
Abstract
The introduction of a -NO2 substituent in o-aminophenol would create an open-unit polymer suitable for different applications. We report the synthesis of poly(5-nitro-2-aminophenol) by aniline-catalyzed oxidative polymerization. The polymer structure was intensively characterized by spectroscopic techniques, elemental analysis, cyclic voltammetry, electrical conductivity and microscopic measurements such as SEM, TEM, TGA and DSC. The hydrophilic and hydrophobic nature of the substituents provided the formation of amphiphilicity, as judged by SEM. Thermal measurements reveal high stability up to 500 °C. Electrical conductivity decreases with increasing temperature at various frequencies. This behavior reflects the semiconducting nature in the extrinsic range, which is characterized by high carriers and low mobility. Dielectric measurements have shown that permittivity decreases gradually at lower levels, mainly due to the transport of charging carriers. Investigating the copolymer as a new scale blocker has resulted in moderate to high performance attributed to the change in polymer geometry because of the presence of different tautomers having equivalent ortho functionalities and could equally coordinate with free calcium ions and thus reduce the nucleation rate and block crystal growth. Thus, the abovementioned features indicate a new potential candidate for application as a scaling inhibitor of CaSO4 and CaCO3 precipitation, a common problem in industry.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Scale formation is commonly encountered and poses problems in water boiler coolers and oil well water, which have a negative impact on operating systems and equipment, reducing heat transfer as an insulating layer and downhole completion equipment [1]. Among the commonly encountered scaling cations in aqueous systems are Ca2+ cations, which deposit calcium carbonate and/or calcium sulfate. Sulfate scales are often attributed to the mixing of incompatible sea and formation waters, where the concentrations of calcium ions are high in both [2]. Carbonate scale, on the other hand, is generally attributed to the process of self-scaling, where the loss of carbon dioxide gas from the water to the hydrocarbon phase occurs as pressure falls [3]. Functionalized organic polymers are green scale inhibitors and have shown excellent properties in delaying, reducing, and/or preventing scale deposition [4]. The effectiveness of these inhibitors depends on the chain length of the polymers, and thus, the efficiency order in most cases was a low chain- > higher chain length, where the polymeric repeating units were nearly 10–15 [5].
Aminophenols are interesting electrochemical materials because they have two oxidizable groups (–NH2 and –OH), enabling them to show electrochemical behavior resembling anilines and phenols [6]. The reported electrochemical and spectroscopic analyses revealed that the only electroactive polymer obtained by the oxidation of the isomers of aminophenol is that obtained with ortho-aminophenol. The structure of poly(o-aminophenol) has been studied by several techniques [7] to obtain better insight into redox reactions. o-Aminophenol has a high electron density in the para-position with respect to the -OH group; thus, the propagation step proceeds to produce a polymer having a ladder-type structure built-in by phenoxazine units [8] however, some open units remain linear in the polymer, and the -OH groups are oxidizable to ortho-quinonimines [9]. The redox response of poly(o-aminophenol) is usually due to the oxidation–reduction of phenoxazine units [10].
Polymerization of o-aminophenol and its copper complex was chemically achieved by oxidative polymerization in aqueous HCl and evaluated as a corrosion inhibitor on mild steel in 1 M HCl [11, 12]. Homogenous poly(o-aminophenol) was also prepared several times by electrochemical methods, and its film was used in the field of bioelectrochemistry [13]. The oxidation mechanism was studied using electrochemical techniques [10b] and chemical synthesis, where the formation of a soluble dimer (3-aminophenoxazone) was proven [14]. Poly(aniline-co–o-aminophenol) has also been synthesized electrochemically [15].
The introduction of –NO2 group substitution into polyaniline chains modifies the electronic and vibrational spectra of the resultant polymer [16]. Generally, anilines containing a strong electron-withdrawing substituent, such as -NO2, produce an unstable radical intermediate, and thus, polymerization does not occur. To solve this obstacle, a trace amount of aniline as an initiator/promoter improves the reaction rate and yield [17].
Since the scale inhibitors prevent the formation of metal salts based on the metal ion concentration in the medium, it rational to conclude that the inhibitor/metal ion interaction should be used as a parameter of the inhibition efficiency [18]. As a continuation of our recent interests [19], the evaluation of chemically prepared poly(5-nitro-2-aminophenol), for the first time, as a new scale inhibitor of CaCO3 and CaSO4 using slandered NACE, electrochemical tests and microscopic examinations was the main objective. The introduction of an electron-rich group such as -NO2 at the meta-position with respect to the -OH group would give an attractive monomeric unit with a number of resonating structures having several high-density sites and thus dominate the open units in the polymer and increase the coordination sites and thereby enhance its inhibition efficiency. The noteworthy, derivative-based monomer used in this work was actively used as an azo-chromophore for nonlinear optical applications and as a highly fungicidal agent for commercial applications [20]. The measurements were performed on as-synthesized samples without the further redoping procedure usually used in the literature.
Methods and materials
Chemicals
Commercial aniline (AlGomhoria Chemicals Co.; Egypt), 5-nitro-2-aminophenol (ICI, Manchester, UK), ammonium persulfate (Oxford Lab Fine Chemicals, India), double distilled water, analytical reagent-grade NaCl, NaHCO3, Na2SO4 and CaCl2 (Al-Gomhorya Chemicals Co., Egypt) were used without further purification.
Measurements
Infrared spectra (IR, KBr pellets; 3 mm thickness) were recorded on a Perkin-Elmer Infrared Spectrophotometer (FTIR 1650). All spectra were recorded within the wavenumber range of 4000–600 cm−1 at 25 °C. Absorption spectra were measured with a UV 500 UV–Vis spectrometer at 16 °C (rt) in DMSO with a polymer concentration of 2 mg/10 mL. Elemental analysis of the as-synthesized copolymer was performed at the Microanalytical Unit, Cairo University. Inherent viscosities (ηinh) were measured at a concentration of 0.5 g/dL in H2SO4 at 30 °C by using an Ubbelohde viscometer. Thermogravimetric (TG) and differential thermogravimetric (DTG) analyses were carried out in the temperature range from 20 °C to 400 °C in a nitrogen atmosphere by a Shimadzu DTG 60H thermal analyser. The experimental conditions were platinum crucible, nitrogen atmosphere with a 30 mL/min flow rate and a heating rate of 10 C/min. Differential scanning calorimetry (DSC-TGA) analyses were carried out using SDT-Q600-V20.5-Build-15 at the microanalytical unit, Cairo University. Cyclic voltammetry was performed using an eDAQ system (www.eDAQ.com, Australia) consisting of an ER466 potentiostat connected to an e-corder that was inputted to eChem software (running on a PC using Microsoft Windows 10). The working electrode was a 3 mm diameter glassy carbon electrode; the reference electrode was Ag/AgCl; and the auxiliary electrode was a 0.25 mm diameter Pt wire. The applied potentials ranged from -500 to + 500 mV, and the scan rate during one cycle was 100 mV s−1. The volume of the voltammetric cell was approximately 15 ml. The dielectric measurements of the polymeric materials were carried out using a high-resolution Alpha analyser (Novo Control, Hundsangen, Germany) in parallel plate geometry over a frequency range from 10−1 Hz to 107 Hz at different temperatures. In this geometry, the sample cell consists of two gold-coated brass disk electrodes. The upper one is 10 mm, and the lower one is 20 mm in diameter. The pressed sample with a 12 mm diameter was sandwiched between the two electrodes. The applied voltage was kept constant at 0.2 V to avoid any nonlinear effects. The empty sample capacitor is used as a reference to eliminate the additional contributions of the cables and the measurement cell. The temperature of the sample is controlled by a Quattro Novo control cryo-system with temperature stability better than 0.2 K, as described in references [21]. The polymer powder was pressed to form discs with diameters of 10 mm and thicknesses of 1 mm. Silver electrodes were deposited on both sides of the sample surface by thermal evaporation, and two copper wires were fixed on the sample using conducting silver paint. The morphologies of the polymers were observed by scanning electron microscopy (SEM) (JEOL-JSMIT 200, Japan) and transmission electron microscopy (TEM) (JEOL-JTM-1400 plus, Japan) at the E-Microscope Unit, Faculty of Science, Alexandria University. The samples were sonicated in deionized water for 5 min, deposited onto carbon-coated copper mesh and allowed to air-dry before examination.
Synthesis of poly(5-nitro-2-aminophenol)
5-Nitro-2-aminophenol 1 (5 g, 32.46 mmol) and aniline (0.5 g, 3.24 mmol) were dissolved in aqueous 1 M HCl (100 ml), and a solution of ammonium persulfate (9.25 g, 40.57 mmol) dissolved in water (50 ml) was subsequently added over a period of 30 min. The mixture was mechanically stirred for 24 h at rt, and the color change pattern during polymerization to a brownish black was observed from t = 10 min to 24 h. Polymerization was stopped by the addition of methanol (50 ml). The resulting precipitate was subsequently washed with water, aqueous 1 M HCl, water and acetone to remove the unreacted starting materials and short oligomers. Finally, the precipitate was dried in a vacuum oven at 50 °C. Yield: 3.1 g (62%). IR (cm−1, υ): 3503, 1628, 1600, 1524, 1439, 1401, 1292, 1212, 1164, 1079, 1041, 880, 819, 776, 743, 715, 648, 582, 544. UV–vis (λmax nm): 255, 415, 500. Calc. for C36H32N10Cl3O13: (919); C, 47.05; H, 3.51; N, 15.24; C/N ratio 3.08; Found: C, 47.88; H, 3.86; N, 15.70; C/N ratio 3.05.
Determination of the scale inhibitor rate
NACE test for CaSO4 scaling
Following the reported method described previously [22], calcium brine: 7.5 g/L NaCl + 11.0 g/L CaCl2.2H2O and sulfate brine: 7.5 g/L NaCl + 10.66 g/L NaSO4, 50 ml of each brine solution was connected in the test cell with different concentrations of sodium alginate and chitosan. Testing cells were placed in the water bath set at 71 °C for 72 h. Then, the concentration level of calcium ions was determined in the solution by titration with EDTA and Murexide indicator. The scale inhibitor percent was calculated following the equation: % inhibition = 100 x (Ca-Cb)/(Cc – Cb), where Ca = Ca2+ concentration in the treated sample after precipitation, Cb = Ca2+ concentration in the blank after precipitation and Cc = Ca2+ concentration in the blank before precipitation.
Electrochemical test for CaCO3 scaling
Monitoring the buildup of calcium carbonate layers on the metal surface was studied by electrochemical methods, including chronoamperometry and electrochemical impedance spectroscopy [19a].
Chronoamperometry test
Cathodic polarization of the steel electrode initializes the scaling process by forcing few nuclei of CaCO3 to be precipitated on the steel surface according to the following Eqs. (1) and (2):
In the chronoamperometry test, cathodic polarization was applied to the steel electrode surface, which increases the local pH at the cathode, as illustrated in Eq. 1, while increasing the hydroxyl ion concentration enables calcium carbonate to precipitate, as given in Eq. 2. The electrochemical measurements were carried out in a cell in three-electrode mode using a platinum sheet and saturated calomel electrode (SCE) as counter and reference electrodes, respectively. The material used for constructing the working electrode was steel that had the following chemical composition (wt.%): C, 0.21; S, 0.04; Mn, 2.5; P, 0.04; Si, 0.35; balance Fe. The steel electrode was polarized to − 1.0 V (vs. SCE) in test solution in the absence and presence of different concentrations of polymer 2 for 3 h using a Gamry instrument G300™ Potentiostat/Galvanostat/ZRA.
Electrochemical impedance spectroscopy
EIS measurements were performed at − 1.0 V (vs SCE) after the scale deposition process. The frequency range for EIS measurements was 0.1 to 1 × 104 Hz with an applied potential signal amplitude of 10 mV. All measurements were performed at 40.0 ± 0.1 °C in solutions open to the atmosphere without stirring. To test the reliability and reproducibility of the measurements, triplicate experiments were performed in each case under the same conditions.
Results and discussion
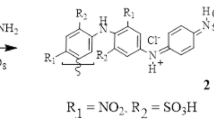
Targeted poly (5-nitro-2-aminophenol) 2 was chemically prepared according to the standard procedure described in the IUPAC technical report [23] using 10 mol % of aniline and 1.25 × equivalent of ammonium persulfate as an oxidizing agent from commercial 5-nitro-2-aminophenol 1 in low pH 1.5 aqueous HCl media (Supplementary 1). In substrate 1 used in this investigation, the presence of a strong electron withdrawal effect exerted by the -NO2 group could partially inhibit the first initiating oxidation step; thus, using a small amount of aniline to ensure the completion of polymerization was our alternative [24]. The percent yield (62%, ηinh = 0.10 dL/g) was calculated by using formula (3) [25]:
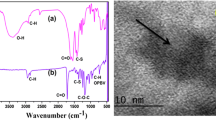
The reaction rate and yield are mainly based on the reactivity of substrate 1 during the oxidation and/or polymerization steps. The mechanistic oxidative polymerization of monomer 1 proceeds through attack of the cation radical at the para-position with respect to the -OH group [8], Scheme 1. The expected high electronic density at that position is mainly due to the directing effect of both -OH and -NO2 substituents. Additionally, the presence of -NO2 at the meta-position with respect to the -OH group would give 1.4 and 1.5 resonating structures, Scheme 1. In the latter resonating structures, the ortho-position with respect to the -OH group has a higher electronic density, favoring polymer propagation at that position. However, the resulting crosslinked polymer seems to be of low probability due to steric hindrance. Further head-to-tail oxidative polymerization can be expected by the unsubstituted comonomer aniline and thereby to achieve a fair polymerization yield, where the polymer contains a higher number of open units. The elemental compositions of polymer 2 were C, 47.88; H, 3.86; N, 15.70; C/N ratio 3.05 [Calc. for C36H32N10Cl3O13: (919); C, 47.05; H, 3.51; N, 15.24; C/N ratio 3.08]. The total amounts of carbon, hydrogen and nitrogen in the resulting polymer were 94%, indicating the contamination of the polymer with chlorine, as speculated for doped emeraldine salt forms [26]. The ratio of C/N was 3.05 (Calc. 3.08) and agrees with the theoretically predicted values for their analogues [27]. The morphology of synthesized polymer 2 investigated by SEM, Fig. 1a, revealed that the polymer has homogenous granular nanostructures with an average particle size of 40 nm. The TEM image, Fig. 1b, exhibited particle microaggregates and clearly indicated the presence of both unsubstituted aniline units (dark nanospheres) and substituted polymeric units (light gray aggregates), attributed to the nature of the substituent and thus the mechanism of the monomer unit interactions [28]. The presence of a monomer hydrophilic -OH group and a hydrophobic -NO2 on one aromatic ring gives the structure amphiphilicity or self-assembly nature, which are surrounded by the aromatic ring forming micelles [29], and the aggregation of such small micelles produces submicron groups that are templates for such morphology.

Chemical synthesis and a proposed formation mechanism of polymer 2

(a) SEM and (b) TEM images of the prepared poly(5-nitro-2-aminophenol) initiated by aniline 2
Infrared spectroscopy
The FTIR spectrum, Supplementary 2, shows a broad absorption at υ 3500–3400 cm−1 that corresponds to overlapping absorptions of adsorbed water and NHvib bonds. Absorption of -NHbend-containing aromatic rings and C = N and C = O str of the quinoid moiety in the polymer chain appeared at υ 1620 cm−1 [30]. Characteristic polymer structure peaks were found at υ 1600 and 1524 cm−1 corresponding to the vibrations of the benzenoid and quinoid moieties, and their intensity ratio was 1.5, confirming the rapid equilibrium between the two forms [31, 32]. The relative intensity of the quinoid/benzenoid bands reflects not only the degree of oxidation of the polymer chain [33] but also confirms the formation of bipolarons in the initial oxidation stage. The absorption bands at υ 1401 cm−1 and υ 1292 cm−1 correspond to the C-N and C-N+ stretching vibrations, respectively. The peak observed at υ 1212 cm−1 corresponds to the N = O symmetric stretching vibration due to NO2 groups as well as stretching vibration of the C-O bonds of phenolic moieties overlapped with the in-phase bending vibration of O–H [7b]. The asymmetric stretching band of the N = O group was not observed and could be buried under C-N absorption. The presence of a peak at υ 1079–1164 cm−1 indicates the doped state of the polymer (-N+/ N+•). The absorption at υ 819 cm−1 corresponds to the bending vibrations of C-H in the plane of the aromatic ring.
UV absorption
The bandgap energy is calculated from the equation ΔE = hc/λ, where ΔE is the bandgap energy (eV), h = 6.625 × 10–34 JS, c = 3 × 108 m/s, and λ is the wavelength. The electronic spectrum of substituted polymer 2, Supplementary 3, shows absorption bands at υ 255 nm, υ 415 nm, and υ 500 nm, and their bandgap energies were 4.86 eV, 2.99 eV, and 2.48 eV, respectively. Unsubstituted polyaniline exhibits a peak at 280–295 nm due to the π-π* transition of the benzenoid ring in the extent of conjugation of the adjacent phenyl rings in the chain. Absorption at 605–635 nm is attributed to the quinoid ring transition, and charge transfer from the HOMO of the benzenoid ring to the LUMO of the quinoid ring. This specific absorption is dependent on the overall oxidation state of the polymer [34]. Homopolymerized o-aminophenol prepared by cyclic voltammetry exhibits an absorption peak at 410 nm [13b]. In our sample, the peak at υ 255 nm corresponds to the π-π* transition of the benzenoid ring, and the absorption peak at υ 418 nm is a characteristic band for the n-π* transition, indicating that the obtained polymer is in the doped state of the emeraldine structure, which further supports the IR data discussed above [18]. The hypsochromic shift of the π-π* transition, the n-π* transition and the quinoid ring transition found at υ 500 nm could be attributed to the presence of random substitution of the monomers of such polymerization, which causes entanglement of the polymer chain and thus disrupts the conjugation.
Cyclic voltammetry
Cyclic voltammetry is a method used for investigating the electrochemical behavior of a system. The measurement is based on varying the applied potential at a working electrode in both the forward and reverse directions (at some scan rate) while observing the current from the plot of current versus applied potential. Unsubstituted polyaniline shows redox activity. Electron transfer processes occur under the action of a potential to an electrode modified with a polymer. The reported electrochemistry of the polymer indicated [35] the presence of three stable oxidation states of polyaniline, namely, the fully reduced state leucoemeraldine (at potentials below + 0.2 V), the fully oxidized state pernigraniline (at a potential above + 0.8 V) and emeraldine (at potentials from + 0.2 to + 0.8 V). Incorporation of various functional groups at the polyaniline aromatic ring shifts the potential of redox transitions in the compound [36], and the effect of the substituent on the redox behavior is determined by its electronic and/or steric effect. Incorporation of more than one functional group at the aniline's aromatic ring not only decreases the oxidation potential in comparison with monosubstituted derivatives but also affects the polymerization rate, which decreases with an increase in the substituent chain length due to steric influence. Generally, the oxidation potentials of all substituted polyaniline derivatives are smaller than that of the unsubstituted polymer, which indicates that the derivatives are oxidized more readily.
The reported redox data of poly(o-aminophenol) are variable and mainly based on the synthetic conditions that gave at least two types of poly(o-aminophenol) differing in their electrochemical properties, and their potential peaks were in the range of 0.1–0.2 V and a slightly pronounced peak at ~ 0.4 V [37]. The latter potential peak is related to the reversible redox center of the -OH of some unconverted liner chains, while only a brood oxidation peak appeared in the forward scan. The former two reduction peaks are attributed to the reduction process of the fully oxidized polymer [38]. Notably, redox data of the o-aminophenol/aniline copolymer exhibited two similar pairs of potential peaks 0.1–0.2 V, and no peaks were observed in the higher potential range.
The electrochemical study of substituted copolymer 2 was conducted by measuring the cyclic voltammogram (Fig. 2). The experiment (one cycle) was conducted at a concentration of 5 mg in 20 ml DMSO solution. The cyclic voltammetry maxima corresponding to transitions between the oxidation forms are postulated in Supplementary 4.

Cyclic voltammogram of substituted prepared polymer 2. Scan rate of 100 mVs−1
The voltammogram wave shape showed two broad cathodic peaks at -0.260 V (i = -2.02 μA), and 0.176 V (i = -1.68 μA) assumed for the reduction of the quinoid structure in the polymer chain, and two broad anodic peaks at 0.338 V (i = 2.03 μA) and -0.132 V (i = 1.73 μA), respectively. The observed redox peaks are due to the different stabilization of the radical cations during the transitions from leucoemeraldine to emeraldine and from emeraldine to the perigraniline state of copolymer 2, respectively [36], Supplementary 4. The redox nature of copolymer 2 was compared with that of poly(o-aminophenol). It is noted that the peak caused by the second redox process had a slightly lower potential (0.338 V) than that observed in the case of poly(o-aminophenol) (0.4 V). The electronic substituent effect of (–NO2) increases the degree of chain conjugation and further stabilizes the polysemiquinone radical cations formed in the first redox process. Since the radical cation of the intermediate is more stable than that of poly(o-aminophenol), a lower potential is required for its formation, and further oxidation with simultaneous deprotonation occurs more readily.
Thermal analysis
TG-DTG and DSC curves for the substituted polymer 2 are shown in Supplementary 5. The TG thermal degradation curve, Supplementary 5a, exhibited an interesting model of the stability of the polymer, and its subsequent weight losses could be divided into four steps. The first minor peak from rt to 143 °C (weight loss 4.15%) could be attributed to the loss of absorbed water. The second minor weight loss from 143 °C – 232 °C (weight loss 8.2%) is attributed to the loss of acid dopant from the polymeric matrix. The third weight loss (major peak) from 233 °C – 409 °C (weight loss 23.35%) (strong differential thermogravimetric endothermic peak at 292 °C) and from 409 °C – 472 °C are likely due to decomposition and/or elimination processes of the side-chain substituents. The oxidative thermal decomposition of polymer backbones was suggested above 500 °C for the remaining polymeric residues (50%) as the final weight residue. In the DSC curve, Supplementary 5b, the polymer shows an endothermic peak with an energy of -7.70 J/g at 200 °C, as well as a broad endothermic peak of energy 45.2 J/g at 350 °C, which corresponds to decomposition or elimination processes of the side-chain substituents and the subsequent morphological change in the polymer [39].
Electrical conductivity
Dielectric measurements
The dielectric loss ε’, permittivity ε” and real part of the complex conductivity σ′ are determined over a frequency range varying from 1 mHz up to 10 MHz at room temperature. The measurements can probe molecular fluctuations and charge transport over broad frequency and temperature ranges. The dielectric spectrum is separated by molecular dynamics at the molecular scale, i.e., charge carrier transport, which is reflected in conductivity mechanisms. The permittivity (ε”) is the measure of a material's response to an electric field and is often expressed as the ratio of its electric displacement to the applied field strength. The permittivity (ε”) was determined at five-spot frequency points and depicted against temperatures in the range of 40 °C to 100 °C for substituted polymer 2 (Fig. 3). In general, two distinguished trends of the real part of the complex permittivity ε′ vs frequency can be assigned. The lower frequency range (1.0 Hz–100 Hz) shows a gradual decrease in ε′ through six orders of magnitudes, mainly due to the charge carrier transport causing the expected high conductivity. At frequencies higher than 1 kHz—> 1000 kHz, a slight effect was observed for frequency on decreasing the permittivity. This effect agrees with the fact that at higher frequencies, the contribution of the alteration of all kinds of polarizations lags the frequency of the applied external electric field [40]. There are three modes of polarization [41]: (1) electronic polarization: slight displacement of electrons with respect to the nucleus, (2) atomic polarization: distortion of atomic position in a molecule or lattice, and (3) orientational polarization for polar molecules: there is a tendency for the permanent dipole to align by the electric field to give a net polarization in that direction. All three modes of polarization contributing to the overall dielectric constant will be dependent on the frequency of the oscillating electric field. The dielectric properties of a polymer are determined by the charge distribution and by the statistical thermal motion of its polar group. The polarization of a dielectric is contributed by ionic, electronic and dipole polarization. However, polymers are gaining wider use as dielectric materials. This is due to the easier processing, flexibility, ability to tailor made for specific uses and better resistance to chemical attack. Physically, this means that the greater the polarization developed by a material in an applied field of given strength, the greater the dielectric constant will be. Therefore, the permittivity increases with increasing temperature, which is likely due to the increase in dipole mobility [42]. The mobility of charge carriers increases with temperature, which increases the polarization and leads to high dielectric loss. The observed higher value of dielectric loss at high temperature is due to charge accumulation at grain boundaries [43].

Effect of temperature on permittivity (ε”) at different frequencies for polymer 2
The variation in conductivity σ’ vs frequency (DC conductivity) and the dissipation factor tan δ vs frequency at a temperature range (40 °C – 100 °C) are illustrated graphically in Fig. 4a and b for polymer 2, respectively. The dissipation factor tan δ represents the energy loss and occurs when the polarization shifts behind the applied electric field caused by the grain boundaries, which is expressed by the ratio between the dielectric loss and the dielectric constant. Figures show a clear sharp peak followed by a small shoulder at lower limit points of frequency. At higher frequencies, it follows the power law: [σ’ (ω) = Aωs], in which A is constant and s characterizes the rate of change of AC conductivity with increasing frequency. The intermediate range of frequency shows a plateau-like behavior that represents the dc conductivity, σdc. It varies slightly between 1 and 2 mS/cm by the variation of the irradiation dose, which confirms the fact that the contribution of the conductivity plays the main role of the permittivity values at the lower frequencies. The accumulation of charge carriers is the origin of electrode polarization, a ubiquitous phenomenon that takes place at the interface between a metallic and an ionic conductor and thus increases by many orders of magnitude by the net dielectric response of the sample cell. Since Coulombic interactions take place here, ion mobility is drastically slowed down at the interfaces.

(a) Conductivity (σ`) vs frequency- and (b) Dissipation factor (tan δ) vs frequency for polymer 2 in the temperature range of 40 °C-100 °C
Figure 5 illustrates the effect of temperature on the conductivity σ’ and dielectric constant ε′ plotted at different frequencies (i.e., 100 Hz, 10, and 100 kHz) for polymer 2. In Fig. 5a, as the temperature increases up to 40 °C, the conductivity at higher frequencies (100 kHz) increases and then steadily decreases. This anomalous metallic to semiconducting behavior change at higher frequencies is indicative of a charge nature in the extrinsic range, which is characterized by high carriers and high mobility at low temperatures and high carriers and low mobility at higher temperatures. At lower frequencies (100 Hz), the conductivity remains constant as the temperature increases. The main two factors affecting the value of conductivity are the density of the charge carriers, n, and their mobility, μ. The charge density and mobility as a function of temperature, and the effect of the change in charge density only plays a limited role in the variation in the DC conductivity. This can be attributed to the fluctuation dynamics of the accumulated charge carriers at the interface. These fluctuation dynamics seem to be the reason for the asymmetric wing of the σ″ peak at lower frequencies, which is accompanied by a shoulder in the tan δ and deviation from the linearity of the conductivity decrease shown in Fig. 4a [44]. The low conductivity of copolymer 2 could be explained by both the reduced conjugation length in distorted chains and the decrease in interchain charge transport induced by the constant tautomeric exchange and the electrostatic interactions between the active functional group and main-chain cationic nitrogen or amine hydrogens. The study of the dielectric constant as a function of temperature and frequency is one of the most convenient methods of studying the molecular orientation behavior and associated relaxation mechanism of polymer structures [45]. In Fig. 5b, the dielectric constant decreases with increasing frequencies. At all frequencies, the dielectric constant increases in the (20 °C-40 °C) temperature range and starts to decrease in the (40 °C - 60 °C) range. The low-temperature dielectric dispersion is attributed to the dielectric response of the side groups, which are more mobile, or the small displacement of the dipoles near the frozen-in position, which is known as the secondary dispersion region or ß-relaxation [46] and may suggest that the dipole–dipole interactions between the different groups or many body interactions suggest lower losses with a higher frequency range. Therefore, the increase in losses at low frequency could be associated with the polarization of the trapped charge carriers, as shown in Fig. 6. With an increase in frequency, polarization decreases and becomes vanishing small at high frequencies [47].

Variation in (a) conductivity (σ′) and (b) dielectric constant (ε′) vs temperature for polymer 2 at 100 Hz, 10 kHz and 100 kHz frequencies

Dielectric loss (ɛʹʹ) vs frequency in the temperature range of 40 °C-100 °C for polymer 2
Jonscher’s power exponent value and its temperature dependence are very important parameters in dielectric data analysis as well as the physical interpretation of conduction mechanisms in disordered materials.
DC conductivity depending on temperature, calculated based on Jonscher’s power law (Table 1):
where (A) is the characteristic parameter, (f) is the frequency and (s) is the exponent dependent on temperature and frequency, with values in the range from 0 for ideal ionic-type crystals to 1 for ideal dielectric dipolar-type crystals. The value of the exponent s in the range 0 < s < 1 is characteristic of hopping conduction phenomena [48, 49]. One observes that the value of (s) increases linearly with temperature. This may be a result of the rise in the electrode polarization contribution with temperature, which leads to a decreasing amount of A. C conductivity.
Assessment of copolymer 2 as a precipitation inhibitor of CaCO3 and CaSO4 in solution
Effect of copolymer 2 on CaSO4 precipitation
To examine polymer 2 as a new scale inhibitor of CaSO4, the free calcium ion concentration was determined according to the NACE test in the absence and presence of different concentrations (ppm) of the polymers. As seen in Table 2, the concentration of free Ca+2 ions increased with increasing polymer concentration, which indicated that the presence of the polymer retarded the calcium sulfate precipitation process. The inhibition efficiency steadily increased with polymer concentration, and the maximum inhibition efficiency achieved 76% in the presence of 100 ppm. The efficiency of polymer 2 is attributed, most likely, to the presence of two ortho active coordinating functionalities (-OH, -NH) on the polymer backbone. Coordination with metal ions would give a stable five-membered ring and thus decrease the probability of CaSO4 precipitation. The inhibition percent of CaSO4 increases with increasing polymer contents, which can be explained either by coordination to the active functional or by the interaction of the polymeric backbone itself with calcium ions, thereby preventing the Ca2+ arrival at the surface of scale nuclei and blocking crystal growth. These observations are compatible with three mechanisms: ionic exchange, dissolution/precipitation, and complexation reactions. The interaction of active functional sites with Ca+2 ions reduces the nucleation rate and blocks crystal growth. Moreover, the polymer may have dispersive properties because of the electrostatic field generated by the extra charges, and thereby the inhibitor chemisorbs onto the crystal surface, producing similar charged crystals [19a]. The high efficiency of polymer 2 could be attributed to a change in polymer geometry because of the expected possible three tautomers having equivalent ortho functionalities and could equally coordinate with metal ions in an aqueous medium (Supplementary 4), thus reducing the nucleation rate and blocking crystal growth.
Effect of copolymer 2 on CaCO3 precipitation
Chronoamperometry measurements
Chronoamperometry curves of the polarized steel electrode in the absence and presence of the polymer are displayed in Fig. 7. As shown, in brine solution, the curve decreases sharply, which indicates fast formation of a calcium carbonate layer on the steel surface and causes a decrease in the current value. On the other hand, the buildup of the calcium carbonate layer on the metal surface was disturbed by the addition of the polymer to the brine solution, which delayed the scaling rate and increased the residual current on the steel surface. Increasing the concentration of the polymer up to 50 ppm increases the residual current values for the steel electrode after polarization from 44 μA to 111 μA. After this concentration, a sudden residual current decrease was observed (Fig. 8). It has been reported that the current density is inversely proportional to the quantity of CaCO3 scales that form on metal surfaces [50].

Chronoamperometry curves for the polarized steel electrode in CaCl2 brine solution in the absence and presence of different concentrations of the studied polymer at –1 V vs SCE and 40 ○C

Variation in residual current for the steel electrode after 3 h of polarization in CaCl2 brine solution in the absence and presence of different concentrations of copolymer 2 at –1 V vs SCE and 40 ○C
Electrochemical impedance measurements
Figure 9 represents Nyquist plots for steel after cathodic polarization in brine solution in the absence and presence of different concentrations of polymer 2. A typical feature of depressed semicircles of impedance spectra of the polymer was shown. Moreover, the size of distorted semicircles decreases in the presence of the polymer due to the reduction of the insulation layer of CaCO3 scale. The equivalent circuit used to fit the experimental data of impedance plots for the scale formation processes in brine solution is illustrated in Supplementary 6.

Impedance spectra of the polarized steel in brine solution in the absence and the presence of different concentrations of polymer 2 at 40 °C
Table 3 shows the computer fit results of the impedance spectra obtained for the steel electrode that was cathodically polarized in the CaCl2 brine solution containing different concentrations of the polymer after 3 h. The percentage of scale inhibition could be determined from the following equation: % scale inhibition = [(Rcto – Rcti)/Rcto] × 100 [19a], where (Rct)o and (Rct)i are charge transfer resistances after polarizing the steel electrode at -1 V (vs. SCE) in a scaling environment for 3 h in the absence and presence of the polymer, respectively. The results indicated that increasing the concentration up to 50 ppm resulted in a decrease in the charge transfer resistance Rct, which is inversely proportional to the formation of scale. Additionally, the presence of the polymer increased the nonideal film capacitance values, Q, due to the formation of the double layer, which reduced the formation of the insulating layer on the metal surface [51]. Further increasing the polymer concentration decreases the nonideal film capacitance because of an increase in the thickness of the electrical double layer associated with the presence of polymer molecules, which increases the charge density near the metal surface and decreases the efficiency of the polymer due to the steric effect of the high molecular weight polymer.
SEM images of CaCO3 crystals after direct precipitation are displayed in Fig. 10. A cubic structure of calcium carbonate crystals is observed in the absence of the inhibitor. No notable crystal modification was observed in the presence of the polymer. The inhibition effect of low concentrations of the polymer towered CaCO3 precipitation can be attributed to two concepts: either the presence of active functional groups such as -NH and -OH acting as multiple adsorption sites and making them easily adorable on the metal surface and hindering crystal attachment [20a] or, these functional groups can interact with calcium ions present in the media and thus prevent arrival at the surface of scale nuclei [51].

SEM images of CaCO3 crystals: (a) in the absence and (b) in the presence of polymer 2
Conclusion
The reported electrochemical and spectroscopic analyses indicated that the only electroactive polymer obtained by the oxidation of the isomers of aminophenol is that obtained with ortho-aminophenol. Introduction of the -NO2 group would create an open-unit analog containing several coordination sites and thereby enhance its scale inhibition efficiency. In this work, we prepared targeted poly(5-nitro-2-aminophenol) using aniline as an initiator. The obtained polymer has a homogenous granular nanostructure, and the presence of hydrophilic/hydrophobic substituents gives the structure amphiphilicity. The absorption bands and their corresponding gab energies clearly pointed out the benzenoid-quinoid ring transition. The voltammogram wave shape showed two broad cathodic peaks assumed for the reduction of quinoid structures and two broad anodic peaks assumed for the oxidation of benzenoid structures. The observed redox peaks are due to the different stabilization of the radical cations during the transitions from leucoemeraldine to emeraldine and from emeraldine to the perigraniline state of copolymer 2. The presence of (–NO2) increased the degree of chain conjugation and further stabilized the radical cation intermediate; therefore, further oxidation with simultaneous deprotonation occurs more readily by a lower potential. The thermal degradation exhibited an interesting model of the stability of the polymer and its subsequent weight losses within four steps up to 500 °C. The AC conductivity and dielectric response measurements indicated that the data could be described by the power low relation: [σ’ (ω) = Aωs] at higher frequencies, where the parameter “S” decreases in the range of study and hence follows a correlated barrier hopping conduction mechanism. The dielectric indicated that the permittivity gradually decreased at the lower frequency range mainly due to the charge carrier transport causing the expected high conductivity, while at a higher frequency range, a slight effect was observed.
As a new scale inhibitor of CaSO4, the inhibition efficiency steadily increased with polymer concentration, and the efficiency achieved 76% attributed to the change in polymer geometry because of the presence of three possible tautomers that could equally coordinate with calcium ions and reduce the nucleation rate. The copolymer exerted moderate CaCO3 inhibition with increasing inhibitor concentration despite the observed anomalous behavior. Thus, the abovementioned features make the polymer a potential candidate for application as a new multifunctional scaling inhibitor, a common problem in industry.
Data availability
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
References
Jamero J, Zarrouk SJ, Mroczek E (2018) Mineral scaling in two-phase geothermal pipelines: two case studies. Geothermics 72:1–14
Kodel KA, Andrade PF, Valença JVD, Souza DN (2012) Study on the composition of mineral scales in oil wells. J Pet Sci Eng 81:1–6
Dyer S, Graham G (2002) The effect of temperature and pressure on oilfield scale formation. J Pet Sci Eng 35:95–107
Amjad Z, Landgraf RT, Penn JL (2014) Calcium sulfate dihydrate (gypsum) scale inhibition by PAA, PAPEMP, and PAA/PAPEMP blend. Int J Corr Scale Inh 3:35–47
a) Majeed N (2011) Polymeric materials for scale inhibition in cooling water systems. Tikritj Eng Sci 18: 1–11. https://doi.org/10.25130/TJES.V1812.111. b) Popov K, Oshchepkov M, Afanasev E, Koltinov K, Dikarev Y, Rönkkömäkic H (2019) A new insight into the mechanism of the scale inhibition: DLS study of gypsum nucleation in presence of phosphonates using nanosilver dispersion as an internal light scattering intensity reference. Colloids Surf A Physicochem Eng Asp 560: 122–129. https://doi.org/10.1016/j.colsurfa.2018.10.015
Gopalasamy T, Gopichand M, Gopalswamy M, Raj J (2014) Poly Meta-Aminophenol: Chemical Synthesis, Characterization and Ac Impedance Study. J Polym ID 827043. https://doi.org/10.1155/2014/827043
a) Guenbour A, Kacemi A, Benbachir A, Aries L (2000) Electropolymerization of 2-aminophenol: Electrochemical and spectroscopic studies. Prog Org Coat 38: 121–126. https://doi.org/10.1016/S0300-9440(00)00085-0. b) Kunimura S, Oshaka T, Oyama N (1988) Preparation of Thin Polymeric Films on Electrode Surfaces by Electropolymerization of o-Aminophenol. Macromolecules 21: 894–900. https://doi.org/10.1021/ma00182a007. c) Jackowska K, Bukowska J, Kudelski A (1993) Electro-oxidation of o-aminophenol studied by cyclic voltammetry and surface enhanced Raman scattering (SERS). J Electroanal Chem 350: 177–178. https://doi.org/10.1016/0022-0728(93)80204-U
Salavagione HJ, Arias J, Garces P, Morallon E, Barbero C, Vazquez JL (2004) Spectroelectrochemical study of the oxidation of aminophenols on platinum electrode in acid medium. J Electroanal Chem 565:375–383
Yano J, Kawakami H, Yamasaki S (1999) Potential response of poly(o-aminophenol) film electrode to dissolved ferric ion. Synth Met 102:1335
a) Ohsaka T, Kunimura S, OyamaN (1988) Electrode Kinetics of Poly(o-Aminophenol) Film Prepared by Electro- Oxidative Polymerization of o-Aminophenol and Its Electrochromic Properties. Electrochim Acta 33: 639–645. https://doi.org/10.1016/0013-4686(88)80062-8. b) Barbero C, Silber JJ, Sereno L (1989) Formation of a Novel Electroactive Film by Electropolymerization or Ortho-aminophenol, Study of Its Electrochemical Structure and Formation Mechanism. Electropolymerization of Analogous Compounds. J Electroanal Chem Interf Electrochem 263: 333–352. https://doi.org/10.1016/0022-0728(89)85103-4
Thenmozhi G, Arockiasamy P, Jaya Santhi R (2014) Isomers of Poly Aminophenol: Chemical Synthesis, Characterization, and Its Corrosion Protection Aspect on Mild Steel in 1 M HCl. Int J Electrochemistry 2 ID 961617
Rivas BL, Sanchez CO, Bernede J-C, Mollinie P (2002) Synthesis, characterization, and properties of poly (2- and 3-aminophenol) and poly(2- and 3-aminophenol)-Cu(II) materials. Polym Bull 49:257–264
a) Tucceri R (2013) Non-Conducting Poly(O-Aminophenol) Films in the Field of the Bioelectrochemistry. Am J Anal Chem 4: 13–26. https://doi.org/10.4236/ajac.2013.46A003. b) Armijo F, Canales II, Del Río R, del Valle MA (2009) Poly-o-Aminophenol Obtained at High Potentials by Cyclic Voltammetry on SnO2: F Electrodes. Application in Quantitative Determination of Ascorbic Acid. J Chil Chem Soc 54: 158–162. https://doi.org/10.4067/S0717-97072009000200014
Kaizer J, Csonka R, Speier G (2002) TEMPO-initiated oxidation of 2-aminophenol to 2-aminophen oxazin-3-one. J Mol Catal A: Chemical 180:91–96
a) Mu S (2004) Electrochemical copolymerization of aniline and o-aminophenol. Synth Met 143: 259–268. https://doi.org/10.1016/j.synthmet.2003.12.008. b) Zhang L, Lian J (2007) Electrochemical synthesis of copolymer of aniline and o-aminophenol and its use to the electrocatalytic oxidation of ascorbic acid. J Electroanal Chem 611: 51–59. https://doi.org/10.1016/j.elechem.2007.08.002. c) A-ul-Haq A, Shah A, Holze R (200) Copolymers and two-layered composites of poly(o-aminophenol) and polyaniline. J Solid State Electrochem 11: 38–51. https://doi.org/10.1007/s10008-005-0064-0
Ibrahim KA (2017) Synthesis and characterization of polyaniline and poly(aniline-co-o-nitroaniline) Using vibrational spectroscopy. Arab J Chem 10:S2668–S2674
a) Rashid M, Sabir S (2009) Oxidative copolymerization of aniline with o- and p-nitroaniline by ammonium persulfate: kinetic and pathway. J Dispers Sci Technol 30:297–304. https://doi.org/10.1080/0193269082540004. b) Waware US, Hamouda AMS, Majumdar D (2020) Synthesis, characterization, and physicochemical studies of copolymers of aniline and 3-nitroaniline. Polym Bull 77:4469–4488. https://doi.org/10.1007/s00289-019-02957-y
Li C, Zhang C, Zhang W (2019) The inhibition effect mechanisms of four scale inhibitors on the formation and crystal growth of CaCO3 in solution. Sci Rep 9:13366
a) Abd-El-Khalek DE, Hassan HHAM, Ramadan SR (2021) Water-soluble sulfonated polyaniline as multifunctional scaling inhibitor for crystallization control in industrial applications. Chem Eng Res Design 169: 135–141. https://doi.org/10.1016/j.cherd.2021.03.004. b) El-Din Z, Hassan AM, HHAM, Abou El-Kheir MM, Youssef RM (2016) Controlling soil surface crust formation using Nanosized sulfonated polyaniline. Unified Journals, J soil water Conserv 1: 001–009.
a) L. Mukhtorov, G. Pestsov, M. Nikishina, E. Ivanova, Y. Atroshchenko, L. Perelomov (2019).Fungicidal Properties of 2-Amino-4-nitrophenol and Its Derivatives Bull. Enviro. Contamin. Toxicol. 102: 880-886. https://doi.org/10.1007/s00128-019-02602-4. b) G. A. Selivanova. Azo chromophores for nonlinear-optical application. Russ. Chem. Bull., Int. Ed. 2021;70: 213—238. https://doi.org/10.1007/s11172-021-3080-z
a) Youssef AM. Abd El-Aziz ME, Abd El-Sayed ES, Moussa MA, Turky GM, Kamel S (2019) Rational design and electrical study of conducting bionanocomposites hydrogel based on chitosan and silver nanoparticles. Int J Bio Macromol 140: 886–894. https://doi.org/10.1016/j.ijbiomac.2019.08.199. b) Kremer F (2002) Dielectric spectroscopy–yesterday today and tomorrow. J Non-Cryst Solids 305:1–9. https://doi.org/10.1016/s0022-3093(02)01083-9. c) Turky GM, El-Adly RA (2017) Study of phase separation and anomalous molecular behavior of Jojoba oil using dielectric spectroscopy. J Mol Liq 242: 1–7. https://doi.org/10.1016/j.molliq.2017.06.126
a) NACE Standard (2001) Laboratory Screening Tests to Determine the Ability of scale Inhibitors to Prevent the Precipitation of Calcium Sulfate. T.M.0374–2001; Item 21208. b) Boris AM, Alla YF, Margarita AK (2007) NACE Corros. 07064
Stejskal J, Gilbert RG (2002) Polyaniline. Preparation of a conducting polymer (IUPAC Technical Report). Pure Appl Chem 74: 857–867
Roy BC, Gupta MD, Ray JK (1995) Studies on Conducting Polymers. 1. Aniline-Initiated Polymerization of Nitroanilines. Macromolecules 28: 1727–1732
Bilal S, Gul S, Holze R, Anwar-ul-Haq AS (2015) An impressive emulsion polymerization route for the synthesis of highly soluble and conducting polyaniline salts. Synth Met 206:131–144
Gizdavic-Nikolaidis MR, Bennett JR, Swift S, Easteal AJ, Ambrose M (2011) Broad spectrum antimicrobial activity of functionalized polyanilines. Acta Biomater 7:4204–4209
Thiemann C, Brett CMA (2001) Electropolymerisation and properties of conducting polymers derived from aminobenzenesulphonic acids and from mixtures with aniline. Synth Met 125:445–451
a) Kazuo A (2009) Advances in Liquid Crystalline Conjugated Polymers. J Polym Sci A Polym Chem 47: 2463–2485. https://doi.org/10.1002/pola.23277. b) Kazuo A (2007) Synthesis and Properties of Liquid-Crystalline-Conjugated Polymers. Bull Chem Soc Japan 8: 649–661. https://doi.org/10.1246/bcsj.80.649
Isakova AA, Gribkova OL, Aliev AD, Indenbom AV, Shevlyakova NV, Tverskoi VA, Nekrasov AA (2020) The Synthesis of Polyaniline in Polyethylene Films with Grafted Sulfonated Polystyrene and Properties of These Films. Prot Met Phys Chem Surf 56:725–733
Socrates G (2001) Infrared and Raman Characteristic Group Frequencies, 3rd edn. Wiley, New York
Zhang Y, Kim JJ, Chen D, Tuller HL, Rutledge GC (2014) Electrospun Polyaniline Fibers as Highly Sensitive Room Temperature Chemiresistive Sensors for Ammonia and Nitrogen Dioxide Gases. Adv Funct Mater 24:4005–4014
Scotto J, Florit M, Posadas D (2018) About the species formed during the electrochemical half oxidation of polyaniline: Polaron-bipolaron equilibrium Electrochim. Acta 268:187–194
Bacon J, Adams RN (1968) Anodic oxidations of aromatic amines. III. Substituted anilines in aqueous media. J Am Chem Soc 90:6596–6599
Zainal MF, Mohd Y (2015) Characterization of PEDOT films for electrochromic applications. Polym Plast Technol Eng 54:276–281
Stejskal J, Sapurina I, Trchova M (2010) Polyaniline nanostructures and the role of aniline oligomers in their formation. Prog Polym Sci 35:1420–2481
Andriianova AN, Biglova YN, Mustafin AG (2020) Effect of structural factors on the physicochemical properties of functionalized polyanilines. RSC Adv 10:7468–7491
Zhuzhel’skiia DV and Malevb VV (2008) Comparison of properties of aniline and o-aminophenol polymers obtained using hydrogen peroxide V. D. Ivanova, Russ. J Electrochem 44(11):1204–1211
Ghanem MA, El-Enany G (2016) Development of conducting poly(o-aminophenol) film and its capacitance behavior. Int J Electrochem Sci 11:9987–9997. https://doi.org/10.20964/2016.12.68
Wang S, Wang F, Ge X (1986) Polymerization of substituted aniline and characterization of the polymers obtained. Synth Met 16:99–104
Gorshkov NV, Goffman VG, Vikulova MA, Burmistrov IN, Kovnev AV, Gorokhovsky AV (2018) Dielectric properties of the polymer–matrix composites based on the system of Co-modified potassium titanate–poly tetra- fluoroethylene. J Compos Mater 52:135–144
Blythe T, Bloor D (2008) Electrical properties of polymers, 2nd Ed. Cambridge University Press
Yang K, Huang X, Huang Y, Xie L, Jiang P (2013) Fluoro-Polymer@BaTiO3 Hybrid Nanoparticles Prepared via RAFT Polymerization: Toward Ferroelectric Polymer Nanocomposites with High Dielectric Constant and Low Dielectric Loss for Energy Storage Application. Chem Mater 25:2327–2338
Das PR, Biswal L, Behera B, Choudhary RNP (2009) Structural and electrical properties of Na2Pb2Eu2W2Ti4X4O30 (X = Nb, Ta) ferroelectric ceramics. Mater Res Bull 44:1214–1218
Moussa MA, Abdel Rehim MH, Turky GM (2021) Anomalous activation behavior of the conductivity mechanisms in polyaniline-doped graphitic carbon nitride. J Phys Chem Solids 158: ID 110243
Scott AH, Curtis DL, Auritzenandj ALL, Res DJ (1962) Dielectric Properties of Semicrystalline Poly chlorotrifluoroethylene. Nalt Bur Stand, Soc A 66: 269–305
Islam N, Gafur MA, Khan AH (2020) Conduction Mechanism and Conductivity Behavior of Pure Polypropylene (PP) and Polypropylene-Banana Fiber (PP-B) Composites. J Build Mat Sci 02:16–20
Yadav VS, Sahu DK, Dhubkarya DC (2010) The Effect of Frequency and Temperature on Dielectric Properties of Pure Poly Vinylidene Fluoride (PVDF) Thin Films, Proceeding of the International Multiconference of Engineers and computer Scientists, Hong Kong
Kolte J, Salame PH, Daryapurkar AS, Gopalan P (2015) Impedance and AC conductivity study of nano crystalline, fine grained multiferroic bismuth ferrite (BiFeO3), synthesized by microwave sintering. AIP Adv 5:097164
Ladhar A, Arous M, Kaddami H, Raihane M, Kallel A, Graça MPF, Costa LC (2015) AC and DC electrical conductivity in natural rubber/nanofibrillated cellulose nanocomposites. J Mol Liq 209:272–279
a) Sahin M, Bilgic S, Yalmaz H (2002) The inhibition effects of some cyclic nitrogen compounds on the corrosion of the steel in NaCl mediums. Appl Surface Sci 195: 1–7. https://doi.org/10.1016/S0169-4332(01)00783-8. b) Ituen E, Akaranta O, James A (2017) Evaluation of performance of corrosion inhibitors using adsorption isotherm models: an overview. Chem Sci Int J 18: 1–34. https://doi.org/10.9734/CSJI/2017/28976
Khamis E, Abd-El-Khalek DE, Abdel-Kawi MA, Anwar JM (2022) New application of brown sea algae as an alternative to phosphorous-containing antiscalant. Environ Tech 43:4, 595-604.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
H.H.: Developed and supervised the research, performed the experimental preparation, analyzed all characterization data, and wrote all data in the final form. D.A.: Performed the experimental part of precipitation inhibition, analyzed the data, and wrote the draft of the discussion of this section. M.A.: Performed the electrical conductivity measurements, analyzed the data, and wrote the draft of discussion of this section. All authors discussed the results and contributed to the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hassan, H.H.A.M., Abd-El-Khalek, D.E. & Fattah, M.A. Synthesis and assessment of poly(5-nitro-2-aminophenol) as a new scaling inhibitor to control the precipitation of CaCO3 and CaSO4 in solution. J Polym Res 29, 255 (2022). https://doi.org/10.1007/s10965-022-03104-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-022-03104-4