
Abstract
This paper reports the synthesis and characterization of a lanthanum–barium–copper oxide, based on a LaBa 2Cu 3 O 7 system, using a wet chemical route that enables the combustion–polymerization of citrate species, in order to generate materials with enhanced surface and textural and morphological properties for potential applications. The synthesized precursor in a form of a coordination complex was characterized by Fourier transform infrared spectroscopy (FTIR) analysis in order to evaluate the formation of homogeneous and soluble citrate species as intermediates of reaction. The morphological and structural characterizations were performed over calcined material with X-ray diffraction (XRD) and electron microscopy (scanning electron microscopy (SEM)-transmission electron microscopy (TEM)) analyses, confirming the obtention of an orthorhombic crystalline phase type Pmmm (47) in the nanometric range ≈8.9 nm. Analyses of the ceramic oxide by Raman spectroscopy and X-ray photoelectron spectroscopy (XPS) allowed to perform qualitative and quantitative assessments of the material composition, showing that the final oxide is closely related to the desired composition, discarding the presence of carbonaceous residues of the synthesis process.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
In recent years, superconductor materials obtained at nanoscale level have attracted a great deal of attention, due to the possibility of generating new and improved properties for potential technological applications. In the case of high critical temperature superconductors, T c, derived from transition and lanthanide oxides, which have shown the most promising effects as a result of their ability to generate strong currents under magnetic fields, important effects have been reported where particle size has provided a significant role in improving the critical current density. In this regard, the synthesis routes based on wet chemical methods allow the generation of solids with textural morphological and topotactic characteristics for the design of innovative materials with these desired properties. Among the most important methods of wet chemistry for the obtention of superconductor materials, combustion technique has recently reported the most amazing results in the consolidation of pure ceramic phases with nanometric structures [1–3].
Alí et al. [4] found that the most common materials which have provided remarkable results in the field of superconductivity are some oxides of classic superconductors, which have a critical temperature above 30 K and perovskite structure, since it has a great flexibility network due to its high critical temperature (T c around 90 K) [5], covering all types of behaviors of advanced ceramics, such as insulators, semiconductors, ionic conductors, conductors, and superconductors, mostly based on LaBa 2Cu 3 O 7, as a derivative of the yttrium-based copper oxide superconductor family.
The crystal structure of LaBa 2Cu 3 O 7 compound is similar to YBa 2Cu 3 O 7 oxide; however, it is clear that an additional number of CuO chains and CuO 2 planes may be identified on the structure to create a superconducting state, which depends on the presence of a high pure phase, as it has been reported previously [6]. Thus, in the present work, we focus on the use of a wet chemical route based on the citrate–nitrate combustion technique, in order to obtain a pure phase of LaBa 2Cu 3 O 7 powders [7–10].
2 Experiment
Solid phase of LaBa 2Cu 3 O 7−δ was prepared by polymerization–combustion technique, starting from corresponding nitrates of La(NO 3)3, Ba(NO 3)2, and Cu(NO 3)2 with monohydrated citric acid (99.99 %) as complexing agent. Stoichiometric quantities of each solid were mixtured as reported in previous literature [11–13]. Once total dissolution was reached, a stoichiometric quantity of solid citric acid was added in a molar ratio 0.5:1 with respect to the total concentration of metal cations in dissolution. The system was kept under reflux at 120 ∘C for 12 h to obtain a viscous liquid, which was then heated in an oven at 150 ∘C under air flux, until complete solvent evaporation and treated at 250 ∘C to initiate the combustion process. The carbonaceous remnant was removed by thermal treatment at 300 ∘C under oxygen flow (5 mL min −1) for 3 h. The sample was kept in a chamber under controlled humidity (20 %) before physicochemical characterization.
The characterization of metal-organic precursor was done by Fourier transform infrared spectroscopy (FTIR) in a Perkin-Elmer FTIR-1000 equipment in order to assess the formation of citrate complexes and allow the verification of the effectiveness of the synthesis process and the chemical composition. FTIR results were evaluated by comparison with the SDBS databases.
The solid material obtained at the end of the previous step was grounded and calcined at 800 ∘C for 2 h under oxygen flow (5 mL min −1) in order to remove remnants of the combustion step. The structure was characterized using X-ray diffraction (XRD) measurements in a PANalytical X’Pert PRO-MPD equipment, using the Cu K α radiation (λ = 1.540(6) Å) at room temperature between 20 ∘ and 90 ∘ , with steps of 0.02. The crystallite size was calculated using the highest diffraction signals, the Debye-Scherrer equation, taking the value of half peak width set by a Lorentzian function and using a constant of 0.89 as reference. XRD results were refined by GSAS and PCW software.
Scanning electron microscopy (SEM) analysis were performed on a LEO 440 microscope (Leica-Zeiss), equipped with an electron gun, while studies of transmission electron microscopy (TEM) were performed on a JEOL 2100 equipment using a LaB 6 thermionic gun equipped with a CCD imaging system and operated with an acceleration voltage of 200 kV. For analysis, the sample was grounded to obtain fine powders, which were sieved to 200 U.S. standard mesh and dispersed in a test tube with 5.0 mL of water. Each tube was then placed in an ultrasonic equipment for 30 min, after which a drop of the top of each tube was taken and dried at 45 ∘C for respective analysis [13].
The X-ray photoelectron spectroscopy (XPS) data was obtained at room temperature in a Thermo Scientific Escalab 250 XI spectrometer, with monochromatic Al K α (hv = 1486.6 eV) radiation. The analyzer was operated at a 25 eV pass energy and a 0.05 eV step size. The pressure in the analytical chamber was 6.3 × 10 −9 mbar. C 1s signal (284.6 eV) was used as an internal energy reference in all experiments. Determination of core-level peak positions was accomplished after background subtraction using CasaXPS software. Peaks in a spectrum were fitted by a combination of Gauss and Lorentz curves, which also allowed overlapping peaks to be separated. Raman spectroscopy measurements were carried out in a HR-UV 800 infinity microprobe (Jobin-Yvon) spectrometer equipped with a CCD detector (−70 ∘C) and a laser power of 10.7 mW. The Raman spectra of the solid was collected between 100 and 800 cm −1, projecting a continuous wave laser of He–Ne supplying the excitation line at 632 nm through the sample exposed to air at room temperature Finally, the magnetization measurements were performed in a VersaLab vibrating sample magnetometer in ZFC mode using 0.5 g of previously dehydrated sample at 300 ∘C overnight.
3 Results and Discussion
The chemical identification of metal-organic precursor of LaBa 2Cu 3 O 7−δ , derived from FTIR results, confirms the presence of characteristic absorption bands associated with citrate species which are shown in Fig. 1. These results suggest that chemical conditions in aqueous medium favor the obtention of soluble metallic coordination compounds as previously reported [13].

Infrared spectrum of LaBa 2Cu 3 O 7−δ precursor in KBr pellet obtained at room temperature with its main transmittance signals
The signal located at 3769 cm −1 is associated with the vibrational mode of OH hydroxyl group, which serves as a link of intermolecularly formed dimers between coordination compounds. At 3113 cm −1, the stretching band is associated with a vibrational mode of N–H and O–H groups, linked by hydrogen bonds and containing information of carboxylic acid and ammonia dissolution used for pH adjustment. The signal located at 2341 cm −1, corresponding mainly to stretching modes associated with the presence of CO 2, derived from the processes of synthesis of the material and occluded during the combustion [14].
The signal at 1729 cm −1 corresponds to the stretch mode of the C =O group present in dimers of aliphatic saturated carboxylic acids. The signal located at 1584 cm −1 is associated with the asymmetric deformation mode of COO– groups of unidentate coordination compounds. The signal in 1390 cm −1 corresponds to a strong stretch mode associated to bidentate coordination compounds and may also contain information related to the functional groups COO − and NH 4+. These ammonium groups were formed by the reaction between the nitrate groups and NH 4OH, used for pH adjustment. Additionally, these signals are related to the potential presence of acetic acid esters (CH 3COOR), which may form in the drying process and heat treatment of citric acid sol. The stretching modes of the CO bonding correspond to signals located at 891 cm −1, which may be associated with the presence of acetals and aliphatic saturated primary alcohols and also due to bends out of the plane of the C–H bond, while signals present in the fingerprint zone correspond to link of the different metal cations of La 3+, Ba 2+, and Cu 2+ with O–H groups and oxygen atoms.
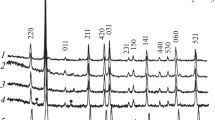
The diffraction pattern of the LaBa 2Cu 3 O 7−δ system is shown in Fig. 2 with its main indexed diffraction signals, whereas Fig. 3 shows the developed Rietveld refinement. The red line in Fig. 3 corresponds to the theoretical model provided by the GSAS software; the crosses represent the experimental pattern and the green line, the difference between theoretical and experimental patterns, confirming the effectiveness of the proposed synthesis method for the obtention of the crystalline phase. The oxide displayed a perovskite structure with space group Pmmm (47). The statistical parameters, atomic positions, and cell volume determined from Rietveld refinement are summarized in Tables 1 and 2.

X-ray pattern of LaBa 2Cu 3 O 7−δ oxide obtained by the polymerization–combustion technique

Rietveld refinement profile for the system LaBa 2Cu 3 O 7−δ
The crystal size of the sample was determined using the highest diffraction signals, the Debye–Scherrer equation, confirming the obtention of a nanometric material (≈8.9 nm), taking a value of half peak width (β), adjusted to a Lorentzian function and using a constant of 0.89 as reference. With this result and a detailed analysis of the X-ray results by PCW software, it was possible to obtain the unit cell of LaBa 2Cu 3 O 6.5 oxide, which is shown in Fig. 4.

Unit cell of LaBa 2Cu 3 O 6.5 obtained from X-ray experimental data and plotted with PCW software
According to Müller-Berdnordz and Pimentel [15, 16], the structure may be identified as a cuprate, whose essential structural feature is a sequence of parallel atomic CuO 2 planes, which were responsible for the electronic properties of superconductor phase. Among these planes, there are several atomic layers that contain La, Ba, and O atoms, which act as a load reservoir. This crystalline structure shows a pronounced planar anisotropy, in which the electrical conductivity acts in a parallel crystal orientation with reference to the CuO 2 planes, and is much larger than in the perpendicular orientation to these atomic layers [16].
At microscopic level (Fig. 5), the solid is composed by irregular aggregates, with sizes ranging from 15 to 150 nm. This result provides evidence about the synthesis route in the formation of larger aggregates which grow and form regular particles, due to low surface energy, associated with the formation of these oxides during the combustion and calcination process [17–20].

Scanning electron microscopy of a secondary and b c and d retrodispersed images for the LaBa 2Cu 3 O 6.5 sample
These pictures are related to the effectiveness of the second thermal treatment and confirm that chemical composition was homogeneous along analyzed surface by secondary (Fig. 5a) and retro-dispersed electron analysis (Fig. 5b, c and d), obtained at 100 nm, respectively.
The TEM images of LaBa 2Cu 3 O 6.5 oxide are shown in Fig. 6 and indicate the obtention of solid material with a narrow size distribution, consistent with nanosized particles. The data derived from checking the lattice parameters of more than 50 images of these samples were evaluated by the ELMIX software: the obtained particles showed an average diameter of 5 to 10 nm in accordance with previous XRD results, oriented along the (1 1 0) facet [21, 22]. The d-spacing values for (1 1 0) and (1 2 3) facets were calculated by the ELMIX software and were 0.31 and 0.28 nm, respectively. Figure 7 displays the histogram of particle size distribution for obtained oxide. The data were collected from a population of 300 particles, showing an asymmetric distribution, revealing the presence of nanometric particles, distributed from 5 to 10 nm, due to the low calcination temperature after the combustion process [23, 24].

Transmission electron microscopy images for LaBa 2Cu 3 O 6.5 sample with its corresponding d-spacings for the main diffraction facets

Particle size distribution estimated from TEM images of LaBa 2Cu 3 O 6.5 oxide
The Raman spectrum of bulk LaBa 2Cu 3 O 6.5 oxide at room temperature consists of several signals, and three of them were identified as the strongest and categorized as vibrational bands: one-phonon (1P) TO at 180, 200, and 740 cm −1 modes [25–27]. The frequency and shape of the phonon bands are consistent with component metal oxides, whereas the scattering intensities are strongly related to the scattering surface present in the volume illuminated by the laser beam. More accurately, the Raman study suggests the presence of additional bands around 450 cm −1 ascribed in presence of La 2 O 3 oxide. The Raman spectrum of bulk BaO displays two bands, located at 180 and 275 cm −1. The band centered at 300 cm −1 is associated with CuO oxide; thus, we conclude that the synthesis method permits the coexistence of metal oxides of La, Ba, and Cu in several planes along unit cells of perovskite, thus allowing its identification as reported previously [28, 29]. The presence of carbonaceous species was discarded since the characteristic band, located at 1310 cm −1, was not found, confirming the effectiveness of the proposed synthesis method as shown in Fig. 8.

Raman spectra for LaBa 2Cu 3 O 6.5 sample obtained for the polymerization–combustion method
The XPS results from 300 to 1200 eV allowed to find all the elements present in the sample. The auger lines were also present in Fig. 9. For reference, the Au 4 f 7/2 peak, at the 83.96 eV of binding energy (BE), was employed, since the difference in the electric potential between the gold reference particles and the sample surface can be expected to be below 0.05 eV for particles of 100 nm diameter, based on electric field considerations around spherical particles [30]. Detailed scans were recorded, in which peak fitting was performed in order to establish precise elemental peak locations.

XPS spectra of LaBa 2Cu 3 O 6.5 powder at 25 ∘C
Figure 10a shows the valence states of La 3+ (5 d) in the perovskite oxide. The analysis of the O 1s spectrum of lanthanum oxide required three components, with one well-defined peak on the low-BE side of the spectrum (520 eV) and other components in a narrow shoulder on the high-BE side, around 834 eV corresponding with La 3 d 5/2 c4f component. The valence band region is characterized by several features, including the band structure near the Fermi level, so the La 5p and 5s peaks, as well as the O 2s peak, reflect the structure of the La 2 O 3 in good agreement with previous works [31].

Slow scan XPS spectra of a La (5 d), b Ba (3 d), c Cu (2 p) and d O (1 s) at 25 ∘C
Figure 10b shows the BE of Ba 2+ located at 778 and 795 eV, confirming that the Ba 3d core lines consist of two components and is consistent with previous references in which these lines are expected [31]. The Cu 2 p 3/2 peak of Fig. 10c shows a single contribution that can be attributed to either Cu(I) [32, 33], but excludes the presence of copper atoms in the oxidation state + 2. The distinction between Cu(I) and Cu(0) can be achieved in the auger region of the spectrum and clearly there is presence of the peak at 930 eV, attributable to Cu(I) species [34], and a lower shoulder lying at 958 eV, ascribable to Cu. In fact, it is possible that the presence of Cu species appears as a broad signal at ∼531.6 eV for hydrated copper oxide and at 530 eV for crystalline Cu 2O species due to possible humidity of the sample [35, 36].
The behavior of magnetization as a function of temperature of the sample is represented in Fig. 11. In this figure, the paramagnetic behavior of the system is observed between 50 and 100 K, showing a characteristic performance of the LaBa 2Cu 3 O 6.5 system without superconducting transition. These results are consistent with those found by Roa et al. [37], who demonstrated that the method of chemical synthesis by this route allows the establishment of grain boundaries that affect the electrical resistance in this temperature range.

Magnetization curve for the LaBa 2Cu 3 O 6.5, system as a function of absolute temperature (× 103)
4 Conclusions
Current synthesis methods, based on a wet chemical route of polymerization–combustion to prepare a perovskite oxide, based on the LaBa 2Cu 3 O 6.5 system, show a strong correlation with the obtention of a nanometric ceramic phase.
The characterization of the metal-organic precursor by FTIR confirms the presence of several vibrational bands, related to citrate functional groups, and enables the obtention of oxides in the nanometric range with structural homogeneity.
Structural characterization was determined by Rietveld refinement from X-ray diffraction data. The results confirm the presence of a perovskite crystalline phase, with space group Pmmm (47); orthorhombic structure with cell parameters a = 3.925 Å, b = 3.934 Å, and c = 11.707 Å; and with a preferential orientation along (1 1 0) facet. The crystal size of 8.9 nm calculated with the Debye–Scherrer equation was confirmed through the analysis of TEMimages.
The microstructural analysis developed by SEM-TEM techniques confirmed the obtention of solid matter with irregular aggregates, ranging from 15 to 150 nm, providing evidence about the effectiveness of synthesis method, ratifying the obtention of nanometric crystals with an average size between 5 and 10 nm and d-spacings of 0.31 and 0.28 nm for the (1 1 0) and (1 2 3) facets respectively, in accordance with a pure perovskite phase of LaBa 2Cu 3 O 6.5.
Raman spectroscopy results, developed over calcined samples, show the absence of carbonaceous deposits or carbonate species and confirm the success of calcination process and the chemical homogeneity of sample, which is a clear evidence of the performed stoichiometric control, consistent with XPS results. Finally, magnetization results are coherent with a non-superconducting behavior with δ values greater than 0.5 between 50 and 100 K.
References
Shahadan, M., Suan, M., Johan, M.R.: Synthesis of Al2O3 nanoparticles highly distributed in YBa 2Cu 3 O 7 superconductor by citrate–nitrate auto-combustion reaction. Physica C: Superconductivity 492, 49–54 (2013)
Shahadan, M., Suan, M., Rafie, M., Chua Siang, J.T.: Synthesis of Y 3Ba 5Cu 8 O 18 superconductor powder by auto-combustion reaction: effects of citrate–nitrate ratio. Physica C: Superconductivity 480, 75–78 (2012)
Mendonça, T.M., Tavares, P.B., Correia, J.G., Lopes, A.M.L., Darie, C., Araújo, J.P.: The urea combustion method in the preparation of precursors for high-T C single phase HgBa 2Ca 2Cu 3 O 8+δ superconductors. Physica C: Superconductivity 471, 1643–1646 (2011)
Ali, A.O., Ekicibil, A., Selda, K.C., Atilla, C., Ali, O.E., Yüksel, U., Tezer, F., Kıymaç, K.: The structural superconducting and transport properties of the compounds Y 3Ba 5Cu 8 O 18 and Y 3Ba 5Ca 2Cu 8 O 18. J. Supercond. Magn. 24, 2243–2252 (2011)
Ekicibil, E.A., Selda, K.C., Ali, O.A., Atilla, C., Tezer, F., Kerim, K: Exploration of the superconducting properties of with and without Ca doping by magnetic measurements. Sol. State Sci. 13, 1954–1959 (2011)
Li, J., Wu, Y., Pan, Y., Guo, J.: Influence of citrate-to-nitrate ratio on the thermal behavior and chemical environment of alumina gel. Ceram. Int. 33, 735–738 (2007)
Chandradass, J., Kim, K.H.: Effect of acidity on the citrate–nitrate combustion synthesis of alumina–zirconia composite powder. Met. Mater. Int. 15, 1039–1043 (2009)
Marinsek, M., Kemperl, J., Likozar, B., Macek, J.: Temperature profile analysis of the citrate–nitrate combustion system. Ind. Eng. Chem. Res. 47, 4379–4386 (2008)
Banerjee, S., Devi, P.S.: Effect of citrate to nitrate ratio on the decomposition characteristics and phase formation of alumina. J. Therm. Anal. Calorim. 90, 699–706 (2007)
Manoharan, S.S., Patil, K.C.: Combustion synthesis of metal chromite powders. J. Am. Ceram. Soc. 75, 1012–1015 (1992)
Gómez Cuaspud, J.A., Schmal, M.: Nanostructured metal oxides obtained by means polymerization combustion at low temperature for CO selective oxidation. Int. J. Hydrogen Energy 38, 7458–7468 (2013)
Gomez, J.A., Valencia, J.S., Carda, J.B.: Preparation and characterization of perovskite oxides by polymerization-combustion. J. Chil. Chem. Soc. 55, 445–449 (2010)
Gomez, J.A., Schmal, M.: Effect of metal oxides concentration over supported cordierite monoliths on the partial oxidation of ethanol. Appl. Catal. B Environ., 148–149, 1–10 (2014)
Cruz Pacheco, A.F., Gómez Cuaspud, J. A. : Synthesis and characterization of the La 0.8Sr 0.2MnO 3 System. Rev. Ciencia en Desarrollo 6, 133–139 (2015)
Muller, J.G., K.A.: physic B. Berlin, vol. 64, pp 189–193. berlin (1986)
Shiohara, Y., Endo, A.: Crystal growth of bulk high-Tc superconducting oxide materials. Mater. Sci. Eng. R19, 1–86 (1997)
Dai, N., Wang, Z., Jiang, T., Feng, J., Sun, W., Qiao, J., Rooney, D., Sun, K.: A new family of barium-doped Sr 2Fe 1.5Mo 0.5 O 6?δ perovskites for application in intermediate temperature solid oxide fuel cells. J. Power Sources 268, 176–182 (2014)
Naeimi, A.S., Dehghan, E., Sanavi Khoshnoud, D., Gholizadeh, A.: Enhancement of ferromagnetism in Ba and Er co-doped BiFeO 3 nanoparticles, vol. 393, pp 502–507 (2015)
Ungelenk, J., Roming, S., Adler, P., Schnelle, W., Winterlik, J., Felser, C., Feldmann, C.: Ultrafine MnWO 4 nanoparticles and their magnetic properties. Solid State Sci. 46, 89–94 (2015)
Verma, S., Bamzai, K.K.: Preparation by chemical co-precipitation, spectral and electrical characteristics of neodymium orthophosphate nanoparticles. J. Rare Earths 33, 535–544 (2015)
Waqee-ur-Rehman, M., Qasim, I., Mumtaz, M., Nadeem, K., Khurram, A.A.: Infield response of (Al 2 O 3)x/CuTl-1223 nanoparticles-superconductor composites. Phys. B Condens. Matter 476, 37–40 (2015)
Swarup, P.M., Seshubai, V., Rajasekharan, T.: A generic process to introduce nanoparticles into powder preforms and its application to infiltration growth processing of REBa 2Cu 3 O 7 superconductor. Mater. Chem. Phys. 161, 59–64 (2015)
Jabbar, A., Qasim, I., Khan, S.A., Nadeem, K., Waqee-ur-Rehman, M., Mumtaz, M., Zeb, F.: Highly coercive cobalt ferrite nanoparticles-CuTl-1223 superconductor composites. J. Magn. Magn. Mater. 377, 6–11 (2015)
Nadeem, K., Naeem, F., Mumtaz, M., Naeem, S., Jabbar, A., Qasim, I., Khan, N.A.: Synthesis and characterization of core–shell Ni/NiO nanoparticles/CuTl-1223 superconductor composites. Ceram. Int. 40, 13819–13825 (2014)
Kourouklis, G.A., Jayaraman, A., Batlogg, B., Cava, R.J., Stavola, M., Krol, D.M., Rietman, E.A., Schneemeyer, L.F.: Raman scattering in MBa 2Cu 3 O 7 with M = Y, Eu and Gd: effect of O18 substitution and oxygen vacancies on the Cu-O vibrational modes. Phys. Rev. B 36, 8320 (1987)
Trolio, A.D., Grimaldi, G., Mattei, G., Testa, A.M.: Structural and superconducting properties of EuBa 2Cu 3 O 7−x thin films grown by off-axis pulsed laser deposition Supercond. Sci. Technol. 17, 1009–1013 (2004)
McMullan, W.G., Gygax, S., Irwin, J.C.: Zero-wavevector vibrational modes in EuBa 2Cu 3 O 7−x Original Research Article. Solid State Commun. 66, 165–169 (1988)
Liu, C.S., Zhang, A.M., Xu, T.F., Wu, W.C.: Theory of two-magnon Raman scattering in alkaline iron selenide superconductors. Physica C: Superconductivity and its Applications 506, 76–82 (2014)
Geng, Z., Feng, S.: Doping and temperature dependence of electronic Raman response in cuprate superconductors. Phys. Lett. A 375, 214–219 (2010)
International Organization for Standardization: ISO15472:2001(E) Surface Chemical Analysis-X-ray Photoelectron Spectrometers-Calibration of Energy Scales. International Organization for Standardization, Geneva (2001)
Lianwei, S., Xianyou, Z.: XPS study on barium lanthanum magnesium niobate. J. Rare Earths 24, 310–313 (2006)
Sinapi, F., Lejeune, I., Delhalle, J., Mekhalif, Z.: Comparative protective abilities of organothiols SAM coatings applied to copper dissolution in aqueous environments. Electrochim. Acta 52, 5182 (2007)
Laibinis, P.E., Whitesides, G.M., Am, J. Chem. Soc 114, 9022 (1992)
Hutt, D.A., Liu, C.: Oxidation protection of copper surfaces using self-assembled monolayers of octadecanethiol. Appl. Surf. Sci. 252, 400 (2005)
Caprioli, F., Decker, F., Marrani, A.G., Beccari, M., Di Castro, V.: Copper protection by self-assembled monolayers of aromatic thiols in alkaline solutions. Phys. Chem. Chem. Phys. 12, 9230 (2010)
He, J.B., Lu, D.-Y., Jin, G.-P.: Potential dependence of cuprous/cupric duplex film growth on copper electrode in alkaline media. Appl. Surf. Sci. 253, 689 (2006)
Roa, J., Landinez, D.A., Corredor, L.T.: Desdoblamiento de la transición de apareamiento en superconductores policristalinos de GdBa 2Cu 3 O 7−d. Revista colombiana de física 35, 303–306 (2003)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Morales Rivera, A.M., Gómez Cuaspud, J.A., Parra Várgas, C.A. et al. Synthesis and Characterization of LaBa2Cu3O7−δ System by Combustion Technique. J Supercond Nov Magn 29, 1163–1171 (2016). https://doi.org/10.1007/s10948-015-3311-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10948-015-3311-3