests, with ΔH and ΔMR values across loci used as an independent blocking variable and local populations used as the independent grouping factor.
To investigate the relationships among genetic and landscape attributes, all genetic parameters were standardized at the metapopulation level to account for the potential differences due to past demographic divergence. A PCA was then carried out on the main matrix of 14 standardized genetic parameters with the prcomp function of the R package stats and using the set of landscape attributes (Table 1) as secondary matrix. The graphical output of the analysis was a ‘triplot’ where landscape attributes were displayed as secondary vectors whose direction and length represented their correlation with the first and second principal components of the PCA on genetic parameters.
Results
The PCA applied to nuclear and chloroplast markers showed that silver fir local populations from northern and central Apennines were clearly genetically separated along both first principal components, which respectively explained about 21% (nSSRs) and 28% (cpSSRs) of the total variance (Fig. 3).

Biplot from Principal Component Analysis (PCA) on data from a nuclear and b chloroplast microsatellite markers. Blue dots represent local populations from the Tuscan-Emilian Apennine (TEA) National Park in northern Apennines, while orange dots represent local populations from the Gran Sasso–Monti della Laga (GSL) National Park in central Apennines. The ‘×’ symbols represent the metapopulation centroids. Labels: see Table 1
Allelic richness (Ar) ranged from 3.41 (CAO) to 4.71 (NER), which means that the most genetically diverse local population had ~ 38% alleles per locus more than the lowest genetically diverse one (Table 2). NER also exhibited the largest values of expected heterozygosity (HE = 0.46), haplotypic richness (Pb = 25.26) and haplotypic diversity (Hk = 0.97), while the lowest values were shown by the locally peripheral populations ORS (HE = 0.36), CAO (Pb = 25.26) and VCL (Hk = 0.81). Among these four measures of within-population genetic diversity, only HE was, on average, significantly different between the two metapopulations, with TEA local populations characterized by larger HE values (HE-TEA = 0.43) than GSL local populations (HE-GSL = 0.39) (t = − 2.64, df = 11.54, P = 0.022). Within-population genetic distances were significantly different between the two metapopulations both in their average (GD: t = − 3.45, df = 11.55, P = 0.005) and standard deviation values (GDsd: t = − 2.91, df = 15.55, P = 0.011). GD values of TEA local populations ranged from 15.89 (NER) to 12.31 (ORS), with an average of 14.29, while GD values of GSL local populations ranged from 13.63 (CAE) to 11.85 (VCL), with an average of 12.83. Genetic differentiation among local populations of the same metapopulation ranged from 0.014 to 0.035 for D values, and from 0.009 to 0.026 for GST values. For both parameters, ORS was the most genetically differentiated local population. The fixation index (FIS) ranged from − 0.04 (COR) to 0.06 (ORS), but it was significantly larger than zero only in the CPL local population (FIS = 0.06 ± 0.027). The effective population size (Ne) ranged from 10 (CAO) to 4251 (CER), meaning that the largest local population was characterized by ~ 400 times the number of reproducing individuals with respect to the smallest one. Eight local populations showed Ne values < 50 (ORS, SCL, PIA, BAL, VCL, SEG, CAO, CAE), while only three local populations had a Ne > 500 (CER, CPL and TOS). Finally, the intensity of spatial genetic structure (Sp) ranged from − 0.002 (TOS) to 0.040 (ORS). Beyond HE, GD and GDsd, no other genetic parameter was significantly different between the two metapopulations.
In almost all local populations, a significant signature of genetic bottleneck was found according to the test for deficiency in M-ratio but not to the test for heterozygosity excess (Table 3). The only local population showing a significant heterozygosity excess was PIA. The Friedman tests revealed that local populations were not heterogeneous with respect to bottleneck intensity (ΔH: Friedman chi-squared = 13.363, df = 16, P = 0.646; ΔMR: Friedman chi-squared = 13.255, df = 16, P = 0.654).
The first two principal components (PC1 and PC2) of the multivariate analysis on standardized genetic parameters explained 62% of the total variance. With reference to the Euclidean space of Fig. 4, nuclear allelic richness (Ar), expected heterozygosity (HE) and average within-population genetic distances (GD) had negative loadings on PC1 while genetic differentiation (GST and D) and parameters describing spatial genetic structure (Sp and F1) had positive loadings. Thus, PC1 can be interpreted as a ‘diversity vs differentiation’ spectrum which runs from highly diverse and scarcely differentiated/spatially structured local populations to poorly diverse and highly differentiated/structured ones. Instead, haplotypic diversity (Hk, Pb and D2sh) and inbreeding (FIS) had strong negative loading values on PC2.

Triplot from the Principal Component Analysis (PCA) on genetic parameters standardized at the metapopulation level. PC1 is mainly related to nuclear genetic variation, while PC2 to haplotypic diversity and overall inbreeding. Local population labels (see Table 1) were coloured according to the metapopulation (blue = Tuscan-Emilian Apennine National Park—TEA; orange = Gran Sasso–Monti della Laga National Park—GSL). Grey arrows are the loadings of each genetic parameter (see Table 2), whereas green arrows indicate the significant correlations of PC1 scores with the landscape attributes (see Table 1)
The scores of PC1 were negatively correlated with AREA (ρ = − 0.450, P = 0.061) and CTI (ρ = − 0.447, P = 0.063), while positively correlated with HLI (ρ = 0.43, P = 0.075), ROU (ρ = 0.425, P = 0.079) and SLOPE (ρ = 0.416, P = 0.086) (Fig. 4). This means, for instance, that CAO and ORS, which mostly suffered the effects of fragmentation (i.e., lowest genetic diversity, highest differentiation and spatial structuring), were the smallest silver fir stands in the two metapopulations, located on the steepest and roughest slopes, with lower soil water availability and higher heat exposure. In contrast, NER and TOS, which minimally suffered the fragmentation effects (i.e., highest genetic diversity, and lowest differentiation from surrounding populations) were the largest silver fir stands, located on the less steep slopes, with higher soil water availability and lower heat exposure. Dividing the set of local populations in two groups (above and below the median value of the distribution of PC1 scores), local populations that retained larger genetic diversity (negative scores in Fig. 4) were, on average, 2.82 times larger than local populations with lower genetic diversity. Accordingly, their Compound Topographic Index was 1.07 times larger, while slope, Roughness Index, and Heat Load Index were, respectively, 1.32, 1.18, 1.14 smaller than local populations with genetic diversity below the median value. Finally, the scores of PC2 were not significantly correlated with any landscape attributes.
Discussion
In this study, we dug into genetic data from two rear edge, highly fragmented silver fir metapopulations testing for a possible mitigating role of landscape features on the genetic consequences of fragmentation. We found that past bottlenecks have left a coherent genetic footprint in the two metapopulations. After removing variance between metapopulations to properly focus on non-idiosyncratic effects of fragmentation, we found a gradient in its impacts on genetic features. Some silver fir local populations went through pronounced effects (i.e., depletion of genetic diversity and increase of genetic divergence) whilst others displayed more resistance in both metapopulations. The latter were the largest local populations (but not the least isolated), located on less steep slopes, with higher soil water availability and lower heat exposure. This suggested the existence of a set of demographic and environmental factors that could have coherently buffered the detrimental effects of fragmentation in both metapopulations. These results could be used in planning landscape restoration for the evolutionary rescue of the potential Apennine montane belt vegetation composed by mixed Fagus-Abies forests.
Genetic consequences of silver fir habitat fragmentation
The two metapopulations (TEA and GSL), within a surface area of a few squared kilometres, include several small silver fir local populations with similar census size ranges (dozens to few thousands of individuals) and levels of isolation (average pairwise distance among local populations of 18 and 13.5 km in TEA and GLS, respectively). Similar levels of fragmentation—which allowed to set up an ideally replicated experimental design for studying the genetic consequences of the process—have apparently left a coherent genetic footprint. Firstly, almost all studied local populations showed consistent signals of past demographic changes. Although both the tests on the deficiency of M-ratio and excess of heterozygosity (Hk) are effective for identifying demographic reductions given a proper sample size in terms of individuals and loci (Peery et al. 2012), they tend to intercept different types of bottlenecks. M-ratio, that is the range in allele size conditioned on the number of alleles, is suitable for identifying more ancient and prolonged bottlenecks; on the contrary, Hk, that is the heterozygosity given the number of alleles, is effective to identify more recent bottlenecks (Williamson-Natesan 2005). Although they could be refined by applying genomic marker set with higher resolution, our results seem to tell that current silver fir local populations are remnants of larger populations that underwent ancient, severe bottlenecks which, according to reconstructions based on different data sources, dated back from 70 to 5k yrs BP (Follieri et al. 1998; Magri et al. 2015; Piotti et al. 2017). The results of such past demographic dynamics and more recent human pressure have determined a generally low effective population size (Ne). In most cases (15 out of 18 populations), Ne estimates were below the critical threshold of 500. It is still debated how to interpret the 50/500 rule of thumb proposed by Franklin (1980) and the reliability of Ne as a suitable indicator of genetic diversity for forest trees, particularly when populations are large and continuously distributed (Santos-del-Blanco et al. 2022). Generally, Ne value > 500 is considered the minimum size for ensuring the viability of populations in the long run (Jamieson and Allendorf 2012).
Although the bottleneck analysis showed a coherent genetic footprint on allele frequencies left by past size reductions, we found that other genetic features were quite heterogeneous among local populations within each metapopulation. For instance, both genetic diversity and differentiation measures showed similar ranges within the two metapopulations. Least diverse and most differentiated local populations exhibited a decrease in allelic richness of 25% (in TEA) and 22% (in GLS) as well as an increase in genetic differentiation of 174% (in TEA) and 92% (in GLS) with respect to most diverse and less differentiated ones. This shows a strong and similar impact of fragmentation on the genetic structure of the two metapopulations, in sharp contrast with the general lack of signals at the basis of the so-called ‘paradox of forest fragmentation genetics’ (Kramer et al. 2008). These differences matched with those obtained in the few studies showing detectable genetic signals of fragmentation in Fagus sylvatica, a wind pollinated species often co-occurring with silver fir in southern European mountain ranges, when comparing continuous and fragmented populations (Jump and Peñuelas 2006; Leonardi et al. 2012). However, such comparison is impossible when studying silver fir in the Apennines due to the lack of continuous populations, and this is the reason why we focused on targeting results on local dynamics in two comparable metapopulations.
We applied a multivariate approach to a set of genetic parameters to find the main axes summarizing the metapopulations’ genetic structure after standardizing the parameters at the metapopulation level to remove potential confounding effects due to different evolutionary histories. There is clear evidence that silver fir populations of northern and central Apennines belong to different genetic lineages that started diverging around 66k yrs ago (Piotti et al. 2017). The first axis of the resulting PCA depicted a ‘diversity vs differentiation’ spectrum, defining a continuum from local populations that were more genetically diverse and less differentiated (i.e., NER and TOS) to those that were more genetically differentiated and less diverse (i.e., CAO and ORS). Since fragmentation is expected to deplete genetic variation and increase genetic divergence, this ‘diversity vs differentiation’ spectrum could be interpreted as a severity gradient of the fragmentation effects. The same gradient was found in both metapopulations, as TEA and GSL local populations were similarly distributed on the first PCA axis. The inherently limited number of local populations within each metapopulation (eight and ten for TEA and GSL, respectively) precluded a separate statistic treatment for each metapopulation to test the consistency between metapopulations, only allowing such a qualitative evaluation. Nonetheless, even when genetic parameters were not standardized within metapopulations, the same gradient emerged along the first PCA axis, with the two metapopulations separated on the second axis (results not shown). Overall, these results point towards the existence of landscape features that enhanced or buffered the genetic consequences of fragmentation at the local scale, leaving a genetic footprint that prevailed over the one left by regional-scale dynamics. The standardization procedure indeed enabled the comparison of the genetic signals between metapopulations. However, in the analysis we could not thoroughly test the effect of possibly different demographic histories within metapopulations, as we have little information about them. There are no historical records available to reconstruct recent demographic dynamics in the two investigated areas. Instead, paleobotanical records indicate that, until 5k years ago, Abies alba was far more locally abundant in northern Apennines, both at high and low elevations, even in sites where today is absent (Follieri et al. 1998; Bertoldi et al. 2004; Vescovi et al. 2010; Magri et al. 2015; Morales-Molino et al. 2021). Unfortunately, the lack of paleobotanical records from the eastern slope of the central Apennines hindered possible comparison based on this layer of information. However, the demographic reconstruction based on genetic data about these two areas by Piotti et al. (2017) showed that the species had similar demographic trajectories in the two investigated areas since the two genetic lineages diverged before the Last Glacial Maximum. Although detailed demographic reconstructions were not performed at the local population level, available evidence suggests that the two metapopulations were two single, large populations that have been shrinking over the last millennia. This is in line with the bottleneck analysis showing homogeneity in the bottleneck intensity among local populations and, overall, provides support to the robustness of our results when based on the comparability between metapopulations. Of course, as pointed out before, deeper genomic investigations will provide more precise estimates of demographic processes at a higher temporal resolution.
Population size and soil water availability mitigate the detrimental effects of fragmentation
We found that local populations least affected by fragmentation were the largest ones, even when large local populations were highly isolated (e.g., NER). Population size is positively correlated with levels of genetic variation in many plant and animal species (Frankham 1996). When fragmentation effects were detected, larger tree populations often exhibited the highest values of genetic diversity as well as the lowest values of differentiation (Prober and Brown 1994; Hall et al. 1996; Petit et al. 2002; Vakkari et al. 2006). Fragmentation acts not only by decreasing the average number and size of populations, but also increasing the average distance among remaining ones (Fahrig 2003). Isolated populations should undergo reduced gene flow and, therefore, face a greater risk of entering trajectories of increasing genetic differentiation and pauperization. However, we found that isolation was not significantly associated with the genetic layout of silver fir local populations. This means that the average distance among remnant fir patches neither prompted nor hindered local genetic dynamics and that levels of gene flow among silver fir remnants were still sufficiently high for counteracting the effects of genetic drift (Hamrick 2004). When studies on the genetic consequences of fragmentation were set up in order to disentangle the relative role of population size and isolation in speeding up genetic drift, they found that highly outcrossing species were more sensitive to demographic reductions (Leonardi et al. 2012). Among conifers, silver fir pollen and seeds are relatively heavy-weighted (Eisenhut 1961), but considerable rates of pollen (62–83%) and seed (48–79%) gene flow were detected by a previous study carried out in the central Apennines within two of our study populations (CPL and CPH) (Leonarduzzi et al. 2016b). Reconstructing the parentage of seedlings and trapped seeds also showed that seed dispersal can occasionally reach up to 200 m (Cremer et al. 2012; Leonarduzzi et al. 2016b; Major et al. 2021). Pollen dispersal can even reach farther distances, and there is substantial evidence on its effectiveness in maintaining genetic connectivity among fragmented populations of wind-pollinated trees (e.g., Bacles et al. 2005; Kamm et al. 2009; Aleksic et al. 2022), with surprisingly high levels of pollen immigration documented also in highly isolated stands of Quercus robur and Pinus sylvestris (Robledo-Arnuncio and Gil 2005; Buschbom et al. 2011). The low correlation between parameters describing chloroplast and nuclear diversity as well as the smaller variance among local populations intercepted by PCA in parameters describing chloroplast diversity point to the non-interruption of gene flow by pollen in the two metapopulations, even in small local populations and at distances up to 40 km (NER-ORS). In addition, it is worth bearing in mind that, in both geographic areas, there are silver fir plantations that could have acted as additional (and unconsidered) pollen sources at different time scales. Nonetheless, if we consider the coniferous forest cover (CONIF) as a proxy of the presence of any plantations in the buffer area of each local population, we found that this was not correlated with genetic diversity and differentiation patterns. Since most of these stands were planted over the last century with material of unknown origin (Urbinati 2014; Santini et al. 2018), characterizing their genetic make-up and monitoring gene flow in natural silver fir regeneration will be pivotal for assessing whether they represent sources of maladapted and/or genetically depleted material (Steinitz et al. 2012; Unger et al. 2016) or instead help to maintain the genetic connection among natural local populations. Besides the potential effects of outbreeding depression, dispersal dynamics may also be affected by genetic erosion itself in the long run (Ouborg et al. 2006). Inbreeding depression effects on plant fecundity, seed germination and seed dispersal capabilities were indeed observed in plants (Mix et al. 2006; Leimu et al. 2010). Unfortunately, no data on the reproductive dynamics of the studied local populations (e.g., average number of cones and seeds produced) are available to be correlated against genetic metrics estimated in the present study.
Besides their large size, local populations that maintained high levels of genetic diversity also exhibited a combination of specific topographic features: they were located on the roughest and steepest slopes, characterized by the lowest values of Heat Load Index (HLI) and the highest values of Compound Topographic Index (CTI). HLI represents a measure of soil heat exposure, while CTI is a measure of soil wetness. Taken together, these variables can be considered a proxy for describing soil water availability. Soil water availability is a key factor for silver fir growth and survival at different life stages (e.g., Nourtier et al. 2014; Sánchez-Salguero et al. 2017; Walder et al. 2021). For instance, Piedallu et al. (2023) found ten times higher mortality rates in silver fir forests with a high level of vulnerability directly related to water stress. Cailleret et al. (2014) found that, at regional scale, more xeric conditions (i.e., south facing, high slopes) can increase silver fir mortality. Although silver fir has a deep taproot system and can use water from relatively deep soil during drought periods (Magh et al. 2019), shallow soils, or any combination of topographic features determining low soil water availability, had an impact on its resistance to drought. In this regard, our results provide evidence that topographic features determining limited soil water availability influenced not only the probability of dieback but also to the capacity of a forest tree population to retain genetic diversity.
Human influence could be a confounding factor when linking genetic diversity and environmental features. However, the role of human activities was accounted by including the proximity of each local population to the closest road (ROAD) in the analysis (Dainese et al. 2017; Vitali et al. 2018; Garbarino et al. 2020) and it was seemingly unrelated to genetic features. Unravelling the reliance of genetic diversity on environmental variables is crucial to model future habitat suitability at a fine scale (Kittlein et al. 2022) and to support actions for management and conservation of forest tree species in a changing environment (Hoban et al. 2021; Vajana et al. 2022), as it is discussed in the following paragraph.
Linking genetic diversity and environmental variables to inform management and conservation actions
A higher number of tree species tends to increase the productivity of forest ecosystems (Vilà et al. 2013; Liang et al. 2016), and their resistance/resilience to biotic and abiotic stresses (Jactel and Brockerhoff 2007; Seidl et al. 2016) as well as to enlarge the range of ecosystem services they can provide compared to monospecific stands (Gamfeldt et al. 2013; Rudow et al. 2020). Besides some still controversial aspects, there is increasing evidence that such benefits occur also in the Fagus-Abies forests (Versace et al. 2021), and silver fir is considered, in some European countries, as the ideal species to steer forests towards greater production and resistance to climate change (Tinner et al. 2013; Vitasse et al. 2019). Despite their ecological relevance, Mediterranean mixed Fagus-Abies forests are on a long-term trajectory of demographic decline (e.g., Magri et al. 2015), in an area where the consequences of the climatic crisis are expected to hit hardest. Therefore, it is critical to take effective actions on extant natural populations of silver fir to enhance their regeneration in areas of potential expansion. Nature-based sylvicultural treatments such as selective thinning to release regeneration core areas (with tree age up to 70 years), gap opening to simulate the natural windthrow, or girdling of a limited number of dominant or concurrent beech trees can progressively improve the conditions for silver fir to establish and grow (Urbinati 2014). Beyond any type of sylvicultural treatment, recent literature calls for management programs to be fine-tuned based on a careful characterization of the ecological requirements of the species (Kittlein et al. 2022; Vejpustková et al. 2023). This study is precisely going in this direction by exploring the dependency of genetic diversity on landscape features and providing an additional, crucial information to the management and conservation of fragmented silver fir local populations. Genetic information is considered essential to assess the conservation status of populations (Laikre et al. 2020) and to design suitable strategies for the management, translocation and conservation of genetic resources (Aitken and Whitlock 2013; Fady et al. 2016a; Andrello et al. 2022). Although our study was based on neutral genetic markers, which are not informative regarding adaptive dynamics (Holderegger et al. 2006), our findings can nonetheless be distilled in few, clear indications for the conservation of shrinking silver fir Apennine forests. Firstly, our results tell which local populations, given their levels of genetic diversity and distinctiveness, should be prioritized for conservation actions. Secondly, they give new insights on which combination of environmental and topographic variables is buffering the detrimental effects of fragmentation. In particular, our findings point to the fact that sylvicultural treatments aimed at making local populations larger, rather than less isolated, could be more beneficial in counteracting the negative effects of genetic drift. They also provided a quantitative link between levels of genetic diversity and a multidimensional topographic space, which could be applied to enhance the chance of silver fir local populations to thrive and, thus, resist the genetic consequences of fragmentation. In fact, mapping this favourable multidimensional space can provide forest managers a tool to define areas where to intensify ad hoc reforestation efforts, obviously keeping in mind that our results can not be extended beyond the space of environmental and topographic parameters explored in the two metapopulations.
Conclusions
Studies on the genetic consequences of habitat fragmentation have often relied on suboptimal and largely descriptive sampling designs. To draw robust conclusions on silver fir rear edge populations, we i) selected two highly fragmented silver fir metapopulations featuring similar geographical extension and ii) set up data analysis to remove variance in genetic data possibly due to different demographic histories. We could then isolate genetic signals which point to population size and soil water availability as the driving factors that mitigated the detrimental effects of fragmentation. As drought is becoming more intense and prolonged in Mediterranean areas, the relationship between soil water availability and genetic diversity is particularly relevant when put in the context of climatic changes predicted at the southern edge of silver fir distribution.
Hopefully, our study would trigger new research on detecting consistent genetic consequences determined by habitat fragmentation, to increase our knowledge on the impact of this process on forest ecosystems. There are several ways to improve the data collection needed for that. First, the use of unmanned aerial vehicles will provide higher resolution spatial data, enhancing the recognition of small patches of adult trees and regeneration and a more accurate characterization of the topographic space where trees grow and regenerate. Including trees in their regeneration stages (seedlings and saplings) would be particularly beneficial to future studies. Since fragmentation is relatively recent in many forest ecosystems, shifting the research focus from adult trees to regeneration would allow a better detection of early genetic consequences of fragmentation in the short term (Lowe et al. 2015).
In addition, next generation sequencing genomic resources will permit a much deeper investigation of the evolutionary processes that produced those signals highlighted in our study. In particular, the recent release of the first version of silver fir genome (Mosca et al. 2019) and the increasing knowledge of the evolutionary significance of specific candidate genes and stress responses (Cailleret et al. 2014; Brousseau et al. 2016; Csilléry et al. 2020) could encourage the transition from landscape genetics to landscape genomics approaches. Landscape genomics investigations would improve the understanding of the functional link between genetic diversity and environmental features, useful grounds for developing genomic-informed conservation actions that could guide afforestation and reforestation initiatives to counteract the negative impact of habitat fragmentation on forest ecosystems.
Data availability
The datasets analysed during the current study will be made available from the corresponding author under request.
References
Aguilar R, Quesada M, Ashworth L, Herrerias-Diego Y, Lobo J (2008) Genetic consequences of habitat fragmentation in plant populations: susceptible signals in plant traits and methodological approaches. Mol Ecol 17:5177–5188
Aitken SN, Bemmels JB (2016) Time to get moving: assisted gene flow of forest trees. Evol Appl 9:271–290
Aitken SN, Whitlock MC (2013) Assisted gene flow to facilitate local adaptation to climate change. Ann Rev Ecol Evol Syst 44:367–388
Aitken SN, Yeaman S, Holliday JA, Wang T, Curtis-McLane S (2008) Adaptation, migration or extirpation: climate change outcomes for tree populations: climate change outcomes for tree populations. Evol Appl 1:95–111
Aleksić JM, Mataruga M, Daničić V, Cvjetković B, Milanović Đ, Vendramin GG, Avanzi C, Piotti A (2022) High pollen immigration but no gene flow via-seed into a genetic conservation unit of the endangered Picea omorika after disturbance. For Ecol Manag 510:120115
Alfaro RI, Fady B, Vendramin GG, Dawson IK, Fleming RA, Sáenz-Romero C, Lindig-Cisneros RA, Murdock T, Vinceti B, Navarro CM, Skrøppa T, Baldinelli G, El-Kassaby YA, Loo J (2014) The role of forest genetic resources in responding to biotic and abiotic factors in the context of anthropogenic climate change. For Ecol Manag 333:76–87
Allen CD, Macalady AK, Chenchouni H, Bachelet D, McDowell N, Vennetier M, Kitzberger T, Rigling A, Breshears DD, Hogg EH, Gonzalez P, Fensham R, Zhang Z, Castro J, Demidova N, Lim JH, Allard G, Running SW, Semerci A, Cobb N (2010) A global overview of drought and heat-induced tree mortality reveals emerging climate change risks for forests. For Ecol Manag 259:660–684
Andrello M, D’Aloia C, Dalongeville A, Escalante MA, Guerrero J, Perrier C, Torres-Florez JP, Xuereb A, Manel S (2022) Evolving spatial conservation prioritization with intraspecific genetic data. Trends Ecol Evol 37:553–564
Bacles CFE, Burczyk J, Lowe AJ, Ennos RA (2005) Historical and contemporary mating patterns in remnant populations of the forest tree Fraxinus excelsior L. Evolution 59:979–990
Bacles CFE, Jump AS (2011) Taking a tree’s perspective on forest fragmentation genetics. Trends Plant Sci 16:13–18
Bertoldi R, Chelli A, Roma R, Tellini C, Vescovi P (2004) First remarks on late Pleistocene lacustrine deposit in the Berceto area (northern Apennines, Italy). Quaternario 17:133–143
Brousseau L, Postolache D, Lascoux M, Drouzas AD, Källman T, Leonarduzzi C, Liepelt S, Piotti A, Popescu F, Roschanski AM, Zhelev P, Fady B, Vendramin GG (2016) Local adaptation in European firs assessed through extensive sampling across altitudinal gradients in southern Europe. PLoS ONE 11:e0158216
Burrough PA, McDonnell RA (1998) Principles of geographical information systems. Oxford University Press, Oxford
Buschbom J, Yanbaev Y, Degen B (2011) Efficient long-distance gene flow into an isolated relict oak stand. J Hered 102:464–472
Cailleret M, Nourtier M, Amm A, Durand-Gillmann M, Davi H (2014) Drought-induced decline and mortality of silver fir differ among three sites in Southern France. Ann for Sci 71:643–657
Carrer M, Nola P, Motta R, Urbinati C (2010) Contrasting tree-ring growth to climate responses of Abies alba toward the southern limit of its distribution area. Oikos 119:1515–1525
Caudullo G, Welk E, San-Miguel-Ayanz J (2017) Chorological maps for the main European woody species. Data Brief 12:662–666
Chybicki IJ, Burczyk J (2009) Simultaneous estimation of null alleles and inbreeding coefficients. J Hered 100:106–113
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Cremer E, Ziegenhagen B, Schulerowitz K, Mengel C, Donges K, Bialozyt R, Hussendörfer E, Liepelt S (2012) Local seed dispersal in European silver fir (Abies alba Mill.): lessons learned from a seed trap experiment. Trees 26:987–996
Csilléry K, Buchmann N, Fady B (2020) Adaptation to drought is coupled with slow growth, but independent from phenology in marginal silver fir (Abies alba Mill.) populations. Evol Appl 13:2357–2376
Dainese M, Aikio S, Hulme PE, Bertolli A, Prosser F, Marini L (2017) Human disturbance and upward expansion of plants in a warming climate. Nat Clim Change 7:577–580
DeSilva R, Dodd RS (2020) Fragmented and isolated: limited gene flow coupled with weak isolation by environment in the paleoendemic giant sequoia (Sequoiadendron giganteum). Am J Bot 107:45–55
Delnevo N, Piotti A, Carbognani M, van Etten EJ, Stock WD, Field DL, Byrne M (2021) Genetic and ecological consequences of recent habitat fragmentation in a narrow endemic plant species within an urban context. Biodivers Conserv 30:3457–3478
Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014) NEESTIMATOR v2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour 14:209–214
Eisenhut G (1961) Untersuchungen über die Morphologie und Ökologie der Pollenkörner heimischer und fremdländischer Waldbäume, 15. Verlag nicht ermittelbar
Fady B, Aravanopoulos FA, Alizoti P, Mátyás C, von Wühlisch G, Westergren M, Belletti P, Cvjetkovic B, Ducci F, Huber G, Kelleher CT, Khaldi A, Kharrat MBD, Kraigher H, Kramer K, Mühlethaler U, Peric S, Perry A, Rousi M, Sbay H, Stojnic S, Tijardovic M, Tsvetkov I, Varela MC, Vendramin GG, Zlatanov T (2016b) Evolution-based approach needed for the conservation and silviculture of peripheral forest tree populations. For Ecol Manag 375:66–75
Fady B, Cottrell J, Ackzell L, Alía R, Muys B, Prada A, González-Martínez SC (2016a) Forests and global change: what can genetics contribute to the major forest management and policy challenges of the twenty-first century? Reg Environ Change 16:927–939
Fahrig L (2003) Effects of habitat fragmentation on biodiversity. Annu Rev Ecol Evol Syst 34:487–515
Follieri M, Giardini M, Magri D, Sadori L (1998) Palynostratigraphy of the last glacial period in the volcanic region of central Italy. Quat Int 47:3–20
Frankham R (1996) Relationship of genetic variation to population size in wildlife. Conserv Biol 10:1500–1508
Franklin IR (1980) Evolutionary change in small populations. In: Soulé ME, Wilcox BA (eds) Conservation biology: an evolutionary-ecological perspective. Sinauer Associates, Sunderland, pp 135–149
Gallucci V, Urbinati C (2009) Growth dynamics and climatic sensitivity of Silver fir (Abies alba Mill.) in the European important site (SIC) at the Alpe della Luna—Bocca Trabaria (PU—Italy). Forest 6:85–99
Gamfeldt L, Snäll T, Bagchi R, Jonsson M, Gustafsson L, Kjellander P, Ruiz-Jaen MC, Fröberg M, Stendahl J, Philipson CD, Mikusiński G, Andersson E, Westerlund B, Andrén H, Moberg F, Moen J, Bengtsson J (2013) Higher levels of multiple ecosystem services are found in forests with more tree species. Nat Commun 4:1340
Garbarino M, Morresi D, Urbinati C, Malandra F, Motta R, Sibona EM, Vitali A, Weisberg PJ (2020) Contrasting land use legacy effects on forest landscape dynamics in the Italian alps and the Apennines. Landsc Ecol 35:2679–2694
Garza JC, Williamson EG (2001) Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10:305–318
Goldstein DB, Ruiz Linares A, Cavalli-Sforza LL, Feldman MW (1995) An evaluation of genetic distances for use with microsatellite loci. Genetics 139:463–471
Hall P, Walker S, Bawa K (1996) Effect of forest fragmentation on genetic diversity and mating system in a tropical tree, Pithecellobium elegans. Conserv Biol 10:757–768
Hampe A, Petit RJ (2005) Conserving biodiversity under climate change: the rear edge matters: rear edges and climate change. Ecol Lett 8:461–467
Hamrick JL (2004) Response of forest trees to global environmental changes. For Ecol Manag 197:323–335
Hansen OK, Kjær ED, Vendramin GG (2005) Chloroplast microsatellite variation in Abies nordmanniana and simulation of causes for low differentiation among populations. Tree Genet Genomes 1:116–123
Hanski I, Gilpin M (1991) Metapopulation dynamics: brief history and conceptual domain. Biol J Linn Soc Lond 42:3–16
Hoban S, Bruford MW, Funk WC, Galbusera P, Griffith MP, Grueber CE, Heuertz M, Hunter ME, Hvilsom C, Stroil BK, Kershaw F, Khoury CK, Laikre L, Lopes-Fernandes M, MacDonald AJ, Mergeay J, Meek M, Mittan C, Mukassabi TA, O’Brien D, Ogden R, Palma-Silva C, Ramakrishnan U, Segelbacher G, Shaw RE, Sjögren-Gulve P, Veličković N, Vernesi C (2021) Global commitments to conserving and monitoring genetic diversity are now necessary and feasible. Bioscience 71:964–976
Holderegger R, Kamm U, Gugerli F (2006) Adaptive vs. neutral genetic diversity: implications for landscape genetics. Landsc Ecol 21:797–807
Jactel H, Brockerhoff EG (2007) Tree diversity reduces herbivory by forest insects. Ecol Lett 10:835–848
Jamieson IG, Allendorf FW (2012) How does the 50/500 rule apply to MVPs? Trends Ecol Evol 27:578–584
Jombart T, Ahmed I (2011) Adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27:3070–3071
Jost L (2008) GST and its relatives do not measure differentiation. Mol Ecol 17:4015–4026
Jost L, Archer F, Flanagan S, Gaggiotti O, Hoban S, Latch E (2018) Differentiation measures for conservation genetics. Evol Appl 11:1139–1148
Jump AS, Peñuelas J (2006) Genetic effects of chronic habitat fragmentation in a wind-pollinated tree. Proc Natl Acad Sci USA 103:8096–8100
Kalinowski ST (2005) HP-RARE 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5:187–189
Kamm U, Rotach P, Gugerli F, Siroky M, Edwards P, Holderegger R (2009) Frequent long-distance gene flow in a rare temperate forest tree (Sorbus domestica) at the landscape scale. Heredity 103:476–482
Karger DN, Conrad O, Böhner J, Kawohl T, Kreft H, Soria-Auza RW, Zimmermann NE, Linder P, Kessler M (2017) Climatologies at high resolution for the earth land surface areas. Sci Data 4:170122
Karger DN, Conrad O, Böhner J, Kawohl T, Kreft H, Soria-Auza RW, Zimmermann NE, Linder HP, Kessler M (2018) Data from: climatologies at high resolution for the earth’s land surface areas. Dryad Digital Repos. https://doi.org/10.5061/dryad.kd1d4
Kittlein MJ, Mora MS, Mapelli FJ, Austrich A, Gaggiotti OE (2022) Deep learning and satellite imagery predict genetic diversity and differentiation. Methods Ecol Evol 13:711–721
Konnert M, Fady B, Gömöry D, A’Hara S, Wolter F, Ducci F, Koskela J, Bozzano M, Maaten T, Kowalczyk J (2015) Use and transfer of forest reproductive material in Europe in the context of climate change. European Forest Genetic Resources Programme (EUFORGEN), Bioversity International, Romel, p 75
Koskela J, Lefèvre F, Schueler S, Kraigher H, Olrik DC, Hubert J, Longauer R, Bozzano M, Yrjänä L, Alizoti P, Rotach P, Vietto L, Bordács S, Myking T, Eysteinsson T, Souvannavong O, Fady B, De Cuyper B, Heinze B, von Wühlisch G, Ducousso A, Ditlevsen B (2013) Translating conservation genetics into management: pan-European minimum requirements for dynamic conservation units of forest tree genetic diversity. Biol Conserv 157:39–49
Kramer AT, Ison JL, Ashley MV, Howe HF (2008) The paradox of forest fragmentation genetics. Conserv Biol 22:878–885
Laikre L, Hoban S, Bruford MW, Segelbacher G, Allendorf FW, Gajardo G, Rodríguez AG, Hedrick PW, Heuertz M, Hohenlohe PA, Jaffé R, Johannesson K, Liggins L, MacDonald AJ, OrozcoterWengel P, Reusch TBH, Rodríguez-Correa H, Russo IRM, Ryman N, Vernesi C (2020) Post-2020 goals overlook genetic diversity. Science 367:1083–1085
Leimu R, Vergeer P, Angeloni F, Ouborg NJ (2010) Habitat fragmentation, climate change, and inbreeding in plants. Ann N Y Acad Sci 1195:84–98
Leonardi S, Piovani P, Scalfi M, Piotti A, Giannini R, Menozzi P (2012) Effect of habitat fragmentation on the genetic diversity and structure of peripheral populations of beech in central Italy. J Hered 103:408–417
Leonarduzzi C, Piotti A, Spanu I, Vendramin GG (2016b) Effective gene flow in a historically fragmented area at the southern edge of silver fir (Abies alba Mill.) distribution. Tree Genet Genomes 12:95
Leonarduzzi C, Spanu I, Labriola M, González-Martínez SC, Piotti A, Vendramin GG (2016a) Development and characterization of three highly informative EST-SSR multiplexes for Pinus halepensis Mill. and their transferability to other Mediterranean pines. Plant Mol Biol Rep 34:993–1002
Liang J et al (2016) Positive biodiversity-productivity relationship predominant in global forests. Science. https://doi.org/10.1126/science.aaf8957
Liepelt S, Kuhlenkamp V, Anzidei M, Vendramin GG, Ziegenhagen B (2001) Pitfalls in determining size homoplasy of microsatellite loci. Mol Ecol Notes 1:332–335
Loiselle BA, Sork VL, Nason JD, Graham C (1995) Spatial genetic structure of a tropical undestory shrub, Psychotria officinalis (Rubiaceae). Am J Bot 82:1420–1425
Lowe AJ, Boshier D, Ward M, Bacles CFE, Navarro C (2005) Genetic resource impacts of habitat loss and degradation; reconciling empirical evidence and predicted theory for neotropical trees. Heredity 95:255–273
Lowe AJ, Cavers S, Boshier D, Breed MF, Hollingsworth PM (2015) The resilience of forest fragmentation genetics—no longer a paradox—we were just looking in the wrong place. Heredity 115:97–99
Magh RK, Bonn B, Grote R, Burzlaff T, Pfautsch S, Rennenberg H (2019) Drought superimposes the positive effect of silver fir on water relations of European beech in mature forest stands. Forests 10:897
Magri D, Agrillo E, Di Rita F, Furlanetto G, Pini R, Ravazzi C, Spada F (2015) Holocene dynamics of tree taxa populations in Italy. Rev Palaeobot Palynol 218:267–284
Major EI, Höhn M, Avanzi C, Fady B, Heer K, Opgenoorth L, Piotti A, Popescu F, Postolache D, Vendramin GG, Csilléry K (2021) Fine-scale spatial genetic structure across the species range reflects recent colonization of high elevation habitats in silver fir (Abies alba Mill.). Mol Ecol 30:5247–5265
Manel S, Schwartz MK, Luikart G, Taberlet P (2003) Landscape genetics: combining landscape ecology and population genetics. Trends Ecol Evol 18:189–197
McCune B, Keon D (2002) Equations for potential annual direct incident radiation and heat load. J Veg Sci 4:603–606
McRae BH, Beier P (2007) Circuit theory predicts gene flow in plant and animal populations. Proc Natl Acad Sci USA 104:19885–19890
Mix C, Pico FX, van Groenendael JM, Ouborg NJ (2006) Inbreeding and soil conditions affect dispersal and components of performance of two plant species in fragmented landscapes. Basic Appl Ecol 7:59–69
Morales-Molino C, Steffen M, Samartin S, van Leeuwen JFN, Hürlimann D, Vescovi E, Tinner W (2021) Long-term responses of Mediterranean mountain forests to climate change, fire and human activities in the northern Apennines (Italy). Ecosystems 24:1361–1377
Mosca E, Cruz F, Gómez-Garrido J, Bianco L, Rellstab C, Brodbeck S, Csilléry K, Fady B, Fladung M, Fussi B, Gömöry D, González-Martínez SC, Grivet D, Gut M, Hansen OK, Heer K, Kaya Z, Krutovsky KV, Kersten B, Liepelt S, Opgenoorth L, Sperisen C, Ullrich KK, Vendramin GG, Westergren M, Ziegenhagen B, Alioto T, Gugerli F, Heinze B, Höhn M, Troggio M, Neale DB (2019) A reference genome sequence for the European silver fir (Abies alba Mill.): a community-generated genomic resource. G3-Genes Genom Genet 9:2039–2049
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70:3321–3323
Nourtier M, Chanzy A, Cailleret M, Yingge X, Huc R, Davi H (2014) Transpiration of silver fir (Abies alba Mill.) during and after drought in relation to soil properties in a Mediterranean mountain area. Ann For Sci 71:683–695
Ottewell KM, Donnellan SC, Lowe AJ, Paton DC (2009) Predicting reproductive success of insect-versus bird-pollinated scattered trees in agricultural landscapes. Biol Conserv 142:888–898
Ouborg NJ, Vergeer P, Mix C (2006) The rough edges of the conservation genetics paradigm for plants. J Ecol 94:1233–1248
Di Pasquale G, Allevato E, Cocchiararo A, Moser D, Pacciarelli M, Saracino A (2014) Late Holocene persistence of Abies alba in low-mid altitude deciduous forests of central and southern Italy: new perspectives from charcoal data. J Veg Sci 25:1299–1310
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28:2537–2539
Peery MZ, Kirby R, Reid BN, Stoelting R, Doucet-Bёer E, Robinson S, Vásquez-Carrillo C, Pauli JN, Palsbøll PJ (2012) Reliability of genetic bottleneck tests for detecting recent population declines. Mol Ecol 21:3403–3418
Petit RJ, Hampe RJA (2006) Some evolutionary consequences of being a tree. Annu Rev Ecol Evol Syst 37:187–214
Petit RJ, Latouche-Hallé C, Pemonge MH, Kremer A (2002) Chloroplast DNA variation of oaks in France and the influence of forest fragmentation on genetic diversity. For Ecol Manag 156:115–129
Petit RJ, Mousadik AE, Pons O (1998) Identifying populations for conservation on the basis of genetic markers. Conserv Biol 12:844–855
Piedallu C, Dallery D, Bresson C, Legay M, Gégout JC, Pierrat R (2023) Spatial vulnerability assessment of silver fir and Norway spruce dieback driven by climate warming. Landsc Ecol 38:341–361
Piotti A, Leonarduzzi C, Postolache D, Bagnoli F, Spanu I, Brousseau L, Urbinati C, Leonardi S, Vendramin GG (2017) Unexpected scenarios from Mediterranean refugial areas: disentangling complex demographic dynamics along the Apennine distribution of silver fir. J Biogeogr 44:1547–1558
Postolache D, Leonarduzzi C, Piotti A, Spanu I, Roig A, Fady B, Roschanski A, Liepelt S, Vendramin GG (2014) Transcriptome versus genomic microsatellite markers: highly informative multiplexes for genotyping Abies alba Mill. and congeneric species. Plant Mol Biol Rep 32:750–760
Prober SM, Brown AHD (1994) Conservation of the grassy white box woodlands: population genetics and fragmentation of Eucalyptus albens. Conserv Biol 8:1003–1013
Riley SJ, DeGloria SD, Elliot R (1999) A terrain ruggedness index that quantifies topographic heterogeneity. Intermt J Sci 5:23–27
Robledo-Arnuncio JJ, Collada C, Alía R, Gil L (2005) Genetic structure of montane isolates of Pinus sylvestris L. in a Mediterranean refugial area: genetic structure of Scots pine montane isolates. J Biogeogr 32:595–605
Robledo-Arnuncio JJ, Gil L (2005) Patterns of pollen dispersal in a small population of Pinus sylvestris L. revealed by total-exclusion paternity analysis. Heredity 94:13–22
Rovelli E (1995) La distribuzione dell’abete (Abies alba Mill.) sull’Appennino. Monti e Boschi 6:5–13
Rudow A, Westergren M, Buiteveld J, Buriánek V, Cengel B, Cottrell J, De Dato G, Järve K, Kajba D, Kelleher C, Lefèvre F, Liesebach M, Nagy L, Stojnić S, Villar M, Yrjänä L, Bozzano M (2020) Decision support tool for the management of dynamic genetic conservation units. European Forest Genetic Resources Programme (EUFORGEN) European Forest Institute, Rome, p 104
Santini F, Andrisano T, Leonardi S, Ciaschetti G, Labriola M, Vendramin GG, Piotti A (2018) Tracking the origin of silver fir plantations along the boundary between different genetic clusters in central Apennines: implications for their management. For Ecol Manag 408:220–227
Santos-del-Blanco L, Olsson S, Budde KB, Grivet D, González-Martínez SC, Alía R, Robledo-Arnuncio JJ (2022) On the feasibility of estimating contemporary effective population size (Ne) for genetic conservation and monitoring of forest trees. Biol Conserv 273:109704
Seidl R, Spies TA, Peterson DL, Stephens SL, Hicke JA (2016) Searching for resilience: addressing the impacts of changing disturbance regimes on forest ecosystem services. J Appl Ecol 53:120–129
Seidl R, Thom D, Kautz M, Martin-Benito D, Peltoniemi M, Vacchiano G, Wild J, Ascoli D, Petr M, Honkaniemi J, Lexer MJ, Trotsiuk V, Mairota P, Svoboda M, Fabrika M, Nagel TA, Reyer CPO (2017) Forest disturbances under climate change. Nat Clim Change 7:395–402
da Silva CC, Ribeiro MC, Côrtes MC, Galetti M, Collevatti RG (2015) Contemporary and historic factors influence differently genetic differentiation and diversity in a tropical palm. Heredity 115:216–224
Sork VL, Smouse PE (2006) Genetic analysis of landscape connectivity in tree populations. Landsc Ecol 21:821–836
Steinitz O, Robledo-Arnuncio JJ, Nathan R (2012) Effects of forest plantations on the genetic composition of conspecific native Aleppo pine populations. Mol Ecol 21:300–313
Sánchez-Salguero R, Camarero JJ, Gutiérrez E, González-Rouco FJ, Gazol A, Sangüesa-Barreda G, Andreu-Hayles L, Linares JC, Seftigen K (2017) Assessing forest vulnerability to climate warming using a process-based model of tree growth: bad prospects for rear-edges. Glob Change Biol 23:2705–2719
Tarquini S, Isola I, Favalli M, Battistini A (2007) TINITALY, a digital elevation model of Italy with a 10 m-cell size (Version 1.0), Istituto Nazionale di Geofisica e Vulcanologia (INGV). https://doi.org/10.13127/tinitaly/1.0
Tinner W, Colombaroli D, Heiri O, Henne PD, Steinacher M, Untenecker J, Vescovi E, Allen JRM, Carraro G, Conedera M, Joos F, Lotter AF, Luterbacher J, Samartin S, Valsecchi V (2013) The past ecology of Abies alba provides new perspectives on future responses of silver fir forests to global warming. Ecol Monogr 83:419–439
Unger GM, Heuertz M, Vendramin GG, Robledo-Arnuncio JJ (2016) Assessing early fitness consequences of exotic gene flow in the wild: a field study with Iberian pine relicts. Evol Appl 9:367–380
Urbinati C (2014) Final report of the project: Caratterizzazione genetica, ecologico-strutturale e dendrocronologica dei popolamenti di abete bianco del Parco Nazionale del Gran Sasso e dei Monti della Laga. Università Politecnica delle Marche
Vajana E, Bozzano M, Marchi M, Piotti A (2022) On the inclusion of adaptive potential in species distribution models: towards a genomic-informed approach to forest management and conservation. Environments 10:3
Vakkari P, Blom A, Rusanen M, Raisio J, Toivonen H (2006) Genetic variability of fragmented stands of pedunculate oak (Quercus robur) in Finland. Genetica 127:231–241
Vejpustková M, Čihák T, Fišer P (2023) The increasing drought sensitivity of silver fir (Abies alba Mill.) is evident in the last two decades. J For Sci 69:67–79
Vekemans X, Hardy OJ (2004) New insights from fine-scale spatial genetic structure analyses in plant populations. Mol Ecol 13:921–935
Vendramin GG, Lelli L, Rossi P, Morgante M (1996) A set of primers for the amplification of 20 chloroplast microsatellites in Pinaceae. Mol Ecol 5:595–598
Versace S, Garfì V, Dalponte M, Febbraro Mirko D, Frizzera L, Gianelle D, Tognetti R (2021) Species interactions in pure and mixed-species stands of silver fir and European beech in Mediterranean mountains. Forest 14:1–11
Vescovi E, Kaltenrieder P, Tinner W (2010) Late-glacial and Holocene vegetation history of Pavullo nel Frignano (Northern Apennines, Italy). Rev Palaeobot Palynol 160:32–45
Vilà M, Carrillo-Gavilán A, Vayreda J, Bugmann H, Fridman J, Grodzki W, Haase J, Kunstler G, Schelhaas M, Trasobares A (2013) Disentangling biodiversity and climatic determinants of wood production. PLoS ONE 8:e53530
Vinceti B, Manica M, Lauridsen N, Verkerk PJ, Lindner M, Fady B (2020) Managing forest genetic resources as a strategy to adapt forests to climate change: perceptions of European forest owners and managers. Eur J For Res 139:1107–1119
Vitali V, Büntgen U, Bauhus J (2017) Silver fir and Douglas fir are more tolerant to extreme droughts than Norway spruce in south-western Germany. Glob Change Biol 23:5108–5119
Vitali A, Urbinati C, Weisberg PJ, Urza AK, Garbarino M (2018) Effects of natural and anthropogenic drivers on land-cover change and treeline dynamics in the Apennines (Italy). J Veg Sci 29:189–199
Vitasse Y, Bottero A, Rebetez M, Conedera M, Augustin S, Brang P, Tinner W (2019) What is the potential of silver fir to thrive under warmer and drier climate? Eur J For Res 138:547–560
Walder D, Krebs P, Bugmann H, Manetti MC, Pollastrini M, Anzillotti S, Conedera M (2021) Silver fir (Abies alba Mill.) is able to thrive and prosper under meso-Mediterranean conditions. For Ecol Manag 498:119537
Williamson-Natesan EG (2005) Comparison of methods for detecting bottlenecks from microsatellite loci. Conserv Genet 6:551–562
Wilson MC, Chen XY, Corlett RT, Didham RK, Ding P, Holt RD, Holyoak M, Hu G, Hughes AC, Jiang L, Laurance WF, Liu J, Pimm SL, Robinson SK, Russo SE, Si X, Wilcove DS, Wu J, Yu M (2016) Habitat fragmentation and biodiversity conservation: key findings and future challenges. Landsc Ecol 31:219–227
Winter DJ (2012) MMOD : an R library for the calculation of population differentiation statistics. Mol Ecol Resour 12:1158–1160
Young A, Boyle T, Brown T (1996) The population genetic consequences of habitat fragmentation for plants. Trends Ecol Evol 11:413–418
Acknowledgements
We wish to thank the personnel of the Tuscan-Emilian Apennine National Park and the Gran Sasso–Monti della Laga National Park for providing authorization and logistic support, and Simone Barbarotti, Elena Bianchi, Catia Boggi, Daniele Di Santo, Valeria Gallucci, Stefano Leonardi, Cristina Leonarduzzi, Alma Piermattei, Willy Reggioni, Francesco Renzaglia, Emidia Santini, Stefano Zanzucchi for helping during field campaigns and laboratory activities.
Funding
Open access funding provided by Università Politecnica delle Marche within the CRUI-CARE Agreement. This research was supported by the Rural Development Programme of the Abruzzo Region (2007–2013), Axe 3—measure n. 323 ‘Conservation and restoration of rural heritage’, by the project ‘Caratterizzazione genetica, ecologico-strutturale e dendrocronologica dei popolamenti di abete bianco del Parco Nazionale del Gran Sasso e dei Monti della Laga’ funded by Gran Sasso–Monti della Laga National Park, and by the project ‘Studio della variabilità genetica delle popolazioni di Abete bianco allo scopo di individuare la forma di gestione migliore per evitare la scomparsa dell’habitat prioritario 9220*’ funded by Tuscan-Emilian Apennine National Park under the ‘Direttiva Ministeriale Biodiversità 2018’ of the Italian Ministry of the Environment and Protection of Land and Sea.
Author information
Authors and Affiliations
Contributions
Camilla Avanzi, Alessandro Vitali, Matteo Garbarino and Andrea Piotti contributed to the study conception and design. Material preparation, data collection and analysis were performed by Camilla Avanzi, Alessandro Vitali, Paolo Piovani, Ilaria Spanu, Matteo Garbarino and Andrea Piotti. Funding was provided by Andrea Piotti and Carlo Urbinati. Giovanni Giuseppe Vendramin and Carlo Urbinati supervised the work. The first draft of the manuscript was written by Camilla Avanzi, Alessandro Vitali and Andrea Piotti and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Avanzi, C., Vitali, A., Piovani, P. et al. Genetic consequences of landscape features in two rear edge, highly fragmented metapopulations of a mediterranean conifer. Landsc Ecol 39, 87 (2024). https://doi.org/10.1007/s10980-024-01887-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10980-024-01887-z