
Abstract
To produce the polymer-powder slurry for injection moulding the thermoplastic polymers such as polypropylene, high-density polyethylene and ethylene-vinyl acetate were used. Depending on skeletal polymers, in every feedstock paraffin wax (PW) was used in the amount of 50% of binder. Application of PW gives the possibility using solvent debinding or thermal debinding at a lower temperature. Then the open porosity into the injected samples reduces the time of thermal debinding of binder residue. Application of one type of skeletal polymer requires using a long isothermal step. The use of a blend of skeletal polymers enables subsequent thermal degradation which consecutively removes skeletal polymers during the heating to the sintering temperature, to minimise the time of isothermal stops. The rheological properties of blends used as a binder and next feedstocks were investigated during mixing in the twin-screw extruder and capillary rheometer. This results gives the information about the possibility to produce feedstocks and then to their injection moulding. Thermal behaviour of samples was determined by differential scanning calorimetry and thermogravimetric analysis to obtain the information about the temperature of mixing the polymers and to propose the thermal debinding cycle.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alloys of iron, cobalt, and titanium alloys including Ti–Ni are the major groups of metal biomaterials. Although they are less biocompatible then ceramic materials, their main advantage is the ability to carry the larger mechanical loads. The test results presented refer to the Co–Cr–Mo alloy. Cobalt-based alloys are a group of metal materials used extensively in dentistry. They are characterized by higher biocompatibility, resistance to pitting and crevice corrosion and the ability to repassivation in physiological solutions than products made of chromium–nickel–molybdenum steel [1, 2].
High biotolerance of the cobalt alloys are caused by the passive layer created on their surface [3,4,5]. The use of chromium as an alloy additive affects the spontaneously forming chromium oxide, which is the main component forming the passive layer. Alloys based on cobalt, chromium and molybdenum, or cobalt, chromium, tungsten and nickel are widely used in dental technology. These materials are used in dentistry for the production of permanent and removable partial denture. Besides, to use in dental prosthetics, they are often used in orthopaedics for the production of hip, ankle and knee joint arthroplasty and the manufacture of plates, wires, tips and bone screws. In addition to biomedical applications, cobalt alloys are used for the production of corrosion-resistant components for high-temperature applications as turbine blades [6].
The analysis of data presented in the literature shows that the best properties for use as biomaterials are sintered cobalt alloys as opposed to casted and forged ones which is related to the segregation of alloying elements during the solidification of liquid metal. As a result of sintering, the consolidation process takes place in a solid state or in the case of local overheating caused by excessive heat, liquid phase sintering occurs. This way of production excludes the possibility of segregation of alloying additives and thus non-homogeneous corrosion resistance.
This article describes the research on biomaterials that have been produced by injection moulding method. The preparation of biomaterials by injection moulding was used for the first time in Europe in 1986 for the manufacture of orthodontic steel claws [7]. This method makes it possible to get ready-made elements with complex shapes that do not allow for other types of moulding. An example may be classic pressing, which is dedicated to the manufacture of simple-form components or hot isostatic pressing requiring additional machining. It would be possible to produce such elements by selective laser sintering (SLS), however, this technology is used for small-scale production.
In this case, the authors proposed a hybrid injection moulding method combined with spraying or low-pressure moulding of top layers for the production of small core/shell-type components of complex shape such as a dental implant. Due to open porosity and a suitable pore size in a top layer (shell) of the proposed implant, it is able to osseointegrate with the bone tissue. Open pores can be used to introduce locally released drugs or other substances that stimulate the process of treatment of the host tissue surrounding the implant. The selection of the suitable binder components and the conditions of injection moulding, degradation and sintering are important in the applied powder injection moulding technology. Properly selected time, temperature, atmosphere of thermal degradation of the binder and subsequent sintering can lead to partial oxidation of the surface layer of the produced part. This process also leads to the formation of open pores in the surface layer without the need for spraying or forming a low-pressure additive layer.
The aim of this research was to produce biomaterials by injection moulding and powder sintering. This technology requires the use of a precisely selected binder which not only plays an important role in injection moulding process but also can not cause contamination resulting from the binder removal process, which would lower the functional properties. Binder degradation products in the injection moulding process often contribute to the sintering process by lowering the solidus temperature of the alloy, e.g., high-speed steel, but excessive carbonation may increase the content of retained austenite [8,9,10]. The part of research results presented in the article mainly concerns the selection of binder components and their share in relation to the powder [11,12,13,14,15,16]. This has a huge impact on the ability to powder injection moulding, followed by the debinding, the sintering process and the final properties of the sinters. In practice, the maximum content of powder is selected based on the torque test of the twin-screw mixer. In the presented work, the maximum content of powder was not tested because the authors’ aim is to produce a feedstock with the lowest viscosity possible, lower than typical thermoplastics such as PP or HDPE. This solution ensures minimal wear of the working surfaces of the injection machine or extruder.
Experimental
For the preparation of biomaterials by injection moulding technology Co–Cr–Mo powder was used. Particle shape and size was tested using scanning electron microscope Zeiss Supra 35 and laser particle size analyser–Analysette 22 MicroTec plus, Fritsch. The density of powder was measured in helium pycnometer AccuPyc 1340, Micrometrics. Powder was mixed with a two-component binder basis on paraffin (PW) and the backbone polymer or blend. Paraffin is used to lower the viscosity of the polymer-powder slurry and possibility use solvent degradation [8]. To improve the wettability of the powder, the effect of the stearic acid (SA) was also tested. The backbone polymer was polypropylene (PP), high-density polyethylene (HDPE), ethylene-vinyl acetate (EVA) copolymer or blends thereof. Regardless of the back bone polymer binder used, the powder volume ratio of the total binder share was up to 64%. The initial selection of the components of the adhesive was based on measuring of the torque of the blades of the mixer Rheomex CTW100p of Haake company which was read both the pure polymers and their blends, and the ready-made polymer-powder slurry (feedstocks) produced at a temperature of 170 °C at a rate of rotary blades 40 r.p.m. At the same time, pure polymers and their blends were tested by differential scanning calorimetry. The experiments were carried out in the Perkin Elmer Diamond calorimeter under nitrogen or air. The test conditions were as follows: heating from 20 to 250 °C with a heating rate of 10 °C min−1 and then cooling at the same rate. To select the thermal degradation conditions of the adhesive, thermogravimetric tests were performed on the Perkin Elmer Pyris 1 thermogravimetric analyzer at a temperature range of 50–600 °C with a heating rate of 10 °C/min under nitrogen or air atmosphere. Rheological experiments were carried out in a capillary rheometer Haake Rheocap S20. The dimensions of diameter and length were respectively 1 and 30 mm To keep an L/D ratio of 30. The shear rate was chosen in a range from 100 to 10,000 s−1 before each test materials were heated during 10 min. In the final stage, the powder together with the binder was mixed in a twin-screw extruder Zamak Mercator. After the first extrusion, the slurry was poured twice more into the extruder for accurate homogenisation. During extrusion, the screwing torque was controlled. Stabilization of the curve showing the screwing moment of the screws guarantees a high homogeneity of the polymer-powder blend. The homogenised mixture was then injected by piston injection moulding machine into a mould with the shape of a rectangular beam. The production of the beams allows performing bending strength tests after injection moulding and sintering. The binder molded in injection-moulded form was first subjected to thermal degradation followed by solvent degradation in pure heptane at 20–40 °C. This process facilitates thermal degradation, the cycle of which is based on a thermogravimetric curve. Both thermal degradation and sintering were performed in a pipe furnace of Czylok company in an atmosphere of flowing gas mixture Ar-10%H2 with the rate 0.1 l/min. The maximum heating rate did not exceed 5 °C per minute, whereas when heated to a temperature of thermal degradation was 1 °C min−1. Sintered at a temperature of 1260 °C beams were subjected to density testing by the hydrostatic method, hardness in the Vickers microhardness tester Future-Tech FM-700 with a load of 100 g and mechanical strength by three-point bending method in a Zwick/Roell Z020 testing machine. Studies of the microstructure were performed in a scanning electron microscope Zeiss Supra 35 with an EDS detector with a predetermined accelerating voltage of 20 kV.
Results and discussion
SEM analysis showed that the Co–Cr–Mo powder used for powder injection moulding has a spherical shape (Fig. 1), which is advantageous due to the wettability of the binder. The average particle size tested in a laser particle size analyser is 24.94 μm. The particle size distribution of the powder along with the cumulative curve is shown in Fig. 2.

Morphology of Co–Cr–Mo alloy powder

Cumulative curve and particle size distribution
Apart from its shape, distribution of particle size is also an important factor. If the particle size distribution characteristics of the investigated powder is relatively wide, the pores originating between big particles may be filled with small particles, which is attested by the curve slope angle coefficient Sw [11].
Powder with the Sw coefficient of ca. 2 is most recommended for powder injection moulding. Moulding of powder with the Sw coefficient equal to 7 is not recommended, as for this powder the particle size distribution characteristics is very narrow. The Sw coefficient of used powder is 2.68. The powder density measured using pycnometer and calculated from the five measurements is 8.33 g cm−3.
To assess the behaviour of the basic binder components, i.e., PP, HDPE and EVA polymers during homogenization with powder, a torque test of the screws was performed during the plasticization of pure polymers at 170 °C. Only pure HDPE was mixed additionally at temperature 180 °C to test the influence of the higher processing temperature on torque. All the investigated blends and feedstocks are placed in Table 1.
During the filling of the chamber of the device within the first few minutes increase and instability of the torque is noticeable. After plasticising, the lowest torque value of the screws reaches HDPE and is 1.1 Nm. This value starts to increase after 22 min, and in the final phase of the study, after 60 min, it increases by about 100%, all the time maintaining an upward trend. This test was repeated at a temperature of 180 °C and observed an even stronger tendency increasing of torque. The torque starts to increase after about 17 min and after about an hour is about 200% higher (Fig. 3). Therefore, increase in temperature by 10 °C is important in the plasticization of HDPE despite the fact that there is no pure thermal degradation as shown by the TGA results shown in the following figures. This effect may be caused by mechanical damage of the polymer chains of HDPE, and further re-combination and crosslinking. Of course, it is a thermally supported process, as has been demonstrated by the test. Analyzing the curve showing the torque during PP plasticization in 1 h, it can be observed continuously decreasing to a minimum value 1.3 Nm after 60 min of the test (Fig. 4). By studying EVA polymer, the minimum torque value was reached after 10 min of plasticization process and stayed constant throughout the test.

Influence of mixing time at temperature 170 or 180 °C on the torque of HDPE

Influence of mixing time on the torque of PP and EVA
On the basis of pure polymer analysis, it can be concluded that the use of HDPE as the main component of the binder, especially at long homogenization times, can increase the torque and viscosity of the polymer-powder slurry. For this reason, EVA and PP polymers should also behave better when blended with the powder. The next step was to study the behaviour of the blend of two polymers and then all three major binder components. A multi-component binder system is preferred during thermal degradation, because individual polymers degrade at different temperatures, allowing degradation during heating itself or with short thermal arrests. In conclusion, this may facilitate the process of degradation of the binder and shorten its time. In each case, the volumetric proportion of the individual polymers was equal to 50/50 or 33/33/33%. After blending the PP and EVA polymers, the torque value reaches 1.7 Nm after about 10 min and is stable throughout the study as with pure EVA. In the case of EVA and HDPE polymer blend, it can be seen that the growing tendency is similar to pure HDPE. The increase of torque starts after approx. 30 min of plasticization (Fig. 5). In a blend of PP and HDPE, polypropylene is more dominant because the torque decreases throughout the test like in the case with pure PP (Fig. 6). Mixing all three polymers causes that during plasticization process the value of the torque initially decreases and then rapidly increases after about 40 min. In this way, we get the same effect as for pure HDPE and HDPE/EVA but delayed.

Influence of mixing time on the torque of blends EVA/PP and HDPE/EVA

Influence of mixing time on the torque of blends HDPE/PP and HDPE/EVA/PP
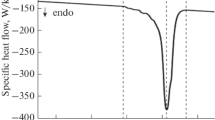
Very important information in the processing of polymers and their blends is to determine their melting temperature, which allows DSC analysis. Figure 7 shows the results of the test of pure polymers and their transition temperature to plasticity. Polymer EVA is characterised by the lowest melting temperature which is 85 °C. The value of this temperature is higher for other polymers, and it is 133 °C for the HDPE polymer and 160 °C for PP polymer. Mixing the two basic polymers HDPE/PP and EVA/PP (Fig. 8) does not significantly affect the changing temperature of transition in a plastic state.

DSC heating curves of pure components

DSC heating curves of HDPE/PP and EVA/PP blends
For both HDPE and PP, these changes are minimal and are about 1 °C. In the case of a blend HDPE/EVA/PP (Fig. 9), no significant changes in the temperature of the plastic transition temperature are observed, for example for PP the temperature peak changes only to the value of 154 °C. Despite the low EVA melting temperature, this polymer does not affect the plastification of PP or HDPE and their melting temperature. The enthalpy of the conversion to the plastic state of the EVA polymer is too small in this case so it is difficult to accurately interpret the melting temperature of this polymer in the blend of HDPE and PP. The same figure shows a blend of EVA/PP/PW where paraffin significantly reduces the melting point of PP to 150.8 °C, which results from the effect of PP plasticization by paraffin. Paraffin also causes plasticization and reduces the melting temperature of the HDPE [14].

DSC heating curves of HDPE/EVA/PP and EVA/PP/PW blends
To understand the viscosity of the polymers used, their blends and polymer-powder thicknesses, and rheological tests were carried out using a capillary rheometer. This type of rheometer enables the viscosity test at the high shear rates typical of injection moulding. The test performed in the shear rate range from 100 to 10,000 s−1 showed that for all polymers, the viscosity is below 1000 Pa s and decreases with the shear rate. This proves that these polymers are dilutions during shared and are suitable for injection moulding (Fig. 10).

Influence of shear rate on the viscosity of pure polymers at 180 °C
Blending basic polymers, e.g., PP/EVA, allows obtaining a blend with an intermediate viscosity between the viscosity of pure polymers. For example, the viscosity of PP and EVA polymers at a shear rate of 1000 s−1 is 78 and 119 Pa s, while the viscosity of the blend at the same shear rate and the temperature is 85 Pa s. The temperature is a significant influence on the viscosity of pure polymers and their mixtures. However, the increase in temperature does not affect the viscosity as much as the addition of paraffin. Figure 11 shows the effect of the test temperature on the viscosity of the PP/EVA blend in the same shear rate as before.

Influence of shear rate on viscosity of PP/EVA blend at the different temperature
Figure 12 shows the effect of the paraffin addition on the viscosity of HDPE and powder. The addition of paraffin strongly reduces the viscosity of the polymer mixture, which means that even the introduction of 64% powder into the HDPE/PW blend does not increase the viscosity to the level of pure HDPE.

Influence of shear rate on viscosity of polymer, blend and feedstock on the basis of HDPE at 180 °C
Also, the addition of stearic acid strongly reduces the viscosity of the mould charge. Figure 13 shows the effect of stearic acid on the viscosity of a polymer-powder slurry containing as much as 68% by volume of powder and a binder in the form of an EVA/PW mixture. At a shear rate of 1000 s−1, the viscosity is about 24 Pa s lower compared to the mixture without the addition of stearic acid. Thus, the composition of the polymer-powder mixture selected in this way may subsequently be more easily injection moulded.

Influence of shear rate and stearic acid on the viscosity of feedstocks at 180 °C
In addition to the melting temperature obtained from DSC, the temperature of the beginning of thermal degradation of the individual binder components is also important. Its selection based on the TGA curve is important for two reasons. The first is the conditions of mixing, extrusion and injection moulding. Too high-processing temperature causes degradation or partial crosslinking of which is likely in the case of HDPE. Particularly important is the correct selection of the thermal degradation conditions of the binder, i.e., time, heating rate, temperature and atmosphere. The results of the TGA curve of pure PP, EVA and HDPE polymers are shown in Fig. 14.

TGA curves of pure components
Due to the fact that the paraffin was subjected to solvent degradation, TGA tests for pure paraffin were not carried out. These results are presented in the article [8], where the temperature of the beginning of PW degradation is 198 °C. Two characteristic temperatures on the thermogravimetric curve of the EVA result from the fact that it is a copolymer of ethylene and vinyl acetate. The beginning of the degradation of one of the components of this copolymer occurs at a temperature of 300 °C, while the second component of the copolymer degradation takes place at a temperature of 444 °C. The end of thermal degradation occurs at 540 °C, which is higher than the temperature of the end of degradation of polypropylene and HDPE. This is advantageous in the case of injection-moulded parts since the task of this polymer is to keep the shape at a high temperature whose value should be close to the sintering temperature. By analysing the TGA curves of the polymer blends presented in Fig. 15, it can be seen that the temperature range of degradation of the PP/EVA blend is narrowed. The degradation start begins at a temperature higher than in the case of pure EVA polymer and reaches about 330 °C. The end of degradation corresponds to a temperature of 490 °C which is lower in the case of EVA polymer. Similarly, the EVA degradation temperature range decreases when mixed with PP and HDPE, which also presents the thermal degradation curve in Fig. 15.

TGA curves of blends
This test was done in an air atmosphere. For comparison, EVA thermal degradation was also performed in a pure nitrogen atmosphere (Fig. 16), which caused that the beginning of degradation temperature. The refraction point of the 444 °C curve did not change, but the temperature of the end of degradation shifted even to 590 °C, which is very beneficial. Unfortunately, due to a necessity of lowering the viscosity, the ingredient such as paraffin has been used. After the addition of paraffin to previously prepared blends, the temperature of the beginning and the end of the degradation decreased significantly, taking the value of 200 and 485 °C for the blend PP/EVA/PW (Fig. 16).

TGA curves of pure EVA under Air, N2 and blends with EVA
The course of the thermal degradation curve is also strongly influenced by the addition of powder to the blend. For example, the powder/PP/PW blend undergoes final thermal degradation at a temperature of about 400 °C, i.e., about 100 °C lower than pure PP. Thus, the analysis of TGA curves of pure polymers only makes sense if we want to examine the effect of the polymer powder ingredients on the value of the degradation temperature of the blend. However, it is not possible to choose the thermal degradation conditions of ready injection-molded components based on the TGA curve of pure polymers or its blends. More reliable information is provided by TGA of feedstocks (Fig. 17).

TGA curves of feedstock
Conclusions
Based on the results of the research, the following conclusions were formulated:
-
1.
Analyzing changes in the torque during plasticization of pure polymers and their blends used as a binder, it can be concluded that high-density polyethylene is the worst of the adhesives tested due to the increasing torque of both pure HDPE and blend EVA/HDPE and EVA/HDPE/PP. In addition, polyethylene is sensitive to the temperature increase, which, like the plasticising time, increases the torque. In the case of pure PP and blends of HDPE/PP, the nature of the curve is declining, based on which it can be assumed that the polymer chains become fragmented. Whereas the most stable polymer is a mixture of EVA and EVA/PP.
-
2.
In addition to the high stability of the torque of the EVA copolymer, undoubtedly the advantageous property in relation to HDPE and PP is its low melting point, amounting only to 85 °C. It’s also allows to significantly reduce the homogenization temperature of the polymer-powder mixture and the costs associated with its production. In addition, DSC studies have shown that HDPE/PP, EVA/PP and HDPE/EVA/PP blends have quite stable melting points of individual components. The melting point of HDPE or PP used in the blend drops only by about 1 °C relative to the pure polymer. Only the addition of paraffin has a stronger effect on the plasticisation of the main ingredient of the blends. For example, in the blend of EVA/PP/PW melting point PP is 150.8 °C and is lower than for pure PP by approx. 10 °C.
-
3.
Rheological investigation of pure polymers and their blends showed a relatively low viscosity decreasing with increasing shear rate. In addition, the increase in temperature causes thinning of polymers and their blends. Examination of pure polymers showed that HDPE has the highest viscosity among the basic components of the binder. Only in the shear rate range up to 200 s−1, the EVA is characterized by a higher viscosity. To preserve the low viscosity of the polymer-powder slurry, it is necessary to use a highly plasticizing component such as PW. In this way, even mixtures with a high powder content of up to 68% are characterized by lower viscosity than pure polymers. In addition, the viscosity of the slurry can be further reduced by covering the powder with a surfactant such as stearic acid.
-
4.
The thermogravimetric test results show a narrow range of homopolymer degradation, and the use of HDPE/PP blends does not change much. The use of the EVA copolymer tends to be more interesting due to the wide temperature range thermal degradation, which depends also on the atmosphere used. In the case of metal powder, it is necessary to use a protective atmosphere. This results in shifting the end of degradation to a higher temperature, which is desirable in view of maintaining the shape of the manufactured element to the sintering temperature. The use of PW also makes it possible to extend the temperature range of degradation mainly by lowering the temperature of the beginning of the binder degradation. It also allows the use of solvent degradation, which initially opens the pores throughout the volume of the element and facilitates thermal degradation. The use of paraffin as well as paraffin and metal powder in the finished polymer-powder mixture reduces the final temperature of degradation of the binder in relation to the temperature of the pure homopolymer or copolymer, which is disadvantageous. For example, the thermogravimetric curve of the finished polymer-powder slurry powder/HDPE/PP/PW/SA showed the final temperature of the binder degradation lower by approx. 25 °C relative to the final degradation temperature of the HDPE/PP blend.
-
5.
The tested polymer-powder slurry is undoubtedly characterised by good rheological properties, and the performed DSC and TGA tests enable the selection of conditions for the homogenization of mixtures and degradation of the binder, respectively. The presented results indicate that it is important to study both individual components of the binder, i.e., pure polymers, copolymer and blends and polymer-powder mixtures, to determine their suitability for use in the powder injection moulding process. The main goal of the research is to choose the components of the polymer-powder slurry that will enable its formation, quick and easy degradation of the binder, which does not leave residual carbon that lowers the properties of the finished implant. However, determining the effect of the binder on the properties of the finished sinter requires further testing of this material.
References
Enderle JD, Bronzino JD. Introduction to biomedical engineering. Burlington: Academic Press; 2012.
Reimann Ł. Electrochemical characteristics of a cobalt alloy with a protective passive layer. Arch Metall Mater. 2016;61(3):1283–90.
Dobruchowska E, Paziewska M, Przybyl K, Reszka K. Structure and corrosion resistance of Co–Cr–Mo alloy used in Birmingham Hip Resurfacing system. Acta Bioeng Biomech. 2017;19(2):31–9.
Alrabeah GO, Bretta P, Knowles JC, Petridis H. The effect of metal ions released from different dental implant-abutment couples on osteoblast function and secretion of bone resorbing mediators. J Dent. 2017;66:91–101.
Dourandish M, Godlinski D, Simchi A, Firouzdor V. Sintering of biocompatible P/M Co–Cr–Mo alloy (F-75) for fabrication of porosity-graded composite structures. Mater Sci Eng, A. 2008;472:338–46.
Luna Ramírez A, Porcayo-Calderon J, Mazur Z, Salinas-Bravo VM, Martinez-Gomez L. Microstructural changes during high temperature service of a cobalt-based superalloy first stage nozzle. Adv Mater Sci Eng. 2016. https://doi.org/10.1155/2016/1745839.
Petzoldt F. Metal injection moulding in Europe: ten facts that you need to know. Powder Inject Mould Int. 2007;1(2):23–8.
Dobrzański LA, Matula G, Varez A, Levenfeld B, Torralba JM. Fabrication methods and heat treatment conditions effect on tribological properties of high speed steels. J Mater Process Technol. 2004;157–158:324–30.
Herranz G, Matula G, Romero A. Effects of chromium carbide on the microstructures and wear resistance of high speed steel obtained by powder injection moulding route. Powder Metall. 2017;60(2):120–30.
Matula G, Dobrzanski LA, Varez A. Comparison of structure and properties of the HS12-1-5-5 type highspeed steel fabricated using the pressureless forming and PIM methods. J Mater Process Technol. 2005;162:230–5.
Hamidi MFFA, Harun WSW, Samykano M, Ghani SAC, Ghazalli Z, Ahmad F, Sulong AB. Review of biocompatible metal injection moulding process parameters for biomedical applications. Mater Sci Eng, C. 2017;78:1263–76.
Dehghan-Manshadi A, Bermingham MJ, Dargusch MS, StJohn DH, Qian M. Metal injection moulding of titanium and titanium alloys: challenges and recent development. Powder Technol. 2017;319:289–301.
Thomas-Vielma P, Cervera A, Levenfeld B, Varez A. Production of alumina parts by powder injection molding with a binder system based on high density polyethylene. J Eur Ceram Soc. 2008;28:763–71.
Hryha E, Borgstrom H, Sterky K, Nyborg L. Influence of the steel powder type and processing parameters on the debinding of PM compacts with gelatin binder. J Therm Anal Calorim. 2014;118:695–704.
Yılmaz E, Gökçe A, Findik F. Assessment of Ti–16Nb–xZr alloys produced via PIM for implant applications. J Therm Anal Calorim. 2017. https://doi.org/10.1007/s10973-017-6808-0.
Sotomayor ME, Krupa I, Várez A, Levenfeld B. Thermal and mechanical characterization of injection moulded high density polyethylene/paraffin wax blends as phase change materials. Renew Energy. 2014;68:140–5.
Acknowledgements
This work was financed by the Ministry of Science and Higher Education of Poland as the statutory financial Grant of the Faculty of Mechanical Engineering SUT.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Matula, G., Tomiczek, B., Król, M. et al. Application of thermal analysis in the selection of polymer components used as a binder for metal injection moulding of Co–Cr–Mo alloy powder. J Therm Anal Calorim 134, 391–399 (2018). https://doi.org/10.1007/s10973-018-7543-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-018-7543-x