
Abstract
The thermal properties together with the identification of the emitted volatiles during heating of the starch-graft-poly(geranyl methacrylate) copolymers with the use of a TG/FTIR-coupled method and some of the physicochemical properties of the copolymers were determined. It was found that the use of the geranyl methacrylate monomer to the graft copolymerization with potato starch allowed to replace ca. 1.46 hydroxyl groups per glycosidic units of starch macromolecule by the poly(geranyl methacrylate) chains under the optimal reaction conditions. Generally, all tested starch graft copolymers exhibited a significant increase in polar solvent resistance, moisture resistance and chemical stability as compared to potato starch. However, the thermal stability of the obtained materials was substantially lower as compared to the thermal stability of potato starch. The beginning of the decomposition of the copolymers was observed below 150 °C. It was due to low thermal stability of the poly(geranyl methacrylate) chains. The decomposition of the prepared materials runs at least four, unseparated stages. The first stage was visible up to 220–240 °C. It was connected with the emission of some aldehyde, acid, alcohol, alkene, ester fragments, H2O and CO2 as a result of the depolymerization, destruction and partial decarboxylation of the poly(geranyl methacrylate) chains. The second stage was spread between ca. 220–240 and 358–375 °C. The emission of organic, saturated, unsaturated, aromatic, oxygen-rich fragments, CO, CO2 and H2O as a result of the decomposition and dehydration of starch was confirmed. Heating of the studied materials between 358–375 and 455–477 °C resulted in subsequent decomposition processes of the residues and the creation of some oxygen-rich saturated and unsaturated fragments, CO, CO2, H2O and CH4. Finally, above 455–477 °C, a minor mass loss as a result of the decomposition processes of the residues formed before was observed. The emission of CO, CO2, H2O, CH4 and some oxygen-rich saturated and unsaturated fragments was confirmed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The chemical modification of native starch with some methacrylate monomers in the graft copolymerization processes is widely studied. It is a simple way to change the properties of starch. Because of native starch properties such as low stability to changes in temperature or pH, high moisture absorption, high swelling in polar solvents or ability to hydrolysis and gelation, its application is often limited in some industrial processes [1,2,3,4,5]. The graft copolymerization allows replacing hydrophilic type groups by hydrophobic type groups or the incorporation of other functional groups to macromolecule of starch. Thus, the significant changes in starch behavior can be generated [6,7,8,9,10]. The radical graft processes may be initiated using different initiating systems. For example, Singh et al. [11] have studied the course of the graft process of starch with acrylonitrile in the presence of peroxydisulfate and under microwave irradiation. In turn, Spychaj et al. [12] have described the preparation of the starch-grafted-N-vinylformamide copolymers manufactured by a reactive extrusion in the presence of radical initiators such as 2,2′-azobis(2-methylpropionamidine)dihydrogen chloride or 2,2′-azobis(isobutyronitrile). The application of cerium (IV) complexes to the graft process of methyl methacrylate, glycidyl methacrylate, butyl acrylate, acrylonitrile, acrylic acid, N,N-diethylaminoethyl methacrylate was also reported [13,14,15,16]. Benzoyl peroxide was successfully used as a thermal initiator to the production of the starch-g-poly(acrylic acid), starch-g-poly(methyl methacrylate) or starch-g-polystyrene copolymers [17,18,19,20]. The graft reaction of vinyl alcohol, acrylic acid, butyl acrylate and starch in the presence of Fenton’s reagent and the graft copolymerization of styrene under irradiation or in the presence of enzymes were also well known [21,22,23,24,25,26].
Meanwhile, potassium or ammonium persulfates are mainly used to initiate the radical “grafting from” processes of a different structure monomer and starch. Among monomers, one can mention a different alkyl chain length methacrylate, styrene, acrylamide, benzyl phenyl methacrylates, etc. [27,28,29,30,31]. Depending on the initiating system, monomer structure and other reaction conditions (time, temperature), the graft copolymers with a different structure and the grafting parameters can be obtained. This type of the graft process includes three steps such as initiation, propagation and termination. Initially, starch macroradicals are formed by a hydrogen transfer to initiator. The created active sites of starch are easily added to the double bonds of monomer to form the starch graft monomer radical which reacts with other monomer molecules to produce the graft copolymer. However, the termination step of the graft copolymerization mainly occurs by the combination or disproportionation processes of two reactive chains. It should be noted that some amount of the non-grafted homopolymer is also produced, but it can be easily separated from the main product by a Soxhlet extraction method [32, 33].
The main objectives of this paper were to study the thermal behavior and the type of the volatiles emitted during the heating of the novel starch-graft-poly(geranyl methacrylate) copolymers in the presence of inert atmosphere by applying the TG/FTIR-coupled method as well as the determination of the effect of the graft reaction conditions on the course of the graft copolymerization of geranyl methacrylate and potato starch and their some physicochemical properties such as the ability of the obtained copolymers to gelate with water, swelling, moisture and solvent resistance and chemical stability. As far as I know, this type of monomer is not used to the synthesis of the starch graft copolymers, to this time. Therefore, the studies on the evaluation of the mentioned above properties are one of the most important items that allow determining their possible areas of application.
Experimental
Materials
Geraniol (purity of 97%. Fluka, Buchs, Switzerland), methacryloyl chloride (purity of 97%, Fluka, Buchs, Switzerland), triethylamine (purity of 99%, Fluka, Buchs, Switzerland), potato starch (purity of 98% assessed by the method given in Ref. [34] and extracted and purified from potato flour, Melvit S.A., Poland) [35], potassium persulfate (purity of ≥ 99%, Fluka, Buchs, Switzerland), tetrahydrofuran, ethanol, chloroform, hexane, toluene, butanol, silica gel (Merck, Germany), sodium carbonate, magnesium sulfate, sodium hydroxide, carbon tetrachloride, hydrochloric acid, magnesium sulfate and buffer solutions with pH 5 (disodium hydrogen phosphate/potassium dihydrogen phosphate, Na2HPO4−/KH2PO4), pH 7 (disodium hydrogen phosphate/potassium dihydrogen phosphate, Na2HPO4−/KH2PO4) and pH 9 (sodium tetraborate/boric acid, Na2B4O7/H3BO3) (POCh, Gliwice, Poland) were used as received.
Synthesis of monomer and the copolymers
The first step of the synthesis of the copolymers included the preparation of the geranyl methacrylate monomer in the esterification process of geraniol and methacryloyl chloride in the presence of triethylamine as a catalyst and its purification according to the method presented in Refs. [36, 37]. The structure of monomer was evaluated and described elsewhere [38]. Such prepared monomer was applied to potassium persulfate initiated copolymerization process with potato starch. The graft process was performed according to the procedure described elsewhere [36, 37].
In order to evaluate the optimal reaction conditions, the effect of the reaction parameters on the grafting was studied, as shown in Figs. 1–4. The grafting parameters such as the grafting percent (G), grafting efficiency (GE), homopolymer percent (H) and overall conversion of monomer (TC) were evaluated according to the following equations:
where m0—the mass of geranyl methacrylate, m1—the mass of starch, m2—the mass of poly(geranyl methacrylate), m3—the mass of the grafted poly(geranyl methacrylate), and m4—the mass of the crude copolymer before the homopolymer extraction.

Influence of the potato starch-to-geranyl methacrylate ratios on the grafting course (the reaction conditions: temperature: 70 °C, time: 150 min, initiator concentration: 2.0% and rmp: 300)
Methods
The structures of the obtained materials were confirmed by the 13C CP/MAS NMR and FTIR methods. In a typical 13C CP/MAS NMR tests, the copolymers were analyzed at the resonance frequency 75.5 MHz (Bruker Avance 300 MSL apparatus). However, the FTIR measurements were taken using KBr disk technique over the region 600–4000 cm−1 and 4 cm−1 resolution (FTIR Tensor 27, Bruker apparatus).
The surface morphology of the copolymers was evaluated using a high resolution, low-vacuum SEM/FIB, the Quanta 3D FEG system (FEI apparatus) at magnification of ×3000 and ×8000.
The selected physicochemical properties such as the ability of the obtained copolymers to gelate with water, swelling in different polarity solvents, moisture resistance, chemical resistance, thermal properties and the decomposition mechanism in the presence of inert atmosphere were tested according to the procedures described below:
the ability of the obtained copolymers to gelate with water when providing heating: the sample (mass ca. 9 mg) composed of distilled water (ca. 6 mg) and the copolymer (ca. 3 mg) was stored at room temperature for 24 h to equilibrate. Then, it was heated in the aluminum pan from 20 to 100 °C with the heating rate 10 °C min−1 (DSC Phoenix 204 apparatus, Netzsch, Germany);
the swelling in different polarity solvents: into the measuring cylinders, 1 cm3 of the samples was poured. The cylinders were filled up to 10 cm3 with the chosen solvents (water, ethanol, butanol, toluene, hexane and carbon tetrachloride). The tests were done at 25 °C in triplicate. After equilibration, the final volumes of the samples were read. The swelling degree (B, %) of the samples was evaluated using the following equation [39]:
$$ B = \left( {V_{1} - V_{0} } \right)/V_{0} \times 100\% $$where V0—the initial, apparent volume of the sample (1 cm3), V1—the final, apparent volume of the swollen sample;
the moisture resistance: ca. 100 mg of the sample was placed into a desiccator and exposed to a water vapor for saturation. The water vapor was produced by a container of liquid water within the desiccator. The tests were done at 25 °C and at the relative humidity (RH) 100%. Each measurement was repeated three times. After saturation, the masses of the samples were evaluated, gravimetrically. The moisture percent absorption (WA, %) was computed from the equation [40]:
$$ {\text{WA}} = m_{1} - m_{0} /m_{0} \times 100\% $$where m0—the initial mass of the sample (ca. 100 mg), m1—the final mass of the sample;
the chemical resistance: ca. 100 mg of each sample was put into the chosen chemical environment (1 mol dm−3 NaOH, buffer solutions with pH 5, 7 and 9 and 1 mol dm−3 HCl) and kept at 25 °C. To each sample, 20 cm3 of the selected solution was added. The tested copolymers were removed from a solvent, dried and weighted weekly. The tests were terminated when the mass of the residues reached a constant value. The residue remaining from the chemical stability tests was filtered, washed with distilled water to neutrality, dried at 60 °C and weighted. Each measurement was repeated three times. The chemical stability (WL, %) of the copolymers was evaluated from the following equation [41]:
$$ {\text{WL}} = \left( {m_{1} - m_{2} } \right)/m_{1} \times 100\% $$where m1—the initial mass of the sample (ca. 100 mg), m2—the final mass of the sample;
the thermal properties: the sample (ca. 10 mg) was heated in the Al2O3 crucible from 40 to 1000 °C with the heating rate 10 °C min−1 and in the presence of inert atmosphere (helium, flow rate 40 cm3 min−1) with the use of a STA 449 Jupiter F1 instrument (Netzsch, Germany);
the type of the gaseous decomposition products emitted under the heating of the copolymers in inert atmosphere was evaluated using a FTIR TGA 585 analyzer (Bruker, Germany) coupled on line to a STA 449 Jupiter F1 instrument. The gaseous FTIR spectra were collected in the range of 600–4000 cm−1 with a resolution 4 cm−1.
Results and discussion
Influence of the reaction conditions on the graft process
Figures 1–4 show the influence of different reaction conditions on the course of the graft process. According to the presented results, the grafting percent (G) values increase but the H, GE and TC values gradually drop as starch-to-monomer ratio increases from 1:0.25 to 1:1.75. The results presented in Fig. 2 indicate that the G and GE values increase as an initiator concentration increases from 0.5% (mass fraction, %) up to 2.0%. When an initiator concentration exceeds 2.0%, the drop of the H, G and TC values is observed. Moreover, one can clearly see that the G, H and TC values obtain almost constant values for an initiator concentration higher than 2.0%. As the reaction temperature grows up to 70 °C, the G, GE and TC values increase (Fig. 3). Meanwhile, the H values significantly decrease. However, the drop of the G, GE and TC values at temperatures higher than 70 °C was seen.

Influence of an initiator concentration on the grafting course (the reaction conditions: temperature: 70 °C, time: 150 min, starch-to-monomer ratio: 1:1.75 and rmp: 300)

Influence of the reaction temperature on the grafting course (the reaction conditions: time: 150 min, initiator concentration: 2.0%, starch-to-monomer ratio: 1:1.75 and rmp: 300)
As the reaction time increases from 30 to 150 min (Fig. 4), the GE, G and TC values also increase but the H values considerably fall. The reaction time higher than 150 min causes the drop of the GE, G and TC values. The obtained results indicate that as the reaction time increases the extent of the propagation of the copolymerization is expected. This leads to the addition of a great number of monomer units to the growing graft chains.

Influence of the reaction time on the grafting course (the reaction conditions: temperature: 70 °C, initiator concentration: 2.0%, starch-to-monomer ratio: 1:1.75 and rmp: 300)
All the presented results show that the optimal reaction conditions for the grafting process of geranyl methacrylate onto potato starch are the following: starch-to-monomer ratio 1:1.75, an initiator concentration 2.0%, the reaction temperature 70 °C and the reaction time 150 min. In these conditions, the rate of the graft process is faster than the rate of the homopolymerization due to the optimal concentration of monomer, initiator and the optimal temperature and reaction time. It may be supposed that under the optimal conditions, the rate of the decomposition of an initiator into radicals is fast. Thus, the creation of macroradicals from starch which can react with monomer molecules is also fast. In addition, the viscosity of the reaction medium is suitable for the fast migration between the reagents, and thus its higher possibility to collision is expected.
Characterization of starch-g-poly(geranyl methacrylate) copolymers
The FTIR and 13C CPMAS/NMR
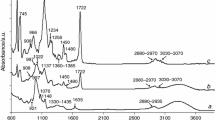
The results obtained from the spectroscopic analyses which confirmed the structures of the prepared starch-g-poly(geranyl methacrylate) copolymers were presented below:
FTIR (KBr disk, cm−1): 3340 (ν OH), 2955 (ν C–H), 2918 (ν C–H), 2878 (ν C–H), 1720 (ν C=O), 1670 (ν C=C), 1445 (δ C–H), 1375 (δ C–H), 1230 (ν C–O), 1146 (ν C–O), 1077 (ν C–O), 1014 (ν C–O), 998 (ν C–O), 832 (γ = C–H), 750 (γ = C–H);
13C CPMAS/NMR (75 MHz, δ ppm): 174.2 (C=O), 100.6 (CH–O), 92.8 (CH–O), 82.6 (CH–O), 71.5 (C–O), 61.1 (CH2–O), 141.8 (=C), 135.7 (=C), 123.9 (=CH), 116.5 (=CH), 37.5–27.4 (CH2), 25.3 (CH3), 22.7 (C–CH3). 21.2 (CH3), 18.5 (CH3), 16.4 (CH3).
The structure morphology
The SEM analyses of the obtained starch-g-poly(geranyl methacrylate) copolymers confirmed their heterogenous, porous surface morphology (Fig. 5). The formation of agglutinated aggregates with a lot of irregular holes was well seen. According to the SEM, the diameter of the holes was between 0.23 and 0.91 μm.

SEM imagines at magnification 3000 times (a) and 8000 times (b) for the obtained copolymers (copolymer4 as an example)
The physicochemical properties
In order to evaluate the physicochemical properties of novel materials, the copolymers with the following G values were selected: 19.8% ± 0.4 (copolymer1), 29.6% ± 0.6 (copolymer2), 38.5% ± 0.4 (copolymer3) and 48.8% ± 0.5 (copolymer4).
On the basis of data presented in Table 1, the obtained starch-g-poly(geranyl methacrylate) copolymers were generally characterized by low swelling in polar solvents. Their swelling decreased as the G values increased. The swelling of the copolymer4 (G = 48.8% ± 0.5) was minimal in polar solvents (water, ethanol and butanol) and did not exceed 8%. The same dependence was observed in the case of the moisture absorption results. All the copolymers absorbed very little moisture as it was well visible from Table 2. The percent of the moisture absorption was between ca. 14.6% (copolymer1) and ca. 5.5% (copolymer4). The grafting process of hydrophobic type monomer (geranyl methacrylate) allowed to the reduction of hydrophilic type groups of starch (hydroxyl groups). As the result, as the G values of the copolymers grew, the formation of hydrogen bonding between copolymer surface and polar solvents and moisture was quite restricted. Nevertheless, very surprising results were obtained from the swelling studies of the copolymers in nonpolar solvents. Due to the incorporation of hydrophobic poly(geranyl methacrylate) chains into starch macromolecule, the copolymers with more hydrophobic structures were prepared. Thus, their swelling in nonpolar solvents supposed to be high. However, the results pointed to their moderate swelling in toluene, CCl4 and hexane. The maximum swelling in these solvents varied between 14 and 15% for the copolymer4 where the maximum of the G value was noted. It was suspected that such low swelling was mainly due to the type of hydrophobic polymer which was covalently linked to starch macromolecule and the length of the graft chains.
The chemical stability of the copolymers directly depended on their G values, (Table 3). The copolymers with lower G values were characterized by lower chemical resistance due to the presence of more, unmodified starch macromolecules which were able to hydrolysis. However, the copolymer4 with the maximum G value exhibited greater chemical stability in neutral, acidic and basic environments as compared to other tested copolymers. The mass loss of the copolymer4 was below 9% in acidic and neutral conditions and below 19% in quite alkaline environment. A high chemical resistance of this material was connected with the type of the polymer bonded with starch macromolecules [38]. According to our previous studies, the poly(geranyl methacrylate) homopolymer was high-resistant material toward different chemical environments [38].
As it was confirmed on the basis of the DSC studies, the obtained copolymers were unable to gelate with water under the applied temperature conditions. The lack of an endothermic peak associated with the gelation process is observed on the DSC curves which show only a straight line in the temperature range being examined. The copolymers were prepared at 70 °C. This temperature was also the gelation temperature of potato starch. As a result, potato starch gelated during the graft process. Further heating with water did not result in the repeated gelation.
The thermal properties
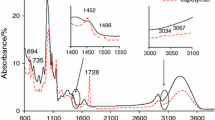
The prepared copolymers decomposed at least four, unseparated temperature ranges under inert conditions (Fig. 6; Table 4). In addition, as it was presented in Fig. 6, the materials were thermally unstable. The beginning of their decomposition happened at temperatures lower than 150 °C. Meanwhile, on the TG curves, a slight mass loss below 100 °C was observed. However, the DTG curves showed a constant increase in the evaporation rate with temperature. This indicated that the adsorbed water was not removed before the decomposition of the samples started. The gaseous FTIR spectrum gathered below Tonset1 (Fig. 7) confirmed a steady, continuous evaporation of the adsorbed water (the absorption bands from ca. 1300 cm−1 to ca. 1800 cm−1 and above 3500 cm−1).

TG/DTG curves for the selected starch-g-poly(geranyl methacrylate) copolymers

Gaseous FTIR spectra gathered at different decomposition temperatures
The first decomposition stage of the studied materials was visible from the beginning of their heating up to ca. 220–240 °C (Tonset2). The determined mass loss from the TG curves was from 18.5 to 34.2%. The first DTG peaks were in the temperature range of 178–190 °C in all the samples (Table 4). According to the gaseous FTIR spectrum collected in this temperature range (Fig. 7), the formation of the mixture of volatiles was observed. It was confirmed by the occurrence of the following absorption bands responsible for the out-of-plane deformation vibrations of (732–980 cm−1), the stretching vibrations of C–O (1114–1268 cm−1), the deformation vibrations of C–H (1367 cm−1 and 1442 cm−1), the stretching vibrations of C=C and/or H–O–H bending in water (1600 cm−1 and 1630 cm−1), the stretching vibrations of C=O (1743 cm−1 and 1800 cm−1), the stretching vibrations of C–H (2700–2973 cm−1), the stretching vibrations of =C–H (3085 cm−1), the stretching vibrations of OH (3250–3720 cm−1) and the bands characteristic for the emission of CO2 (670 cm−1 and 2306–2356 cm−1) [42,43,44,45,46,47,48,49,50]. The presence of those absorption bands may indicate the creation of the fragments from the poly(geranyl methacrylate) chains as a result of its depolymerization, destruction and partial decarboxylation. Consequently, it led to the emission of some aldehyde, acid, alcohol, alkene and ester fragments.
The second and the third decomposition stages were between ca. 220–240 °C (Tonset2) and 358–375 °C (Tonset3). The DTG signal showed the maximum value in the temperature range of ca. 255–285 °C in all the samples. The mass loss was from 34.4 to 24.2%. The gaseous FTIR spectrum collected in this temperature range differed from the previous FTIR spectrum, although similar vibrations were distinguished. The absorption bands from 782 to 941 cm−1 (the out-of-plane deformation vibrations of =C–H and CAr–H), from 1108 to 1282 cm−1 (the stretching vibrations of C–O), the bands at 1357 cm−1 and 1442 cm−1 (the deformation vibrations of C–H), the band at 1630 cm−1 (the stretching vibrations of C=C), the bands from 1743 to 1795 cm−1 (the stretching vibrations of C=O), the bands from 2710 to 2967 cm−1 (the stretching vibrations of C–H), the band at 3014 cm−1 (characteristic for CH4), the band at 3068 cm−1 (the stretching vibrations of =C–H and CAr–H) and the bands within the range 3570–3725 cm−1 (the stretching vibrations of OH) were clearly observed. In addition, the creation of CO (two bands at 2100 cm−1 and 2063 cm−1) and CO2 (bands at 670 cm−1 and 2310–2375 cm−1) was also confirmed. The data indicated the decomposition process of starch combined with its dehydration at this decomposition stage. This led to the formation of some organic, saturated, unsaturated and oxygen-rich fragments, presumably aldehyde, acid, furane, aromatic, alkane and alkene fragments [50,51,52,53].
The next stage occurred from ca. 358–375 °C (Tonset3) up to ca. 455–477 °C (Tend3), with Tmax3 at 404–420 °C and with a mass loss from 23.8 to 29.2%. The FTIR spectrum collected in this temperature range was very similar to the FTIR spectrum gathered at Tmax2. The presence of the out-of-plane deformation vibrations of =C–H (780–985 cm−1), the stretching vibrations of C–O (1210–1310 cm−1), the deformation vibrations of C–H (two bands at 1365 cm−1 and 1455 cm−1), the stretching vibrations of C=C (1635 cm−1), the stretching vibrations of C=O (1720–1790 cm−1), the stretching vibrations of C–H (2760–2980 cm−1), the stretching vibrations of =C–H (3070 cm−1) and the stretching vibrations of OH (3570–3725 cm−1) was confirmed, in addition to the formation of CO (two bands at 2100 cm−1 and 2063 cm−1. The results indicated a subsequent decomposition of the created residues from starch and poly(geranyl methacrylate). As a result, the emission of other alkane, alkene, aldehyde and acid fragments than those observed at Tmax2 was expected.
The heating of the copolymers above Tend3 caused their further, slow decomposition with a mass loss between 1.5 and 2.3%. This mass loss was connected with the emission of mainly inorganic fragments such as CO2 (the bands at 670 cm−1 and 2310–2375 cm−1), CO (two bands at 2100 cm−1 and 2063 cm−1), H2O (the bands above 3500 cm−1), organic fragments such as CH4 (the band at 3014 cm−1) and some oxygen-rich saturated and unsaturated compounds as a result of the decomposition processes of the created residues at lower temperatures. Among them, the formation of some aldehyde and acid fragments was confirmed on the basis of the presence of the stretching vibrations of OH (ca. 3750 cm−1), the stretching vibrations of C–H (2760–2910 cm−1), the stretching vibrations of =C–H (3100 cm−1), the stretching vibrations of C=O (1715–1810 cm−1), the stretching vibrations of C=C (1635 cm−1), the deformation vibrations of C–H (two bands at 1370 cm−1 and 1468 cm−1), the stretching vibrations of C–O (1215–1320 cm−1) and the out-of-plane deformation vibrations of =C–H (720–993 cm−1).
In addition, the TG/DTG data show that the decomposition process of the studied copolymers is not completed under inert conditions. Heating of the copolymers up to 1000 °C resulted in the creation of some carbon residues. The amount of the residues was from 11 to 21% and depended on the G value of the copolymers.
Conclusions
The obtained results indicated that the copolymers with the different G values were prepared. These materials formed the agglutinated aggregates with heterogenous, porous surface morphology. The most interesting and valuable results were received for the copolymer with the maximum G value (48.8% ± 0.5). This copolymer was synthesized under the optimal reaction conditions (starch-to-geranyl methacrylate ratio 1:1.75, potassium persulfate concentration 2.0%, the reaction temperature 70 °C and the reaction time 150 min). The copolymer4 possessed excellent resistance toward polar solvents (water, ethanol, butanol), high resistance to moisture absorption and great chemical resistance in neutral, acidic and basic environments as compared to the properties of other copolymers and unmodified potato starch. Meanwhile, as it was proved, the starch-g-poly(geranyl methacrylate) copolymers were thermally unstable materials. The beginning of their decomposition below 150 °C was observed. It was due to the depolymerization, destruction and partial decarboxylation of the poly(geranyl methacrylate) chains up to 220–240 °C (Tonset2). The heating of the copolymers above 220–240 °C (Tonset2) resulted in the decomposition and dehydration of starch and thus the emission of some organic, saturated, unsaturated, aromatic, oxygen-rich fragments, CO, CO2 and H2O. Above the temperatures of 358–375 °C (Tonset3), mainly the decomposition processes of the residues connected with the emission of some oxygen-rich saturated and unsaturated fragments, CO, CO2, H2O and CH4 were observed.
References
Robyt J. Starch: structure, properties, chemistry, and enzymology. In: Fraser-Reid BO, Tatsuta K, Thiem J, editors. Glycoscience. Berlin: Springer; 2008.
Jane J. Starch properties, modifications, and applications. J Macromol Sci A Pure Appl Chem. 1995;32:751–7.
Alcazar-Alay SC, Meireles MAA. Physicochemical properties, modifications and applications of starches from different botanical sources. Food Sci Technol. 2015;35:215–36.
Li G, Zhu F. Quinoa starch: structure, properties, and applications. Carbohydr Polym. 2018;181:851–61.
Polesi LF, Silveira Sarmento SB, Canniatti-Brazaca SG. Starch digestibility and functional properties of rice starch subjected to gamma radiation. Rice Sci. 2018;25:42–51.
Men Y, Du X, Shen J, Wang L, Liu Z. Preparation of corn starch-g-polystyrene copolymer in ionic liquid: 1-ethyl-3-methylimidazolium acetate. Carbohydr Polym. 2015;121:348–54.
Kumar ThakurV. Biopolymer grafting, synthesis and properties. Amsterdam: Elsevier; 2018.
Hu Y, Tang M. Synthesis of starch-g-lactic acid copolymer with high grafting degree catalyzed by ammonia water. Carbohydr Polym. 2014;118:79–82.
Xiao CM, Tan J, Xue GN. Synthesis and properties of starch-g-poly(maleic anhydride-co-vinyl acetate). Express Polym Lett. 2010;4:9–16.
Worzakowska M. Starch-g-poly(benzyl methacrylate) copolymers. Characterization and thermal properties. J Therm Anal Calorim. 2016;124:1309–18.
Singh V, Tiwari A, Pandey S, Singh SK. Peroxydisulfate initiated synthesis of potato starch-graft-poly(acrylonitrile) under microwave irradiation. Express Polym Lett. 2007;1:51–8.
Spychaj T, Schmidt B, Ulfig K, Markowska-Szczupak A. Starch-grafted-N-vinylformamide copolymers manufactured by reactive extrusion: synthesis and characterization. Polimery. 2012;57:95–100.
Rahman L, Silong S, Zin WM, Ab. Rahman MZA, Ahmad M, Haron J. Graft copolymerization of methyl acrylate onto sago starch using ceric ammonium nitrate as an initiator. J Appl Polym Sci. 2000;76:516–23.
Nagaty A, Abd-Et.-Mouti F, Mansour OY. Graft polymerization of vinyl monomers onto starch by use of tetravalent cerium. Eur Polym J. 1980;16:343–6.
Thanh Tung N, Van Khoi N. Preparation and characterization of starch-g-poly(acrylic acid) copolymers using ceric ammonium nitrate as initiator. J Chem. 2009;47:148–54.
Kalia S, Avérous L. Biopolymers: biomedical and environmental applications. Scrivener: Wiley; 2011.
Pimpan V, Thothong P. Synthesis of cassava starch-g-poly(methyl methacrylate) copolymers with benzoyl peroxide as an initiator. J Appl Polym Sci. 2006;101:4083–9.
Djordjevic S, Nicolic L, Kovacevic S, Milijkovic M, Djordjevic D. Graft copolymerization of acrylic acid onto hydrolyzed potato starch using various initiators. Chem Eng. 2013;57:55–61.
Celik M. Preparation and characterization of starch-g-polymethacrylamide copolymers. J Polym Res. 2006;13:427–32.
Keleş H, Çelik M, Saçak M, Aksu L. Graft copolymerization of methyl methacrylate upon gelatin initiated by benzoyl peroxide in aqueous medium. J Appl Polym Sci. 1999;74:1547–56.
Zhu Z, Zhuo R. Slow release behavior of starch-g-poly(vinyl alcohol) matrix for 2,4,5-trichlorophenoxyacetic acid herbicide. Eur Polym J. 2001;37:1913–9.
Li M, Zu Z, Pan X. Effects of starch acryloylation on the grafting efficiency, adhesion, and film properties of acryloylated starch-g-poly(acrylic acid) for warp sizing. Starch/Starke. 2011;64:826–34.
Mou J, Li X, Wang H, Fei G, Liu Q. Preparation, characterization and water resistance of cationic acetylated starch-g-poly(styrene-butyl acrylate) surfactant-free emulsion. Starch/Starke. 2012;64:826–34.
Fanta GF, Burr RC, Doane WM, Russell CR. Graft polymerization of styrene onto starch by simultaneous cobalt-60 irradiation. J Appl Polym Sci. 1977;21:425–33.
Kiatkamjornwong S, Sonsuk M, Wittayapichet S, Prasassarakich P, Vejjanukroh PC. Degradation of styrene-g-cassava starch filled polystyrene plastic. Polym Degrad Stabil. 1999;66:323–35.
Shogren RL, Willett JL, Biswas A. HRP-mediated synthesis of starch-polyacrylamide graft copolymers. Carbohydr Polym. 2009;75:189–91.
Worzakowska M, Grochowicz M. Effect of some parameters on the synthesis and the physico-chemical properties of new amphiphilic starch-g-copolymers. Carbohydr Polym. 2015;130:344–52.
Qudsieh IYM, Fakhru’l-Razi A, Muyibi SA, Ahmad MB, Ab Rahman MZ, Wan Yunus WMZ. Preparation and characterization of poly(methyl methacrylate) grafted sago starch using potassium persulfate as redox initiator. J Appl Polym Sci. 2004;94:1891–7.
Gao J, Yu J, Wang W, Chang L, Tian R. Graft copolymerization of starch–AN initiated by potassium permanganate. J Appl Polym Sci. 1998;68:1965–72.
Samara SH, Nasr HE, Hebeish A. Synthesis and characterization of starch-poly(vinyl acetate) graft copolymers and their saponified form. J Polym Res. 2005;12:343–53.
Worzakowska M. Thermo-oxidative decomposition behavior of starch-g-poly(citronellyl methacrylate) and starch-g-poly(citronellyl acrylate) copolymers. J Therm Anal Calorim. 2018;132:543–51.
Meimoun J, Wiatz V, Saint-Loup R, Parcq J, Favrelle A, Bonnet F. Modification of starch by graft copolymerization. Starch/Strake. 2018;70:1600351.
Berlin AA, Kislenko VN. Kinetics and mechanism of radical graft polymerization of monomers onto polysaccharides. Prog Polym Sci. 1992;17:765–825.
Krishnaswamy KG, Sreeenivisan A. Separation and determination of the amylose and amylopectin fractions of starch. J Biol Chem. 1948;176:1253–61.
Lim ST, Lee JH, Shin DH, Lim HS. Comparision of protein extraction solutions for rice starch isolation and effects of residual protein content on starch pasting properties. Starch/Starke. 1999;51:410–5.
Worzakowska M, Torres-Garcia E. Kinetics of the pyrolysis process of terpene acrylate homopolymers. Polym Degrad Stabil. 2016;133:227–33.
Worzakowska M. Starch-g-poly(phenyl acrylate) copolymers-synthesis, characterization, and physicochemical properties. Starch/Starke. 2017;69:1700027.
Worzakowska M. High chemical and solvent resistant, branched terpene methacrylate polymers-preparation, thermal properties, and decomposition mechanism. Polym Adv Technol. 2018;29:1414–25.
Tuncel K, Ecevit K, Kenesci K, Piskin E. Nonswellable and swellable ethylene glycol dimethacrylate–acrylic acid copolymer microspheres. J Polym Sci A Polym Chem. 1996;34:45–55.
Pathania D, Sharma R. Synthesis and characterization of graft copolymers of methacrylic acid onto gelatinized potato starch using chromic acid initiator in presence of air. Adv Mater Lett. 2012;3:136–42.
Kaith BS, Singha AS, Grupa SK. Graft copolymerization of flax fibres with binary vinyl monomer mixtures and evaluation of swelling, moisture absorbance and thermal behavior of the grafted fibres. J Polym Mater. 2003;20:195–9.
Sokrates G. Infrared and raman characteristic group frequencies, tables and charts. New York: Wiley; 2001.
NIST Chemistry Webbook, NIST Standard Reference Data, 2011. http://webbook.nist.gov. Accessed 20 Feb 2019.
Sternik D, Gładysz-Płaska A, Grabias E, Majdan M, Knauer W. Study of effect of phosphate and uranium ions on the thermal properties of surfactant-modified natural red clay using TG–FTIR–MS techniques. J Therm Anal Calorim. 2019;136:425–39.
Sidi-Yacoub B, Oudghiri F, Belkadi M, Rodríguez-Barroso R. Characterization of lignocellulosic components in exhausted sugar beet pulp waste by TG/FTIR analysis. J Therm Anal Calorim. 2019. https://doi.org/10.1007/s10973-019-08179-8.
Nunes WDG, Teixeira JA, Nascimento ALCS, Ekawa B, Ionashiro M, Caires FJ. Thermal analysis (TG–DSC, DSC–microscopy and EGA) and characterization of heavy trivalent lanthanides and yttrium isonicotinates. J Therm Anal Calorim. 2019. https://doi.org/10.1007/s10973-019-08112-z.
Pinto BV, Ferreira APG, Cavalheiro ETG. Thermal degradation mechanism for citalopram and escitalopram. J Therm Anal Calorim. 2018;133:1509–18.
Zhu X, He Q, Hu Y, Huang R, Shao N, Gao Y. A comparative study of structure, thermal degradation, and combustion behavior of starch from different plant sources. J Therm Anal Calorim. 2018;132:927–35.
Wang K, Deng J, Zhang Y, Wang CP. Kinetics and mechanisms of coal oxidation mass gain phenomenon by TG–FTIR and in situ IR analysis. J Therm Anal Calorim. 2018;132:591–8.
Worzakowska M. Thermal behavior, decomposition mechanism and some physicochemical properties of starch-g-poly(benzyl acrylate) copolymers. J Therm Anal Calorim. 2016;126:531–40.
Greenwood CT. The thermal degradation of starch. Adv Carbohydr Chem. 1967;22:483–515.
Puddington IE. The thermal decomposition of carbohydrates. Can J Res. 1948;26b:415–31.
Chen X, Xie J, Mei S, He F, Yang H. RDF pyrolysis by TG-FTIR and Py-GC/MS and combustion in a double furnaces reactor. J Therm Anal Calorim. 2019;136:893–902.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Worzakowska, M. The preparation, physicochemical and thermal properties of the high moisture, solvent and chemical resistant starch-g-poly(geranyl methacrylate) copolymers. J Therm Anal Calorim 140, 189–198 (2020). https://doi.org/10.1007/s10973-019-08801-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-019-08801-9