
Abstract
Commercial strong anion exchange resins provided by Purolite, namely MTA4801PF, and MTA4601PFSO4, were utilized for extracting uranium from sulfate leach liquor. The study focused on investigating the kinetics, isotherms, and thermodynamics of the sorption process. Both resins demonstrated similar kinetics and isotherm performance, with sorption capacities of approximately 66 and 71 mg g−1 respectively. The sorption process was proven to be endothermic, feasible, and spontaneous, and U(VI) desorption of around 95% was achieved using 0.5 M sulfuric acid. Commercial resins showed excellent sorption performance when applied to sulfate leach liquor of Egyptian ores despite the complexity of the solution.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Nuclear energy, known for its clean and renewable features, has experienced significant growth and widespread adoption worldwide [1]. However, the use of uranium, the primary fuel for nuclear energy, results in the generation of substantial amounts of uranium-containing wastewater during uranium mining and utilization processes, posing an urgent environmental pollution challenge that requires resolution [2, 3]. Hence, there is an immediate need to explore efficient methods for extracting U(VI) from liquid solutions. This includes solutions produced during uranium ore mining and treating, as well as those found in tailing deposits and lake waters, which exemplify potential uranium sources [3].
Several approaches are now being used to extract U (VI) from liquid solutions [4]. These ways encompass chemical precipitation [5, 6], ion exchange [7], solvent extraction [1, 8], catalytic processes [9, 10], and sorption techniques [11,12,13,14,15].
Adsorption is currently the most effective method for separating uranium ions from aqueous solution due to its several advantages over other techniques. It does not produce secondary pollutants after the procedure is completed, is easy to handle, and can remove uranium ions even in very low concentrations [16, 17]. Researchers from around the world use various adsorbents to treat uranium-contaminated wastewater [18, 19]. With advancements in science and technology, new and versatile adsorbents such as carbon-based materials [20], functionalized fibrous materials [21], porous polymers such as hydrogels [22], magnetic nanoparticles [23], metal–organic frameworks (MOFs) [24], and covalent-organic frameworks (CoFs) [25] as novel adsorbents have been developed that can efficiently treat uranium-contaminated wastewater. However, most of these adsorbents have some drawbacks, such as poor chemical stability in water, low adsorption efficiency in low-concentration regions, complicated synthesis methods, and high costs, which limit their use for the extraction/removal of uranium ions from seawater/wastewater. To implement the sorption method effectively, it is essential to utilize modern sorption materials that exhibit high uranium sorption capacity, improved kinetic properties, and low consumption. These materials should be suitable for simple and complete regeneration. Due to these reasons, numerous studies have investigated the use of various anion exchange resins for treating leach liquors containing uranium. Some of the commonly examined resins include Amberlite IRA-400 [26, 27], Amberlite IR120 [28], AMn resin [29], Dowix 1 [30], C100H resin [31], Amberjet 1200 H [32], Duolite ES-467 [33], Ambersep 920U [34], Amberlyst A-27 and Dowex 21K [35].
This research concentrates on investigating the uptake characteristics of uranium from acidic solutions through solid-phase extraction utilizing commercially accessible anion exchange resins, specifically MTA4801PF and MTA4601PFSO4. The main objective is to understand the factors that influence the rate of uranium extraction from sulfuric acid solutions, which serve as indicators of the performance of the selected resins. Additionally, the optimal loading conditions are determined by analyzing sorption kinetics and adsorption isotherm models.
Experimental
Materials
Purolite strong anion exchange resins namely: MTA4801PF (labeled as R1), and MTA4601PFSO4 (labeled as R2) were obtained from King of Prussia, PA, USA. The key characteristics of the operated resins, as provided by commercial data, are outlined in Table 1. The resins were pre-conditioned before being applied in this work. Analytical grade of hydrochloric, Sulfuric, and nitric acids was gained from Sigma-Aldrich (Saint-Louis, MO, USA) and employed in the desorption experiments. Uranium (UO2(NO3)2·6H2O) salt was employed to create a synthetic stock solution of U(VI) at a concentration of 1.0 g L−1. Fresh U(VI) standard solutions with specific U(VI) ion concentrations were created by diluting the stock solution with double-distilled water. To regulate the pH of the prepared solution, solutions of NaOH and/or HCl (0.1 M) were employed.
Preparation of El Erediya sulfate leach liquor
The uranium source for this study was sourced from El Erediya uranium ore, located in Egypt's eastern desert. To separate uranium from the El Erediya ore, sulfuric acid was employed under the following preferred conditions [36]: a concentration of 1.5 M H2SO4, a uranium ore-to-acid ratio of 1:3, a stirring duration of 2h, utilizing ore particles with a size of 63 μm, stirring at 600 rpm, and the addition of a 1.0 M H2O2 solution at room temperature. The chemical analysis of the resulting sulfate leach solution from El Erediya was carried out using a UV–vis spectrophotometer (SP-8001 UV-, Metretech Inc. version 1.02, 2000/10/01) equipped with a 10 mm glass cell and Arsenazo III indicator. The determined uranium concentrations in the solution are presented in Table 2.
Processes of sorption and desorption
The sorption investigations were accompanied by a batch reactor. A fixed volume (V, L) of solution holding an initial concentration of 50 mg L−1 U(VI) species (Co, mg L−1) was mixed for 10 h to confirm whole equilibrium, with a fixed quantity of resin (m, g) under agitation at 150 rpm. Typically, the sorbent dosage (SD = m/V, g L−1) was kept at 1.0 g L−1. When needed, the sorbent dosage was adjusted within the range of 0.2–1.2 g L−1 to explore the influence of the sorbent dosage, and the initial concentration (Co mg L−1) was varied between 20–160 mg L−1 for the examination of sorption isotherms. After filtration using a filter membrane with a pore size of 1.2 µm, the concentration of the remaining metal (Ce, mg L−1) was determined using a modified version of the Davies and Gray titrimetric method, in addition to a spectrophotometer. U(VI) sorption efficiency (R%), sorption capacity (qe, mg g−1), and the distribution constant (Kd) were calculated using Eqs. 1–3 respectively, operating the data that was supplied.
In the examination of desorption, a comparable experimental procedure was utilized. The sorbents that had captured the metal were blended with the eluent for 10 h, using a sorbent dosage of 0.5 g L−1. Uranium loading was conducted using the previously described procedure, and the actual quantity of U(VI) present in the resin was determined through the mass balance equation. Following filtration, the concentration in the eluate was examined to quantify the released amount and to estimate the desorption efficiency.
Modeling of sorption process
Adsorption kinetic models
The kinetics of uptake were examined to gain a deeper understanding of the adsorption rate and potential adsorption mechanisms of U(VI) ions using MTA4801PF and MTA4601PFSO4 resins. In this case, the Lagergren, pseudo-second-order kinetic equations, as well as the intraparticle diffusion (Weber and Morris) equation, were employed to analyze the adsorption data [37,38,39,40,41].
Pseudo-first order equation (PFO)
Pseudo-first order equation (PFO) was introduced by Lagergren in 1898 to characterize the sorption rate in solid–liquid phase adsorption systems concerning the adsorption capacity. The non-linear form of the PFO model is as follows: [37,38,39,40,41].
where q1 (mg g−1) is the estimated adsorption capacity by PFO model and k1 (min−1) is the Largergren equation rate constant.
Pseudo-second order model (PSO)
Pseudo-second order model (PSO) also referred to as the McKay equation, is founded on the sorption capacity of the solid phase. Furthermore, it supposes that chemisorption constitutes the rate-determining step. The non-linear form of the PSO model is as follows: [37,38,39,40,41]:
The initial adsorption rate, h (mol g−1 h−1), and the half-equilibrium time, t1/2 (h) was obtained from Eqs. 9 and 10 [39, 40].
where k2 (min−1) is the McKay equation rate constant and q2 (mg g−1) is the estimated adsorption capacity by PSO model.
Intra-particle diffusion model (IPD)
Intra-particle diffusion model (IPD) also known as the Weber and Morris model, was announced in 1963 to elucidate the rate-limiting step in adsorption. According to this model, solute adsorption encompasses three processes: (1) film diffusion, (2) surface diffusion, and (3) pore diffusion. Surface and pore diffusion can occur concurrently, while film diffusion is independent. The IPD equation is represented as follows: [37,38,39,40,41]:
where Kid (mg g−1 min−0.5) is a rate constant, and C is the thickness of the boundary layer.
Adsorption isotherm models
The sorption isotherm, which represents the equilibrium properties of the adsorbate-sorbent interaction, is a crucial parameter for optimizing the adsorption system and designing the plant. In this context, the equilibrium data has been analyzed using the Freundlich, Langmuir, and Sips models [37,38,39,40,41].
Freundlich isotherm equation
Freundlich isotherm equation has been employed to characterize sorption on surfaces with heterogeneity [37,38,39,40,41]. This model posits that the sorption sites are distributed exponentially concerning sorption heat. The non-linear form of the Freundlich isotherm model can be formulated as follows: [37,38,39,40,41]:
where KF (mg g−1) corresponds to the Freundlich constant, and 1/nF refers to the heterogeneity of the adsorbate sites.
Langmuir isotherm model
Langmuir isotherm model is one of the most widely employed equations for describing sorption phenomena [37,38,39,40,41]. This model suggested that the sorption occurs through monolayer sorption on a homogeneous surface without any binding between the adsorbed molecules. The non-linear form of the Langmuir model is written as follows: [37,38,39,40,41]:
where qm (mg/ g) is the maximum sorption capacity of the applied resins, and kL (L mg−1) is the Langmuir constant which, refers to the energy of adsorption and reflects the affinity of resin towards the metal ions.
Sips isotherm equation
The Sips model, which combines aspects of the Langmuir and Freundlich models, was developed to predict sorption in heterogeneous systems and overcome the limitation of the Freundlich isotherm regarding the increase in adsorbate concentration. The non-linear form of the Sips model is typically represented as follows: [37,38,39,40,41]:
where qS is the maximum sorption capacity (mg g−1) of Sips model. KS (L/mg) represents the constants of Sips model, mS is Sips constant.
Fitting the kinetic and isotherm models
By using the Coordination coefficient (R2) (Eq. 12) and the non-linear regression Chi-square (x2) (Eq. 13), the fitting of the kinetic and isotherm models has been evaluated. [40, 41]:
where \(q_{\exp }\) and \(q_{{{\text{pred}}}}\) are the experimental and model predicted equilibrium capacity (mg g−1) respectively, \(R^{2}\) and \(x^{2}\) are the coordination and Chi-square coefficients respectively, and n is the number of test points.
Adsorption thermodynamics
To gain a deeper insight into the thermodynamic performance of sorption, the correlation between MTA4801PF and MTA4601PFSO4 resins and their interaction with U(VI) at various temperatures was investigated. The following equations were employed to compute the thermodynamic parameters, namely Gibbs free energy change (ΔG°), standard enthalpy change (ΔH°), and standard entropy change (ΔS°) [40, 41]:
where \(K_{{\text{C}}}\) is a non-dimensional equilibrium constant and it equals Kd X 1000 X ρ [12, 13, 42]; \(T\) is the temperature (K), R is the universal gas constant (8.314 J mol−1 K−1), ρ is solution denisty g/ L, and \(A\) is a constant.
Results and discussion
Effect of pH
To develop accurate kinetic adsorption models, it is crucial to thoroughly understand the mechanisms governing adsorption processes. The surface charge and distribution of species on adsorbents are intricately linked to the pH (acidity or alkalinity) of the surrounding environment. The sorption efficiency of MTA4801PF and MTA4601PFSO4 resins was examined over a range of solution pH values from 1 to 5. Under the fixed condition (mixing time: 10 h; room temperature; U(VI) initial concentration: 50 mg L−1; and sorbent dose: 1 g L−1). This investigation aimed to gain insights into the practical applications of these resins. Based on the data presented in Fig. 1, it is evident that solution pH positively influences the sorption properties of both sorbents, with this influence being beneficial up to a pH of 4.0. For R1 and R2 resins, the maximum sorption efficiency percentages were found to be 84.0% and 94.0%, respectively. However, as the pH of the solution continues to increase, there is a gradual decline in sorption capacity, indicating a negative impact. The maximum sorption capacity at pH 4.0 is 42.0 and 47.0 for R1 and R2 respectively.

U(VI) sorption efficiency as a function of solution pH (mixing time: 10 h; room temperature; U(VI) initial concentration: 50 mg L−1; and sorbent dose: 1 g L−1)
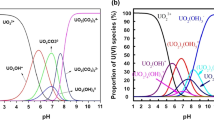
The changes in uranium speciation and alterations in the chemical properties of the resin surface as the solution pH varies may collectively account for the sorption behavior observed in both sorbents across different solution pH levels [25,26,27, 37]. So far, the aqueous speciation distribution of uranium according to pH performed and displayed in Fig. 2. The explored results obvious that the predominant uranium species at the pH range from 0.0–6.0 are the complexes of UO22+, UO2SO4, and UO2(SO4)22−. However, at pH intervals from 6 to 12, UO2(OH)2·H2O complex became the only predominant species. At pH 12, the negative U-hydroxide complexes, UO2(OH)3− and UO2(OH)42− are appeared [43]. At low pH, UO22+ repelled with the cationic sites of the resin, with a little anion exchange for UO2(SO4)22− ions (its percent is very small) for the sorption sites of resin. However, at higher pH, the greater percent of UO2(SO4)22− favors electrostatic attraction to cationic sites on the resin, easily replacing the anions onto the resin. So, with increasing the pH, the percent of UO2(SO4)22− increases and so the ability to exchange with the anions of the resin increases [25,26,27, 37].

Expected aqueous speciation of uranium (50 mg/L) as a function of pH in 0.1 M H2SO4 using Medusa/Hydra program
Effect of contact time and reaction kinetics
One of the crucial factors that can influence the scaling-up of the sorption process is believed to be the reaction rate. To address this, the impact of contact times, ranging from 3.0 to 600.0 min, on uranium sorption from aqueous solutions was investigated using MTA4801PF and MTA4601PFSO4 resins under fixed conditions (1.0 g sorbent L−1; initial concentration 50 mg L−1; temperature 298 K; pH 4.01). The efficiency of metal ion sorption as a function of reaction time is depicted in Fig. 3. The findings indicate that the sorption of uranium using R1 and R2 resins initiated rapidly, reaching equilibrium at 120 min. Subsequently, the reaction proceeded at a slower pace. In numerical terms, the sorption efficiency for R1 and R2 resins was approximately 77.8% and 93.0%, respectively. Following the equilibrium time, there is no significant increase in sorption efficiency. This is attributed to the presence of active groups on both resins, leading to the rapid progression of the initial phase of the reaction. However, as the equilibrium is reached, most of the active sites on the resins' surface become nearly fully occupied by U(VI) ions. Consequently, the ions must then interact with the active groups within the resin, which takes more time to occur [38,39,40].

Impact of shaken time on U(VI) sorption efficiency, % (1.0 g sorbent L−1; initial concentration 50 mg L−1; temperature 298 K; pH 4.01)
In the meantime, To explore the dynamics of U(VI) behavior, pseudo-first-order (PFO) and pseudo-second-order (PSO) models were utilized (Fig. 4). Table 3 displays the associated kinetic parameters. The uranium (VI) adsorption data for MTA4801PF and MTA4601PFSO4 resins were most accurately described by the PSO model. This model exhibited the lowest Chi-square coefficient (0.04 for R1 and 0.09 for R2) and the highest correlation value (R2 = 0.99) for both resins, as presented in Table 3. The experimental data values of 43.0 mg g−1 and 46.1 mg g−1, respectively, closely aligned with the calculated qe values for R1 and R2, which were 44.2 mg g−1 and 46.1 mg g−1, respectively. As a result, the PSO model effectively elucidated the U(VI) sorption mechanism. It indicated that the rate-controlling chemisorption process involved a chemical reaction, electron sharing, or electron transfer between the resins and the uranium species [12, 38,39,40,41]. It's important to note that the MTA4801PF resin exhibits a slower reaction rate (0.0013 min−1) compared to the MTA4601PFSO4 resin (0.0029 min−1). Additionally, the initial adsorption rates for MTA4801PF and MTA4601PFSO4 were 2.5 and 6.5 mol g−1 h−1, while their respective half-equilibrium times were 17.1 and 7.2 h. These findings indicate that R2 has a greater affinity for U(VI) ions than R1, as evidenced by its higher rate constant, initial adsorption rate, and shorter half-equilibrium period.

The kinetic plot for uranium sorption using R1 and R2 resins
The intra-particle diffusion (IPD) model, as shown in Fig. 5, is commonly employed to describe the movement of microparticles within the adsorbent material. The diffusion rate constants are arranged in descending order. Table 4 illustrates a primary external surface diffusion or rapid adsorption followed by a relatively slower intraparticle diffusion process [40, 41]. The observation that this particular stage of the overall adsorption process is the rate-limiting step suggests that U(VI) sorption on both resins is primarily governed by chemisorption or strong surface complexation, rather than mass transport [44, 45].

Morris–Weber illustration for uranium adsorption using R1 and R2 resins
To understand the diffusion mechanism of the U(VI) ion sorption process, the intraparticle diffusion kinetic equation (IPD) was employed, as Lagergren and McKay's reaction kinetic models were not suitable for this purpose [41, 43]. The IPD analysis comprises three sequential steps (represented as multi-linear connections) for the resins under study, as revealed by the data analysis using the Morris-Weber model (Fig. 5). This suggests that there were additional mechanisms at play in the sorption process [37,38,39] beyond the intraparticle diffusion of U(VI) species through the resin bead. This phenomenon can be attributed to the difference in mass transfer rates during the initial and final stages of sorption, highlighting the influence of the boundary layer [37,38,39].
Usually, each linear segment within the graph signifies a dominant mechanism or a combination of control mechanisms that are concurrently in operation [37,38,39]. The diffusion rate constants are presented in descending order. Table 4 illustrates a relatively delayed intraparticle diffusion alongside rapid (immediate) adsorption on the external surface, with these processes approaching zero as equilibrium is reached [40]. The observation that this stage of the overall adsorption process is the rate-limiting step suggests that mass transfer is not the primary mechanism governing U(VI) sorption on either resin. Instead, this process is predominantly controlled by chemisorption or strong surface complexation [41].
Effect of initial concentration of metal ion
Adsorption isotherms are commonly employed to depict the accumulation of metal ions on adsorbent surfaces. In this context, experiments were carried out using the commercial resins MTA4801PF and MTA4601PFSO4 to remove U(VI) from synthetic solutions containing various ions, with initial concentrations range 20 to 160 mg L−1. Figure 6-I presents the obtained results, illustrating the relationship between the initial ion concentration and the sorption percentage. The predicted data indicate that as the initial metal ion concentration increased, the sorption efficiency of metal ions significantly decreased. In detail, For R1 and R2, the uranium adsorption percentage declined from 93.5% to 41.3% and from 97.8% to 56.6%, respectively, when the starting ion concentration rose from 20 to 160 mg L−1. The greater initial ion concentration had a favorable impact on the resin's sorption capacity even though the efficiency of uranium ion sorption decreased with the increase in starting ion concentration, as shown in Fig. 6-II. The information provided demonstrates unequivocally that the resins' maximal experimental capacities are roughly 66.7 mg/g for MTA4801PF and 71.4 mg/g for MTA4601PFSO4, respectively. Langmuir, Freundlich, and other popular two-parameter models, as well as the three-parameter Sips model, were tested to see if they could adequately describe the properties of U(VT) adsorption onto MTA4801PF and MTA4601PFSO4 resins.

Effect of U(VI) initial concentration on uranium removal percent, % (I), and sorption capacity (II) (room temperature; shaking time 120 min; sorbent dose 1 g/ L; pH 4.01)
Figure 7 displays the isotherm curve, and Table 5 provides the corresponding parameters. Considering the values listed for these model parameters, it can be inferred that the Langmuir and Sips models, which suggest that uranium ion sorption is homogeneous and each molecule has a constant sorption activation energy and enthalpies, closely fit the experimental data [38,39,40,41]. The Langmuir model indicated that MTA4601PFSO4 had a slightly higher adsorption capacity than MTA4801PF (66.7 mg/g, R2 = 0.99), with the difference being attributed to the modification procedure. The Pearson Hard and Soft Acids and Bases (HSAB) theory, which builds upon the Lewis theory of acids and bases, can be employed to elucidate the variations in U(VI) binding strength. According to this theory, metal ions are regarded as acids (electron acceptors), while functional groups in resins are seen as bases (electron donors) [37, 38]. Strong acids tend to react more favorably with strong bases, whereas weak acids exhibit the opposite behavior. According to the HSAB theory, chloride is classified as a borderline base, while the sulfate group is considered a hard base [37, 38]. Since uranium ions are considered strong acids, the sulfate group exhibits a higher affinity for them compared to the chloride group. Consequently, the following ranking of the resin's affinity towards uranium ions is observed: MTA4801PF < MTA4601PFSO4. This ranking aligns with the order found in the experimental results, providing evidence that the HSAB theory is consistent with the experimental findings.

The isotherm graph for U(VI) sorption using R1 and R2 resins
The sorption capacity of the applied resins was displayed in Table 6 in comparison with the sorption capacity of other sorbents from the literature. The anticipated data declares that the investigated resins possess sorption capacity within the common resin in literature.
Impact of sorbent dose
The dependence of U(VI) sorption percent on the MTA4801PF and MTA4601PFSO4 resins amount of addition was explored by conducting a set of experiments at resin dose range 0.2–1.2 g L−1. The explored results in Fig. 8 exhibit that the sorption percent is directly proportional to the resin dose which means a higher probability for interaction with uranium species. Furthermore, the MTA4601PFSO4 displays a higher sorption efficiency than MTA4801PF resin all over the investigated resin dose. In numerical terms, the removal percentage for MTA4801PF resin changed from 52.4% to 90.5%, while for sorbent doses ranging from 0.2 to 1.2 g/L, it ranged from 65.7% to 96.2%. This behavior can be explained by the growth in the number of active binding sites accessible due to the higher sorbent dosage, which also enriches the rate of adsorption [50]. Remarkably, U(VI) uptake capacity is described by a ramp down all over the studied resin dose which could be due to the low uranium species concentration not equivalent to the resin sorption capacity and thereby decreasing the qe [15, 44, 45].

The variation of uptake percent with resin dose (U(VI) initial concentration 50 mg L−1; shaking time 120 min; temperature 298 K; pH 4.01)
Effect of temperature on U(Vl) adsorption
The thermodynamic parameters as well as the sorption nature could be declared by investigating uranium species uptake using MTA4801PF and MTA4601PFSO4 resins at different reaction temperatures (20–50 ± 1 °C). Other parameters, such as resin dosage (1.0 g/L), beginning U(VI) concentration (50 mg L−1), and reaction time (120 min), were kept constant. The attained results were treated using the thermodynamic equations to illustrate Van’t Hoff plot (Fig. 9) and evaluate the thermodynamic variable (i.e. entropy (∆S), enthalpy (∆H), and Gibbs free energy (∆G)). The determined thermodynamic variables (Table 7) obvious that the enthalpy change (∆H) has positive values (24.4 and 34.7 kJ mol−1) for R1 and R2 resins respectively which indicates that the U(VI) capture process is endothermic [45, 50]. Furthermore, both applied resins show a negative Gibbs free energy change (∆G) which indicates a feasible and spontaneous sorption process [15, 51]. The increment of the negative values of ∆G with the temperature variation from 20 to 50 reflects that the uranium uptake process is more favorable at the higher reaction temperature. The positive values of the entropy change (∆S) (152.9 J mol−1 K−1 for R1 and 194.5 J mol−1 K−1 for R2 resins, respectively) indicate that the solid-solution mass transfer interface led to a greater degree of molecular disorder throughout the adsorption process [51, 52].

Van’t Hoff plot for U(VI) sorption using MTA4801PF and MTA4601PFSO4 resins (U(VI) initial concentration 50 mg L−1; shaking time 120 min; sorbent dose 1.0 g L−1; pH 4.01)
Scanning electron micrograph (morphology)
The scanning electron microscopy (SEM) image pattern of the applied resins before and after the sorption of uranium (Fig. 10) was employed to confirm U(VI) uptake process. The resin particles appear spherical with a constant smooth surface, as shown in Fig. 10a. However, as can be seen in Fig. 10b, the surface became wrinkly and rough after the adsorption experiment. The release of ions from the solution, which were then adsorbed onto the resin surface, is responsible for this change in surface topography.

SEM micrographs of the both resins (R1 and R2) before (a, b respectively), and after (c, d respectively) U(VI)sorption process
Energetic dispersive X-ray (EDX) analysis
The presented adsorbent's structural features were analyzed through elemental analysis using EDX, as shown in Fig. 11. Indicative peaks can be observed in the given spectrum for Cl and S, which are the main constituents of the introduced adsorbent. The strong signal for Cl indicates the presence of Purolite MTA4801PF (R1) as a major component in the resin adsorbent structure as shown in Fig. 11a. In addition, the strong signal for S indicates the presence of Purolite MTA4601PFSO4 (R2) as a major component in the resin adsorbent structure as shown in Fig. 11b. Variations were observed in the EDX spectrum of the spent adsorbent after removing Uranium species from sulfate media (Fig. 11c, d). Specifically, an additional EDX signal with low intensities was detected, indicating the presence of U(VI) ions. This confirms the successful removal of uranium from the sulfate media through commercial anion exchange resins.

EDX of the both resins (R1 and R2) before (a, b respectively), and after (c, d respectively) U(VI)sorption process
FTIR analysis
The FT-IR spectra of both resins, both before and after uranium adsorption, are given in Fig. 12a–d. The interaction between U(VI) and the groups in the resin, forming a uranyl complex, was indicated by a minor shift in band positions to smaller wavenumbers and a rise in the relative intensity of certain bands following uranium adsorption. Certain transmittance bands are common to both spectra, including the band associated with O–H stretching at 3600 cm−1, two bands at 1600 and 1500 cm−1 attributed to the bending of the N–H group, and the band at 1380 cm−1 related to the stretching of the C-N bond in the vinyl group. The C-N–C stretching is connected to the two bands at 1219 and 1117 cm−1, while the bending of the alkene C = C bond is associated with the two bands at 977 and 888 cm−1. In contrast, the C-H stretching band was linked to the uranium adsorption process, shifting from 704 to 616 cm−1. Additionally, a Cl− stretching band was evident at 761 cm−1. After the adsorption experiment, a prominent band at 1122 cm−1 was observed, aligning with the resin's absorption of U(VI).

FT-IR spectrum of both resins (R1 and R2) before (a, b respectively), and after (c, d respectively) U(VI)sorption process
Desorption and reusability investigation
The maximum sorption capacity resin, MTA4601PFSO4, was loaded, and uranium(VI) desorption was performed using 0.5 M solutions of sulfuric, nitric, and hydrochloric acids at room temperature. The sorbent dose was 0.5 g L−1, and the shaking period was 10 h. The information in Table 8 makes it abundantly evident that the sulfuric acid solution had the best elution. The stability of the MTA4601PFSO4 resin for consecutive six sorption/ desorption cycles was investigated. The anticipated data in Table 9 illuminates that the applied resin possesses stable performance over the investigated six cycles whereas the sorption and desorption percent slightly changed from 93.5 to 93.1% for the sorption process, and from 95.0 to 94.6% for the desorption process.
Case study: El Erediya sulfate leach liquor
Purolite MTA4601PFSO4 strong anion exchange resin was applied for uranium recovery from El Erediya sulfate leach liquor. By introducing a buffer solution, the working leach liquor's original pH was changed. Then, it was added to the uranium recovery procedure utilizing MTA4601PFSO4 resin at the recommended conditions of 1.0 g/L resin dose, room temperature, and 120 min of shaking time. According to the data, 64.0 mg g−1, or almost 90% of the resin's theoretical capacity, was successfully attained. It is possible that the struggle between uranium and other ions present in the sample, such as iron ions, is what caused the decline in Purolite MTA4601PFSO4 capacity.
Conclusion
This study focuses on the solid–liquid method for uranium adsorption from aqueous solutions. Purolite strong anion exchange resins in particular were used for this. According to the data, these resins have a propensity for recovering uranium from acidic solutions. The order of the sorption capacity was MTA4601PFSO4 > MTA4801PF. The sorption process for the used resins followed the pseudo-second-order model, as shown by kinetic modeling of the experimental results. The Langmuir isotherm model offered a good fit for the acquired data, according to the isotherm analysis. The process is also viable, spontaneous, and an endothermic reaction, according to the thermodynamic characteristics. Using 0.5 M sulfuric acid, uranium desorption was completed with a 95% recovery rate. Commercial resins have been shown to work well in sorption, despite the complexity of the solution, when used on the sulfate leach liquid from Egyptian ores.
References
Tang X, Zhou L, Le Z et al (2020) Preparation of porous chitosan/carboxylated carbon nanotube composite aerogels for the efficient removal of uranium(VI) from aqueous solution. Int J Biol Macromol 160:1000–1008. https://doi.org/10.1016/J.IJBIOMAC.2020.05.179
Cheira MF, Kouraim MN, Zidan IH et al (2020) Adsorption of U(VI) from sulfate solution using montmorillonite/polyamide and nano-titanium oxide/polyamide nanocomposites. J Environ Chem Eng 8:104427. https://doi.org/10.1016/J.JECE.2020.104427
Wang B, Deng C, Ma W (2021) Modified nanoscale zero-valent iron in persulfate activation for organic pollution remediation: a review. Environ Sci Pollut Res 2826(28):34229–34247. https://doi.org/10.1007/S11356-021-13972-W
Yin N, Ai Y, Xu Y et al (2020) Preparation of magnetic biomass-carbon aerogel and its application for adsorption of uranium(VI). J Radioanal Nucl Chem 326:1307–1321. https://doi.org/10.1007/S10967-020-07392-2/FIGURES/13
Hussein AEM, Youssef WM, Taha MH, El-Maadawy MM (2017) Uranium adsorption from aqueous nitric acid solution by solvent impregnated polypropylene. Arab J Nucl Sci Appl 50(4):156–170
Taha MH, El-Maadawy MM, Hussein AEM, Youssef WM (2018) Uranium sorption from commercial phosphoric acid using kaolinite and Metakaolinite. J Radioanal Nucl Chem 317:685–699. https://doi.org/10.1007/s10967-018-5951-9
Yin L, Hu B, Zhuang L et al (2020) Synthesis of flexible cross-linked cryptomelane-type manganese oxide nanowire membranes and their application for U(VI) and Eu(III) elimination from solutions. Chem Eng J 381:122744. https://doi.org/10.1016/J.CEJ.2019.122744
Abdel-Samad AA et al (2020) Synthesis and characterization of functionalized activated carbon for removal of uranium and iron from phosphoric acid. J Basic Environ Sci 7:140–153
Sahu A, Vincent T, Shah JG, Wattal PK (2014) Catalytic reduction of U(VI) to U(IV) using hydrogen with platinum loaded on alumina and silica. J Radioanal Nucl Chem 300:163–167. https://doi.org/10.1007/S10967-014-2996-2/FIGURES/10
BADR, Magd M., et al (2023) Investigating the efficiency of bisphenol-A diglycidyl ether/resole phenol formaldehyde/polyamine blends as Cerium adsorbent from aqueous solution. J Environ Chem Eng 11(6):111475. https://doi.org/10.1016/j.jece.2023.111475
Youssef WM (2020) Characterization and application of olive stones activated carbon for uranium (VI) sorption from wet-process phosphoric acid: kinetic, equilibrium and thermodynamic studies. J Basic Environ Sci 7:1–12
Masoud AM, El-Maadawy MM, Taha MH, Meawad A (2023) Uranium capture from aqueous solution using cement kiln dust; equilibrium and kinetic studies. J Radioanal Nucl Chem 332:2487–2497. https://doi.org/10.1007/S10967-023-08937-X/FIGURES/14
Masoud AM, El-Zahhar AA, El Naggar AMA et al (2023) Soya bean derived activated carbon as an efficient adsorbent for capture of valuable heavy metals from waste aqueous solution. Radiochim Acta 111:105–115. https://doi.org/10.1515/RACT-2022-0098/DOWNLOADASSET/SUPPL/J_RACT-2022-0098_SUPPL.DOCX
Elzoghby AA, Haggag ESA, Roshdy OE et al (2023) Kaolinite/thiourea-formaldehyde composite for efficient U(VI) sorption from commercial phosphoric acid. Radiochim Acta 111:91–103. https://doi.org/10.1515/RACT-2022-0091/MACHINEREADABLECITATION/RIS
Youssef WM (2022) Solid–liquid extraction of uranium from aqueous solution using Marathon C as a strong cation exchanger resin: kinetic, and isotherm studies. Int J Environ Anal Chem. https://doi.org/10.1080/03067319.2022.2134995
Mittal H, Alfantazi A, Alhassan SM (2024) Recent developments in the adsorption of uranium ions from wastewater/seawater using carbon-based adsorbents. J Environ Chem Eng 12:111705. https://doi.org/10.1016/J.JECE.2023.111705
Zhang X, Fan H, Yuan J et al (2022) The application and mechanism of iron sulfides in arsenic removal from water and wastewater: a critical review. J Environ Chem Eng 10:108856. https://doi.org/10.1016/J.JECE.2022.108856
Elhefnawy OA, Elabd AA (2023) Highly efficient elimination of uranium (VI) and thorium (IV) from aqueous solution using activated carbon immobilized on polystyrene. Pigment Resin Technol. https://doi.org/10.1108/PRT-09-2022-0107/FULL/PDF
Li H, Zhang X, Luo C et al (2024) Pomelo peel derived phosphorus-doped biochar for efficient disposal of uranium-containing nuclear wastewater: Experimental and theoretical perspectives. Sep Purif Technol 333:125947. https://doi.org/10.1016/J.SEPPUR.2023.125947
Bi C, Nian J, Zhang C et al (2023) Efficient uranium adsorbent prepared by grafting amidoxime groups on dopamine modified graphene oxide. Prog Nucl Energy 155:104515. https://doi.org/10.1016/J.PNUCENE.2022.104515
Ma F, Gui Y, Liu P et al (2020) Functional fibrous materials-based adsorbents for uranium adsorption and environmental remediation. Chem Eng J 390:124597. https://doi.org/10.1016/J.CEJ.2020.124597
Chen D, Zhang D, Zou S et al (2023) A robust polyethyleneimine-based supramolecular hydrogel towards uranium adsorption and deposition. Colloids Surf A Physicochem Eng Asp 660:130886. https://doi.org/10.1016/J.COLSURFA.2022.130886
Rasheed T (2022) Magnetic nanomaterials: Greener and sustainable alternatives for the adsorption of hazardous environmental contaminants. J Clean Prod 362:132338. https://doi.org/10.1016/J.JCLEPRO.2022.132338
Guo X, Liu Q, Liu J et al (2019) Magnetic metal-organic frameworks/carbon dots as a multifunctional platform for detection and removal of uranium. Appl Surf Sci 491:640–649. https://doi.org/10.1016/J.APSUSC.2019.06.108
Cui WR, Xu W, Bin QW (2023) Constructing an ultrastable imidazole covalent organic framework for concurrent uranium detection and recovery. Ecotoxicol Environ Saf 252:114639. https://doi.org/10.1016/J.ECOENV.2023.114639
Guettaf H, Becis A, Ferhat K et al (2009) Concentration-purification of uranium from an acid leaching solution. Phys Procedia 2:765–771. https://doi.org/10.1016/J.PHPRO.2009.11.023
Ser M, Khawassek YS (2014) Production of commercial uranium concentrate from El-Sela shear zone mineralized ore material, south eastern desert—Egypt, AT Inshas Pilot Plant Unit. Nucl Sci Sci J 3:169–179. https://doi.org/10.21608/NSSJ.2014.30972
Ahmed SH, Gammal EME, Amin MI, Youssef WM (2021) Studying the effect of potassium amyl xanthate surfactant on Fe, Cu and U ions for the pretreatment of Abu zeneima sulphate leach liquor. J Period Polytech Chem Eng 65(3):408–415. https://doi.org/10.3311/PPch.15795
Mirjalili K, Roshani M (2007) Resin-in-pulp method for uranium recovery from leached pulp of low grade uranium ore. Hydrometallurgy 85:103–109. https://doi.org/10.1016/J.HYDROMET.2006.08.006
Stoliker DL, Kaviani N, Kent DB, Davis JA (2013) Evaluating ion exchange resin efficiency and oxidative capacity for the separation of uranium(IV) and uranium(VI). Geochem Trans 14:1–9. https://doi.org/10.1186/1467-4866-14-1/FIGURES/3
Elzoghby AA (2021) Kinetic and equilibrium studies for U(VI) and Cd(II) sorption from commercial phosphoric acid using C100H resin. J Radioanal Nucl Chem 329:899–911. https://doi.org/10.1007/S10967-021-07832-7/TABLES/6
Khawassek YM, Masoud AM, Taha MH, Hussein AEM (2018) Kinetics and thermodynamics of uranium ion adsorption from waste solution using Amberjet 1200 H as cation exchanger. J Radioanal Nucl Chem 315:493–502. https://doi.org/10.1007/S10967-017-5692-1/FIGURES/13
Zidan IH, Cheira MF, Bakry AR, Atia BM (2022) Potentiality of uranium recovery from G. Gattar leach liquor using Duolite ES-467 chelating resin: Kinetic, thermodynamic and isotherm features. Int J Environ Anal Chem 102:2102–2124. https://doi.org/10.1080/03067319.2020.1748613
Youssef WM, El Sheikh AS, Ahmed SH, Morsy AM (2020) Polyacrylic acid/polyaniline composite as efficient adsorbent for uranium extraction from nuclear industrial effluent. J Radioanal Nucl Chem 324:87–96. https://doi.org/10.1007/s10967-020-07060-5
Massoud A, Masoud AM, Youssef WM (2019) Sorption characteristics of uranium from sulfate leach liquor by commercial strong base anion exchange resins. J Radioanal Nucl Chem 322:1065–1077. https://doi.org/10.1007/S10967-019-06770-9/FIGURES/12
Haggag ESA, Abdelsamad AA, Masoud AM (2020) Potentiality of uranium extraction from acidic leach liquor by polyacrylamide-acrylic acid titanium silicate composite adsorbent. Int J Environ Anal Chem 100:204–224. https://doi.org/10.1080/03067319.2019.1636037
Taha MH (2021) Sorption of U(VI), Mn (II), Cu(II), Zn(II), and Cd(II) from multi-component phosphoric acid solutions using MARATHON C resin. Environ Sci Pollut Res 28:12475–12489. https://doi.org/10.1007/S11356-020-11256-3/TABLES/7
Hassanein TF, Masoud AM, Mohamed WS et al (2021) Synthesis of polyamide 6/nano-hydroxyapatite hybrid (PA6/n-HAp) for the sorption of rare earth elements and uranium. J Environ Chem Eng 9:104731. https://doi.org/10.1016/J.JECE.2020.104731
Elzoghby AA, Bakry A, Masoud AM et al (2021) Synthesis of polyamide-based nanocomposites using green-synthesized chromium and copper oxides nanoparticles for the sorption of uranium from aqueous solution. J Environ Chem Eng 9:106755. https://doi.org/10.1016/J.JECE.2021.106755
Marques BS, Frantz TS, Sant’Anna Cadaval Junior TR, et al (2019) Adsorption of a textile dye onto piaçava fibers: kinetic, equilibrium, thermodynamics, and application in simulated effluents. Environ Sci Pollut Res 26:28584–28592. https://doi.org/10.1007/S11356-018-3587-5/FIGURES/9
Kang HJ, Kim JH (2019) Adsorption Kinetics, mechanism, isotherm, and thermodynamic analysis of paclitaxel from extracts of Taxus chinensis cell cultures onto sylopute. Biotechnol Bioprocess Eng 24:513–521. https://doi.org/10.1007/S12257-019-0001-1/METRICS
El Didamony AM, Youssef WM, Abdou AA (2019) Modification of amberlite XAD-2 resin for U(VI) adsorption from Egyptian crude phosphoric acid. Egypt J Pet 28:71–76. https://doi.org/10.1016/j.ejpe.2018.11.001
Masoud AM (2022) Sorption behavior of uranium from Sulfate media using purolite A400 as a strong base anion exchange resin. Int J Environ Anal Chem 102:3124–3146. https://doi.org/10.1080/03067319.2020.1763974
Youssef WM, Hagag MS, Ali AH (2018) Synthesis, characterization and application of composite derived from rice husk ash with aluminium oxide for sorption of uranium. Adsorpt Sci Technol 36:1274–1293. https://doi.org/10.1177/0263617418768920/ASSET/IMAGES/LARGE/10.1177_0263617418768920-FIG16.JPEG
Younes AA, Masoud AM, Taha MH (2018) Uranium sorption from aqueous solutions using polyacrylamide-based chelating sorbents. Sep Sci Technol 53:2573–2586. https://doi.org/10.1080/01496395.2018.1467450
Haggag ESA (2021) Cellulose hydrogel for enhanced uranium (VI) capture from nitrate medium: preparation, characterisation and adsorption optimisation. Int J Environ Anal Chem. https://doi.org/10.1080/03067319.2021.2005791
Roshdy OE (2021) Removal of uranium, cadmium and iron ions from phosphoric acid solution using amberjet 1200 H resin: an experimental, isotherm and kinetic study. J Radioanal Nucl Chem 329:85–101. https://doi.org/10.1007/S10967-021-07792-Y/TABLES/8
Youssef WM, Hussein AEM, Taha MH, El-Maadawy MM (2022) Uranium(VI) sorption from liquid waste solution using functionalized polyurethane polymer: kinetic and isotherm characterizations. Russ J Inorg Chem 67:1058–1068. https://doi.org/10.1134/S0036023622070245/METRICS
Hussein AEM, Youssef WM, El-Sheikh AS (2019) Adsorption of uranium from aqueous solutions by expanded perlite. Radiochemistry 61:592–597. https://doi.org/10.1134/S1066362219050114/METRICS
Semnani F, Asadi Z, Samadfam M, Sepehrian H (2012) Uranium(VI) sorption behavior onto amberlite CG-400 anion exchange resin: Effects of pH, contact time, temperature and presence of phosphate. Ann Nucl Energy 48:21–24. https://doi.org/10.1016/J.ANUCENE.2012.05.010
Younes AA, Masoud AM, Taha MH (2021) Amino-functionalised cross-linked polyacrylamide for the adsorption of U(VI) ions from contaminated aqueous solutions. Int J Environ Anal Chem. https://doi.org/10.1080/03067319.2021.2003348
Hussein AEM, Taha MH (2013) Uranium removal from nitric acid raffinate solution by solvent immobilized PVC cement. J Radioanal Nucl Chem 295:709–715. https://doi.org/10.1007/S10967-012-2158-3/TABLES/2
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Youssef, W.M., Masoud, A.M., Elmaadawy, M.M. et al. Uranium (VI) sorption from aqueous solution using commercial anion exchange resins; kinetics, isotherm, and thermodynamic investigations. J Radioanal Nucl Chem 333, 1975–1989 (2024). https://doi.org/10.1007/s10967-024-09438-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-024-09438-1