
Abstract
In this work, polypropylene (PP)/carboxylated acrylonitrile butadiene rubber (XNBR) binary blends were prepared with the elastomer component dosed in its suspension (latex) form into the polymer matrix during melt compounding. For this purpose, samples containing 0-20 wt.% rubber were prepared using two different PP grades as matrices with lower and higher viscosity. Analogous reference samples with the same composition were also fabricated using traditional melt mixing by introducing the rubber in its dry, bulk form in order to analyze the efficiency of the latex route. Mechanical, thermomechanical and morphological analyses were used to investigate the structure-property relationships of the blends. Based on the SEM images the average domain size of the dispersed XNBR domains became markedly smaller when the rubber was introduced in its suspension form into the PP. Based on the Charpy impact tests and the tensile test results, the decreased rubber domain size led to improved ductility and toughness. The improvement was more prominent when the difference between the viscosity of the PP matrix and the XNBR rubber was higher.
Similar content being viewed by others


Introduction
Polypropylene (PP) is one of the most versatile commodity thermoplastics and, as such, it is widely used in various fields, including the automotive, packaging, medical and other engineering industries. It has numerous advantageous properties such as low price, good chemical resistance, low density and good processability [1-4]. On the other hand, the areas of its potential applications are greatly limited by its low impact toughness, especially at room temperature and below. Accordingly, a high number of studies have been devoted to the toughening of PP in order to extend the range of its applications. The most common approach to increase the impact resistance of thermoplastic polymers is to blend them with other polymers, especially rubber-like elastomers, which is a simple, effective and economical method [5-7]. To date, PP was toughened by pairing it with various rubbers such as natural rubber (NR) [8, 9], ethylene-propylene-diene rubber (EPDM) [5, 10], ethylene-butene random copolymer (EBR) [11], acrylonitrile butadiene rubber (NBR) [12, 13], carboxylated acrylonitrile butadiene rubber (XNBR) [14], and also with ground waste rubber obtained from vehicle tires (GTR) [15-17]. In the currently available literature, there are rather few studies on toughening PP using NBR and its hydrogenated (HNBR) or carboxylated variations, which can be attributed to the difficulty of blending polar NBR with nonpolar PP [18].
Rubber-toughened PP-based materials are used in various fields including the packaging, automotive, medical and optical fields. PP/nitrile rubber blends can also be applied in a number of fields, especially where toughness and oil resistance limits are required since NBR has strong polarity and excellent oil resistance [19]. Meanwhile, the carboxylation of the nitrile rubber phase enables the blends to have enhanced abrasion resistance as well.
When preparing PP/rubber blends, there are some major challenges that need to be addressed for the desired improvement of toughness. The most important aspects of rubber-toughening are the small-enough average particle size and the proper dispersion of the rubber domains in the PP matrix [20]. These factors are mostly influenced by the viscosity ratio (VR) of the components and the shear stresses being present throughout the blending process. Generally, a VR value between the components closer to “1” means a better distribution of the secondary phase within the matrix [21]. Several methods were introduced in the literature to improve the above-mentioned factors in order to achieve optimal toughening. A viable strategy for achieving a finer dispersion of the rubber phase is the incorporation of nano-sized reinforcement particles into the matrix, which helps to decrease the viscosity difference between the matrix and the rubber component [22]. This latter technique is also suitable for property-tailoring purposes of such materials [23]. Another feasible approach is the preparation of rubber powder from latex through spray-drying prior to its melt blending with the polymer matrix [24]. It has to be noted, that the final morphology and mechanical behavior of polymer/rubber blends depend on a number of other variables as well, including the presence of any compatibilizers, the blend composition and the degree of dispersion of blended constituents as well [18].
A relatively new strategy in the field of nanocomposite preparation is the so-called water-assisted melt mixing, during which the reinforcement particles are introduced into the molten polymer matrix as an aqueous suspension [25]. Technically speaking, rubber lattices are also aqueous suspensions, with the submicron-scaled rubber particles dispersed in a water-based liquid. Therefore, it seems straightforward, that the idea of water-assisted melt mixing could be applied to the preparation of polymer/rubber blends as well in order to achieve an improved dispersion of elastomer particles. Water-assisted techniques may offer multiple advantages. Firstly, throughout the melt compounding process, there is no need to decompose the larger coagulated bulk rubber domains, since the small particulates are situated individually within the suspension. Secondly, there is a ‘blow-up’ phenomenon generated by water that boils intensively when exposed to the polymer melt at its processing temperature, which is also suggested to improve the dispersion [25].
In a previous work [26] PP/NR blends were prepared with the so-called fast evaporation mixing technique, which is about introducing the latex dropwise into a molten polymer during melt mixing. An NR-based latex with a solid content of 60 wt.% was dosed into molten PP. The results indicated that a much smaller NR domain size and a finer dispersion can be obtained with the fast evaporation mixing method compared to the traditional melt compounding (in which case the rubber is introduced in its dry bulk form). When using the water-assisted technique, the improved dispersion of rubber domains was also accompanied by a higher toughness and ductility of the blends.
In order to get more insight into the efficiency of the water-assisted preparation of rubber-toughened PP blends, in this current study the fast evaporation mixing method was applied to prepare XNBR toughened PP samples. Two different kinds of PPs with different molecular weights and viscosities were used to also analyze the effectiveness of the method at different VRs. Structure–property relationships of the prepared blends were analyzed using morphological, mechanical and thermomechanical measurements.
Materials and methods
Materials
Polypropylenes of type Tipplen H145F and Tipplen H543F were used as matrix material, both grades were kindly supplied by MOL Petrochemicals Ltd. (Tiszaújváros, Hungary). The two PPs have a melt flow rate (MFR) of 29 g/10 min and 4 g/10 min, respectively (measured at 230 °C and 2.16 kg). Carboxylated acrylonitrile butadiene rubber latex, brand Chemigum Latex 550, with a solid content of 41 wt.% was obtained from OMNOVA Solutions Inc. (Beachwood, OH, USA).
Preparation of PP/XNBR blends
The PP/XNBR samples with different rubber content (0-20 wt.%) were produced by melt blending the components in a Brabender PL-2000 type internal mixer (Duisburg, Germany) equipped with a kneading chamber of 50 cm3. The temperature of the chamber was set to 180 °C and the nominal rotational speed of the kneading elements was 60 rpm. The duration of the melt compounding was 5 minutes after both the components were introduced into the chamber. Processing was performed as follows: (i) firstly the PP pellets were added into the mixer, and (ii) once the PP was molten the XNBR was dosed either in its latex or dry form. When adding the XNBR as latex, the so-called fast evaporation mixing technique was applied, during which the latex was dosed dropwise into the molten PP using a syringe. The setup and the detailed procedure of this method are described in a previous work [26]. Dry rubber was achieved by drying the latex at 40 °C for 24 h in a vacuum oven before melt mixing, and subsequently cutting it into pieces of ca. 5x5x5 mm3 before adding into the compounder.
After that, the blends were compression molded into sheets of 2 mm thickness using a Dr. Collin Teach-Line Platen Press 200E hot press (Munich, Germany) at 200 °C and a pressure of 25 bar for 3 mins. The specimens for the characterizations were cut from the compression molded sheets by a Mutronic Diadisc type disc saw (Rieden, Germany). The designations and compositions of the prepared samples are collected in Table 1.
Characterization and testing
The average size of XNBR solid globules within the applied latex was determined using a Horiba Partica LA-950V type laser scattering particle size diffraction analyzer (Kyoto, Japan).
The viscosity of the PPs and the XNBR rubber was determined using a MonTech D-RPA 3000 rheometer (Buchen, Germany). The parameters of the analysis were set so that they represented those circumstances (temperature, time, shear rate) that were present during melt mixing in the kneading chamber. Accordingly, the measurement was performed at 180 °C for 5 mins at a shear rate of 57 1/sec.
The morphology of PP/XNBR blends was evaluated by scanning electron microscopy (SEM) using a JEOL JSM 6380LA microscope (Tokyo, Japan), with an acceleration voltage of 15 kV. The samples were cryogenically fractured and then sputter-coated with gold/palladium alloy before the SEM analysis.
The mechanical properties of PP/XNBR blends were analyzed using tensile tests and Charpy impact tests. The tensile tests were performed on a universal testing machine (Zwick Z005, Ulm, Germany) equipped with a 5 kN force sensor. Tests were run at 5 mm/min crosshead speed, the gripped length was 50 mm. In the case of specimens showing a necking phenomenon, tests were stopped at 150% elongation. Charpy-tests were performed on notched specimens using a Ceast Resil Impactor (Pianezza, Italy) pendulum-type impact testing machine with a 2 J impact energy hammer. The 2 mm notch was prepared with a Mutronic Diadisc saw (Rieden, Germany) equipped with a V-type saw blade. All the mechanical tests were performed at room temperature at a relative humidity of 50% ± 10%. The results represent the average values of five consecutive measurements.
Dynamic mechanical analysis (DMA) was performed using a TA DMA Q800 testing unit (New Castle, DE, USA) equipped with a dual-cantilever bending fixture at a frequency of 1 Hz. The DMA tests were conducted at a strain of 0.02% using a 3 °C/min temperature ramp from -50 to +70 °C.
The whole procedure of sample preparation and characterization is shown as a flow chart in Fig. 1.

Flow chart of the PP/XNBR blend preparation and characterization
Results and discussion
Particle size analysis of XNBR within the latex
The initial size of XNBR particles in the latex suspension was determined by laser scattering analysis. The reason for performing this measurement was to determine if the diameter of rubber globules remained the same after incorporating them into the PP or it increased because of coalescence. Figure 2 shows the results of the particle size analysis. The size of the rubber particles in the latex was in the range of 115-229 nm (0.115-0.229 µm), while the average diameter was 190 nm (0.190 µm).

Size distribution of XNBR rubber globules
Viscosity measurement
The viscosity values of all the components used in this research are presented in Fig. 3. As expected, the H145F type PP with its higher MFR has lower viscosity (288 Pa·s) under the melt mixing circumstances compared to the H543F type (635 Pa·s). Rubbers generally have a considerably higher molecular weight compared to commodity thermoplastics, which explains the significantly higher viscosity of XNBR (2655 Pa·s) than that of the PPs. Based on these results, the VRs of the component pairings can already be determined: the VR between H145F PP and the XNBR is ~9.2, while the VR between H543F PP and the XNBR is ~4.2. Since this latter one is much closer to “1”, a smaller rubber domain size and a better dispersion were expected in this case.

Viscosity values of the two applied PP grades and the XNBR under the melt compounding circumstances
Phase morphology
The morphological features of PP/XNBR blends were observed as per SEM analysis. The ultimate properties of rubber-toughened polymers are highly dependent on the morphology of the blends. Figure 4 shows the morphology of the 10 wt.% rubber containing H145F PP/XNBR blends prepared using the dry dosing method (Fig. 4a, b) and the latex route (Fig. 4c, d). The position of XNBR phases can be identified either directly as amorphous domains within the PP matrix or indirectly as dark holes, where the rubber was situated before the specimen broke. The images reveal a matrix-droplet morphology in both cases. It was expected, since according to the minimum energy dissipation principle, the phase with the higher viscosity tends to be present in the form of droplets within the less viscous matrix [27]. There is an obvious difference in the size of XNBR domains between the two preparation methods. According to Fig. 4a, when using traditional melt mixing, the size of some rubber particles was in the range of a few hundred microns, while barely any submicronic droplets could be observed at higher magnification (Fig. 4b). Apparently, the high difference in the viscosity between the components did not allow the shear stresses to break the XNBR particles into smaller pieces than that. On the other hand, the latex route enabled the rubber to be dispersed in the form of much finer particles, mostly in the range of 0.5-10 µm. It has to be noted though that this is still considerably larger compared to the original size of the XNBR globules that was determined by the laser scattering analysis in the latex. It refers to the fact that during the melt compounding a certain level of coalescence occurred resulting in the agglomeration of rubber. Based on the literature, the optimum size of rubber particles in rubber-toughened plastics is in the range of 0.1 to 5 µm, above which the rubber particles are considered to be too large to interact with the stress field at the tip of the crack and therefore they cannot contribute to energy dissipation. On the other hand, rubber domains below 0.1 µm are too small to cavitate, making the shear banding of the polymer matrix unlikely, which is also rather disadvantageous regarding the toughening [28]. Figure 5 displays the SEM images of the H543F PP-based samples prepared with the different dosing methods. The average size of XNBR domains is significantly smaller compared to the H145F PP-based samples regardless of the preparation route, which can be attributed to the higher viscosity of the H543F grade PP, which is closer to that of XNBR’s, resulting in a VR closer to “1”. The more optimal VR facilitated the rubber droplet break up by exerting an efficient shear stress transfer towards the dispersed phase, which then led to a finer diameter and better dispersion of the dispersed phase [21]. Accordingly, even the traditional melt mixing resulted in rubber domains of a few microns, even though some slightly larger particles (~ 10 µm) can still be observed (Fig. 5a, b). Applying the water-assisted method (Fig. 5c, d) improved the dispersion in this case as well, resulting in much smaller rubber domains. In the SEM image recorded at lower magnification (Fig. 5c), the XNBR droplets are barely visible since they are dispersed in the size range of 0.3-2 µm as seen in Fig. 5d. It should be underlined that the fracture surface morphology does not necessarily agree with that of the bulk though this is generally accepted in the first approximation. To confirm the agreement between the surface and bulk properties auxiliary experiments, such as transmission electron microscopy (TEM) would be needed.

SEM images of samples H145F_10XNBR_D (a, b) and H145F_10XNBR_L (c, d)

SEM images of samples H543F_10XNBR_D (a, b) and H543F_10XNBR_L (c, d)
Image analysis was applied to determine the size dispersion of the XNBR domains within the PP and further morphological parameters, based on SEM images. Over 200 unique rubber particles were measured in all samples containing 10 wt% XNBR. The only exception for that was the H154F_10XNBR_D blend, in which only a small amount of particles were found due to their large size and poor dispersion. Therefore, in that case, only ~50 unique particles were considered. The number average diameter (\(\overline{{D }_{n}}\)) of the XNBR droplets was calculated according to Eq. (1) [29, 30].
where Ni is the number of XNBR droplets with Di diameter.
Further, the inter-particle distance (IPD) and interfacial area per unit volume (Ai) were also determined using Eqs. (2) and (3) [29, 30].
where ϕR represents the volumetric concentration of XNBR rubber within the blend.
The calculated values of \(\overline{{D }_{n}}\), IPD and Ai for the analyzed samples are collected in Table 2. The calculated values of \(\overline{{D }_{n}}\) confirm the assumptions made based on the SEM images, namely that both the technique of XNBR addition and the viscosity of the PP matrix affect the average diameter of the resulting rubber domains. It needs to be noted, however, that the average diameter values were in all cases larger than those measured within the latex by laser scattering analysis (0.19 µm), which refers to some extent of coagulation even in the case of fast evaporation mixing. Nevertheless, when the VR ratio between the components was less favorable (H145F PP-based blends), the difference in the resulting average rubber diameter values between the two preparation methods was over thirtyfold with the water-assisted way being the more advantageous (98.2 µm > 2.7 µm). In H543F PP-based blends the occurring shear stresses were mostly sufficient to break up the rubber domains, exhibiting a much less difference between the two preparation methods (1.6 µm > 0.5 µm), but still proving the superior characteristics of fast evaporation mixing. The morphology refinement owing to the latex route is evident in the other determined morphological parameters too. For instance, there is a corresponding decrease in inter-particle distance (IPD) and a growth in interfacial area per unit volume (Ai) as well.
Tensile mechanical properties
The tensile mechanical properties (Young’s modulus, tensile strength and elongation at break) of the produced PP/XNBR blends were analyzed and compared. Data on the tensile properties of all the samples are summarized in Table 3. Based on the results it can be assumed that the type of PP greatly influenced the modulus of the samples. The H145F PP-based samples generally exhibited a higher modulus than those with H543F PP. This can be attributed to the higher tensile stiffness of the former PP grade. The Young’s modulus of the blends decreased with increasing concentration of XNBR as expected, due to the soft, elastomeric nature of the rubber component. Meanwhile, the dosing technique of XNBR had little to no effect on Young’s modulus of the samples. Apparently, the modulus of the samples mostly depended on the composition of the blends, rather than the dispersion of components.
Similar to Young’s modulus, the tensile strength values also decreased with higher rubber loading. In this case, however, the rate of decrement was greatly affected by the applied dosing method of the XNBR component. The strength of neat H145F PP (32.9 MPa) decreased relatively by 13%, 22% and 44%, at 5 wt.%, 10 wt.% and 20 wt.% XNBR loading, respectively, when the rubber was introduced as latex during the melt compounding. Meanwhile, when incorporated into the PP in its dry, bulk form, the relative decrement was 49%, 67% and 70%. This major drop in the strength values when applying the dry dosing method can be attributed to the poor dispersion of the rubber domains, which was also observed on the SEM images; the presence of rubber domains with a size of >100 µm supposedly acted as failure sites, thereby leading to an early failure of the specimens during the tensile tests. This assumption is also supported by the fact that the elongation at break of samples prepared by latex dosing continuously improved with increasing rubber content, while the ones prepared with dry rubber exhibited a rapid break at low elongation values. Considering these facts, the melt compounding of PP/XNBR blends using the latex-route seems highly favorable for this material combination.
Interestingly, no such advantage of this technique was observed in the tensile strength of the blends with H543F type PP; in this case, the samples with identical compositions exhibited quite similar tensile strengths (the measured values were within the deviation range) regardless of the preparation method. This can be explained by the much better dispersion of rubber particles, which is due to the better VR between the components. Apparently, the shear stresses during the melt compounding were suitable to disperse the dry rubber to small enough domains so they did not act as failure sites anymore. Thereby, it can be concluded that the smaller particles generally resulted in higher strength and elongation values, while the modulus of the samples was barely affected.
Charpy impact performance
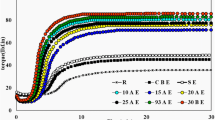
Figure 6a shows the Charpy impact strength of the H145F grade PP and its XNBR toughened blends, while the impact strength of the H543F PP and its blends can be seen in Fig. 6b. According to Fig. 6a, the addition of 5-10 wt.% dry XNBR to the PP H145F matrix led to slightly higher impact strength values; however, with 20 wt.% XNBR a drop was observed. This relatively poor toughening efficiency of dry dosed XNBR can be attributed to the large rubber domains that were observed on the SEM images (Fig. 4a) as well. Dosing XNBR as latex, on the other hand, led to a gradually increasing toughness, peaking at 7.3 kJ/m2, which is relatively 61% higher than that of neat H145F PP (4.5 kJ/m2). Accordingly, applying the water-assisted mixing for this pairing seems a favorable technique, since lower particle size can be achieved, which promotes the toughness of the blends. Based on Fig. 6b the toughness of the H543F-based blends continuously improved with increasing rubber content regardless of the applied dosing method. Even though the impact strength values measured on the samples prepared with latex addition were slightly higher than the dry dosed ones, the differences were within the deviation range for all concentrations. Apparently, the better VR of the H543F-based blends resulted in a more favorable rubber dispersion even for the dry dosed samples and therefore the effect of the water-assisted technique was less remarkable, which is in good accordance with the tensile test results. These results are also in good accord with the SEM analysis of the samples. Similar observations were made for PP/NR blends in an earlier study [26]. Generally, for both PPs there was a relatively low improvement in the impact strength – if any – as a result of XNBR-toughening compared to other elastomer types analyzed in the literature [26, 31]. This can be ascribed to the poor interfacial adhesion between the non-polar PP and polar XNBR phases.

Charpy impact strength of H145F PP/XNBR (a) and H543F PP/XNBR blends (b) containing different amounts of XNBR
Dynamic mechanical properties
The dynamic mechanical properties of the two PP types and their XNBR-toughened blends prepared with solid and liquid dosing are presented in Fig. 7. Figure 7a displays the storage modulus (E’) and loss factor (tan δ) curves of the samples based on the H145F grade PP. Apparently, the presence of XNBR rubber has a significant impact on the dynamic mechanical properties of PP, making the polymer matrix softer in the entire temperature range of analysis, which is manifested in a markedly lower storage modulus. Meanwhile, there is barely any difference between the samples H145F_10XNBR_L and H145F_10XNBR_D. These findings are in good agreement with those results obtained by the tensile tests. The tan δ plot of H145F PP exhibits one major peak, corresponding to its glass transition temperature (Tg), which is located in the range of -20 °C to 20 °C (peaking at 3 °C). When observing the loss factor curves of the XNBR-containing samples there is an obvious difference, namely the presence of a second characteristic peak between -40 °C and -20 °C. This is a typical range of Tg for rubbers [32], including XNBR. Above the glass transition temperature of XNBR, the tan δ curves of the filled and unfilled samples start to separate and there is a constant relative difference of ~25-30% in the whole temperature range after that with the XNBR-filled samples exhibiting higher values. It indicates a softening effect of XNBR on the H145F PP matrix. Apparently, the glass transition of PP did not shift to lower temperatures when being blended with XNBR, which supports the incompatibility between the components.

Storage modulus and loss factor of H145F PP/XNBR blends (a) and H543F PP/XNBR blends (b) prepared using different methods
As shown in Fig. 7b, a very similar dynamic mechanical behavior was found for the H543F PP-based samples. Here, however, the difference between the neat PP and its blends was less significant. The reason for that is the softer nature of the H543F PP matrix material in comparison with the H145F. As a consequence of its higher viscosity, the chain molecules of H543F PP are expected to align into crystalline domains more difficultly than those of H145F’s, which results in lower modulus values, and this is in good agreement with the tensile tests’ findings as well. Below -30 °C, where the XNBR enters its glassy state and becomes highly rigid, the storage modulus of PP/XNBR blends is slightly higher than that of neat PP. Interestingly, the separation of the XNBR-filled samples’ tan δ curve compared to neat PP above the rubber’s Tg is less prominent compared to the H145F PP-based samples, which also refers to a less intense softening effect of the rubber on the PP due to the initially softer nature of the H543F matrix.
Conclusions
PP/XNBR blends with up to 20 wt.% of rubber content were prepared using a water-assisted method, introducing XNBR in its latex form into the molten PP. Analogous reference samples with the same compositions were also fabricated using traditional melt mixing to serve as control samples, in order to analyze the efficiency of the latex route. According to the results, the incorporation of the XNBR into PP in the form of latex was an efficient way to achieve better dispersion and smaller average droplet size of the elastomer component within the blends compared to traditional melt mixing. This was attributed to the fact that latex contains rubber in the form of submicronic-sized globules and therefore, it was not required for shear forces to be present during the melt compounding to break up the bulk rubber. The improved dispersion and the small enough size of rubber domains obtained by the latex dosing ultimately led to enhanced toughness and ductility. Those blends prepared with the novel, water-assisted method consequently outperformed the control samples with respect to elongation at break, tensile strength and Charpy impact strength values. These advantages of the latex route were especially notable when the VR of the XNBR and the PP matrix was high (~9.2), in which case all the analyzed mechanical properties were superior compared to the samples prepared with dry dosing, especially at high (10 wt.%+) rubber concentration. When the VR between the components was closer to “1” (~4.2), the mechanical properties were found to be similar, regardless of the preparation method. The shear forces, in this case, were presumably adequate to achieve a proper dispersion of XNBR, even though the actual size of the rubber domains was still smaller when the latex route was applied.
Overall, water-assisted fast evaporation mixing has proven to be an effective method to prepare PP/XNBR blends with improved toughness and ductility. It needs to be underlined though that there are still challenges that are required to be analyzed further before this technique could be utilized in large-scale production. These include the adaptation of the technique to large-scale production machinery and the recycling of such PP/XNBR blends while also avoiding the potential coalescence of the incorporated rubber domains.
Data availability
The data are available from the author upon reasonable request.
References
Karger-Kocsis J, Bárány T (2019) Polypropylene Handbook: Morphology, Blends and Composites (eds). Springer International Publishing, Cham, Switzerland
Karger-Kocsis J (1995) Polypropylene Structure, Blends and Composites (eds), Chapman and Hall, London, United Kingdom
Singh T, Fekete I, Jakab SK, Lendvai L (2023) Selection of straw waste reinforced sustainable polymer composite using a multi-criteria decision-making approach. Biomass Conv Biorefinery
Lendvai L, Patnaik A (2022) The effect of coupling agent on the mechanical properties of injection molded polypropylene/wheat straw composites. Acta Technica Jaurinensis 15:232–238
Mahallati P, Rodrigue D (2015) Effect of Feeding Strategy on the Properties of PP/Recycled EPDM Blends. Int Polym Proc 30:276–283
Ma LF, Wang WK, Bao RY, Yang W, Xie BH, Yang MB (2013) Toughening of polypropylene with beta-nucleated thermoplastic vulcanizates based on polypropylene/ethylene-propylene-diene rubber blends. Materials & Design 51:536–543
Liu J, Zhu X (2019) Isotactic polypropylene toughened with poly(acrylonitrile-butadiene-styrene): compatibilizing role of nano-ZnO. Polymer-Plastics Technol Mater 58:2007–2018
Bendjaouahdou C, Bensaad S (2018) Aging studies of a polypropylene and natural rubber blend. Int J Industrial Chem 9:345–352
Sharika T, Abraham J, Arif PM, George SC, Kalarikkal N, Thomas S (2019) Excellent electromagnetic shield derived from MWCNT reinforced NR/PP blend nanocomposites with tailored microstructural properties. Compos Part B: Eng 173
Lv F, Fan JF, Huang JR, Cao LM, Yan XS, Ge L, Abubakar S, Chen YK (2020) Preparation of polypropylene/ethylene-propylene-diene terpolymer/nitrile rubber ternary thermoplastics vulcanizates with good mechanical properties and oil resistance by core-shell dynamic vulcanization. Polym Adv Technol 31:2161–2171
Han IS, Park C, Jeon H, Ahn S, Lee HG, Yoo J, Yoo Y (2023) Polypropylene/polyolefin elastomer composites with enhanced impact strength: the effect of rubber domain size on toughness. J Polym Res 30:224
George S, Joseph R, Thomas S, Varughese KT (1995) Blends of isotactic polypropylene and nitrile rubber - Morphology, mechanical-properties and compatibilization. Polymer 36:4405–4416
Soares BG, de Oliveira M, Meireles D, Sirqueira AS, Mauler RS (2008) Dynamically Vulcanized Polypropylene/Nitrile Rubber Blends: The Effect of Peroxide/Bis-Maleimide Curing System and Different Compatibilizing Systems. J Appl Polym Sci 110:3566–3573
Tian B, Li ZG, Li JF, Yao G, Dong W, Liu YG, Di MW (2020) The effects of rubber nanoparticles with different polarities on thermal properties and foaming performance of polypropylene blends. RSC Adv 10:31355–31362
Halász IZ, Kocsis D, Simon DA, Kohári A, Bárány T (2020) Development of Polypropylene-based Thermoplastic Elastomers with Crumb Rubber by Dynamic Vulcanization: A Potential Route for Rubber Recycling. Periodica Polytechnica-Chem Eng 64:248–254
Garcia PS, Gouveia RF, Maia JM, Scuracchio CH, Cruz SA (2018) 2D and 3D imaging of the deformation behavior of partially devulcanized rubber/polypropylene blends. Express Polym Lett 12:1047–1060
Rathi MK, Singh T, Chauhan R (2018) Dynamic mechanical analysis of waste tyre rubber filled brake friction composite materials. In: AIP Conf Proc p. 090082
Guezzout Z, Boublia A, Haddaoui N (2023) Enhancing thermal and mechanical properties of polypropylene-nitrile butadiene rubber nanocomposites through graphene oxide functionalization. J Polym Res 30:207
Peng T, Lv F, Gong Z, Cao LM, Yan XS, Ge L, Abubakar S, Chen YK (2020) Design of PP/EPDM/NBR TPVs with tunable mechanical properties via regulating the core-shell structure. Polym Test 90:8
Liang JZ, Li RKY (2000) Rubber toughening in polypropylene: A review. J Appl Polym Sci 77:409–417
Maria HJ, Lyczko N, Nzihou A, Joseph K, Mathew C, Thomas S (2014) Stress relaxation behavior of organically modified montmorillonite filled natural rubber/nitrile rubber nanocomposites. Appl Clay Sci 87:120–128
Bakhtiari A, Ghasemi FA, Naderi G, Nakhaei MR (2020) An approach to the optimization of mechanical properties of polypropylene/nitrile butadiene rubber/halloysite nanotube/polypropylene-g-maleic anhydride nanocomposites using response surface methodology. Polym Compos 41:2330–2343
Yazid H, Anwar UA, Zaubidah AS, Nurulizzati M, Sabtu M, Andrianny MJ, Nurazila MZ, Zin MRM, Chen RS, Ahmad S (2022) A combined method to probe the behaviour of the filler in polymer blend nanocomposites via X-ray diffraction and thermal measurement. Nano-Struct Nano-Objects 32
Li D, Xia H, Peng J, Zhai M, Wei G, Li J, Qiao J (2007) Radiation preparation of nano-powdered styrene-butadiene rubber (SBR) and its toughening effect for polystyrene and high-impact polystyrene. Radiat Phys Chem 76:1732–1735
Karger-Kocsis J, Kmetty Á, Lendvai L, Drakopoulos S, Bárány T (2015) Water-assisted production of thermoplastic nanocomposites: A review. Materials 8:72–95
Lendvai L (2021) A novel preparation method of polypropylene/natural rubber blends with improved toughness. Polym Int 70:298–307
Remanan S, Ghosh S, Das TK, Das NC (2020) Nano to microblend formation in poly(ethylene-co-methyl acrylate)/ poly(vinylidene fluoride) blend and investigation of its anomalies in rheological properties. Nano-Struct Nano-Objects 23
Pearson RA (2000) Introduction to the Toughening of Polymers. In: (eds.) Toughening of Plastics. Am Chem Soc 1-12
Francis B, Thomas S, Asari GV, Ramaswamy R, Jose S, Rao VL (2006) Synthesis of hydroxyl-terminated poly(ether ether ketone) with pendent tert-butyl groups and its use as a toughener for epoxy resins. J Polym Sci, Part B: Polym Phys 44:541–556
Chandran N, Chandran S, Maria HJ, Thomas S (2015) Compatibilizing action and localization of clay in a polypropylene/natural rubber (PP/NR) blend. RSC Adv 5:86265–86273
Fasihi M, Mansouri H (2016) Effect of rubber interparticle distance distribution on toughening behavior of thermoplastic polyolefin elastomer toughened polypropylene. J Appl Polym Sci 133
Peidayesh H, Špitalský Z, Chodák I (2022) Electrical Conductivity of Rubber Composites with Varying Crosslink Density under Cyclic Deformation. Polymers 14:3640
Acknowledgments
The work reported here was supported by the Hungarian Research Fund [Project No. OTKA K 109409]. The author thanks MOL Petrolkémiai Ltd. for kindly providing the PPs used as matrix material. The author would like to express his deep sense of gratitude to his late supervisor, Prof. J. Karger-Kocsis✝. The author thanks Dr. J. Móczó for his help with the laser diffraction particle size analysis.
Funding
Open access funding provided by Széchenyi István University (SZE).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
No potential conflict of interest is reported by the author.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lendvai, L. Mechanical and morphological properties of PP/XNBR blends produced with rubber latex. J Polym Res 30, 276 (2023). https://doi.org/10.1007/s10965-023-03660-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-023-03660-3