
Abstract
The demand for transportation diesel fuels has been increasing in most countries for the last decade. As is one of the primary sources for diesel distillates, light cycle oil (LCO), having the advantages of inheriting similar density and boiling point range to diesel fuels, but high contents of aromatics, sulfur, and nitrogen, needs to be upgraded before being utilized. This work reports a trinary porous SiO2–Al2O3–TiO2 (SAT) synthesized by sol–gel method and used as the NiMo catalyst support for the LCO hydro-upgrading. NiMo/Al2O3-zeolite (AZ) was used as a reference to evaluate the performances of the NiMo/SAT. The catalysts were characterized by XRD, BET, Pyridine-FTIR, and TEM; and evaluated in a 20-mL fixed-bed microreactor using a Fluid Catalytic Cracking LCO as feedstock. It was found that the SAT support possessed a high surface area, high pore volume, and good acid properties. The NiMo/SAT catalyst exhibited even better hydrodesulfurization, hydrodenitrogenation, and hydrodearomatization performances than NiMo/AZ when the reaction was performed at 375 °C and 1100 psi. Compared with the zeolite addition catalyst, the SAT catalyst has the advantages of simple synthesis procedures and low input cost.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The demand for transportation diesel fuels has been increasing in most countries for the last decade. Overall, globally, the diesel demand increases year by year, from 25.6 Mb/d (million barrels per day) in 2000, to 28.6 Mb/d in 2017, and estimated to 30.8 Mb/d in 2020 [1], but maybe decline as being affected by COVID-19. For the first 10 months of 2017, European demand for diesel grew by 2.4% over 2016, the US did by 2%, and China did by 1.5% [2]. Producing clean transportation fuel, especially ultra-low sulfur diesel (ULSD), is of importance in many countries due to the strict environmental legislation. At present, the allowable sulfur content and the aromatics content in diesel fuels are restricted to 10 ppm and 7%, respectively, in China. Compared with gasoline, diesel has the characteristics of low volatile and high combustion efficiency; therefore, the demand for diesel has kept an increase. Producing sufficient high-quality clean fuels is becoming a challenge for the refineries than ever before. As is one of the primary sources for diesel distillates, light cycle oil (LCO), has the advantages of inheriting similar density and boiling point range to diesel fuels, but also the disadvantages of containing too many undesired components—sulfur/nitrogen compounds, aromatics, and so on.
In general, hydro-upgrading technology [3] are the most effective choices for the refinery to produce ULSD. However, it is difficult to produce ULSD with conventional hydrotreating catalysts and processes. For example, when the sulfur content in diesel was reduced from 500 to 50 ppm, the active amount of the traditional HDS catalyst had to increase fourfold [4].
We investigated the hydrotreating performances of NiMo, CoMo, and NiW catalysts supported by alumina-zeolite (AZ) using both model compounds and a real light cycle oil (LCO) and found that NiMo bimetallic supported catalyst is most suitable for LCO hydro-upgrading [3, 5,6,7,8,9,10]. Adding zeolite into the hydrotreating catalyst greatly enhances the refractory sulfur compound conversion; however, it leads to lower product yields as well [5]. Further, the procedures for synthesis of zeolite are complicated, reulting in high cost. It was also found that the supports altered the metal-support interaction (MSI), promoted the dispersion of the active components, and further improved the catalyst activity [11], which include active carbon, SiO2, TiO2, MgO, and binary oxides, such as SiO2–Al2O3, Al2O3–TiO2 [11]. These supports provided the catalysts with good pore structures, adjustable MSI and tailored acid properties and further improved the catalyst activity.
MoO3 forms the bond with titania only when TiO2 is loaded more than one layer [11]. SiO2 has a smaller interaction with the sulfided phase and lower activity than the alumina. Furthermore, pure TiO2 or SiO2 supports usually have small specific surface areas compared to alumina, and it is difficult to impregnate enough metal components on the catalyst. Therefore, some researchers focus their work on the binary oxides, such as TiO2–Al2O3, SiO2–Al2O3, and so on [12, 13]. These binary oxides possessed acidic properties [14], which enhanced the HDS reaction [12]. With the advantages of homogeneous dispersion of components, controllable particle size, and low density, the sol–gel method was frequently used to synthesize binary oxides, such as Ti–Si mesoporous materials applied in water treatment [15], Co-catalysts supported on Si–Ti in CO oxidation[16], Si/Al–TiO2 in the n-heptane dehydrogenation to cyclics [17], Pt–Pd/ATS in HDA process[18], MoO3–SiO2–Al2O3 mixed oxides in light olefin metathesis[19], and titania-doped mixed oxide supported vanadia catalysts in alkane adsorption process [20].
In our previous work, it was found that SiO2–Al2O3–TiO2 (SAT) supported catalyst had efficient hydrodeoxygenation (HDO) capability [21,22,23,24]. In this study, a trinary porous SAT was synthesized using sol–gel method. Then an NiMo/SAT catalyst was synthesized for the LCO hydro-upgrading process. A zeolite-alumina (AZ) supported NiMo catalyst with good hydrotreating performances from our previous work was used as a reference, of which HDS, HDN, and HDA performances were compared with those of the NiMo/SAT catalyst using LCO as feedstock. Their textural and crystallized properties, the acidic properties, and the morphology were analyzed by BET, XRD, Pyridine-IR, and TEM. The purpose of this work is to upgrade LCO by means of HDS, HDN, and HDA into a premium diesel blending component over a NiMo catalyst supported by SAT trinary oxide.
2 Experimental
2.1 Catalyst preparation
The SAT was prepared by the following process: 3% NH3·H2O was dropped into the TiCl4 solution until forming a white precipitate with a pH of around 7. After being filtrated, the precipitate was washed with distilled water to remove the Cl− (HNO3, with a concentration of 1.6 M and an amount of H/Ti at 0.5, was used to neutralize the suspension) and then the suspension was stirred at 70 °C until forming a titania sol. Na2SiO3 was added into this gel to form a Ti–Si suspension. The pH was adjusted to 9 with HNO3 followed by adding Al(NO3)3 to the solution slowly. It was then stirred at 60 °C and the pH was adjusted to 7 with concentrated NH3·H2O to form a SAT slurry. The hydrogel was separated, kept for 3 days, dried at 120 °C overnight, and calcined at 500 °C for 6 h.
NiMo/SAT was prepared by mixing 1.94 g Ni (NO3)2·6H2O, 1.5 g MoO3 and 8 g SAT. The mixture was extruded, dried and calcined under the same conditions as the SAT synthesized procedure.
NiMo/AZ (alumina-zeolite) catalyst was prepared using the same method as our previous publication [5,6,7,8,9,10]. The catalyst was prepared by mixing 1.5 g zeolite HY (ammonia exchanged Zeolite CBV-300) with 0.5 g Zeolite beta (Zeolyst CP814E. The product was then calcined and hydrothermally treated). Afterwards, it was mixed with 4.0 g alumina, 1.94 g Ni(NO3)2·6H2O, and 1.5 g MoO3 with 2.0 g binder (SASOL, CAPAPAL B), which was then extruded, dried, and calcined.
2.2 Catalyst characterization
The catalysts were characterized using the same method as our previous publication [5,6,7,8,9,10]. The catalyst surface physical properties were detected by a Quantachrome Autosorb-1. X-ray powder patterns were obtained by a Bruker D8 Advance diffractometer. Pyridine-FTIR were determined by a Thermo Scientific NICOLET 6700 FT-IR Spectrometer. A JEOL 2010 STEM was used to test catalyst micro morphologies and structures.
2.3 Catalyst evaluation
Catalyst evaluation and the product analysis were performed using the same method as our previous publication [5,6,7,8,9,10]. The sulfur and nitrogen content of the LCO feed was 14,930 ppm and 140 ppm, respectively. The catalyst stability was also evaluated with a 20 mL fixed-bed microreactor. The hydrogen flow rate was 30 Sccm. Liquid hourly space velocity (LHSV) was 0.6 h−1. The reaction conditions were 375 °C, 1100 psi; 375 °C, 550 psi; and 300 °C, 1100 psi, respectively.
A GC/MS and an Agilent GC-450 equipped with an FID and an PFPD detectors were used to test the compositions of the hydro-upgrading liquid products. The total nitrogen and sulfur contents were measured by an Antek NS-9000 according to ASTM D 4629 and ASTM D 4294 standards, respectively. The simulated distillation results of the feed and the hydro-upgrading liquid products were obtained by a GC/FID. The aromatic contents were analyzed according to ASTM D 6591 standard by an HPLC.
3 Results and discussion
3.1 Catalyst crystal structure and surface properties
Figure 1 shows the XRD pattern of the sulfided NiMo/SAT and NiMo/AZ catalysts. For the NiMo/AZ, some diffraction peaks can be found as marked in Fig. 1 [6]. SAT may be formed as an amorphous structure showing no sharp peaks on the XRD patterns. For both catalysts, the active components, Ni and Mo, are uniform distributed on the catalyst support as only trace MoS2 peaks are shown in Fig. 1.

XRD profiles of the sulfided NiMo/SAT and NiMo/AZ catalysts NMSAT-NiMo/SAT, NMAZ-NiMo/AZ
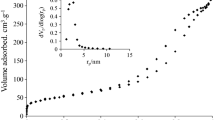
Figure 2a, b, and c show the pore-size distributions and the isotherm curves of SAT, NiMo/SAT, AZ, and NiMo/AZ catalysts. The SAT support shows a three-pore structure system where their pore sizes are distributed around 4 nm, 6 nm, and 15 nm, respectively. This is associated with the SAT supports preparation process. Briefly, Ti-Si was formed first and then SAT was formed later. The small size pore structure might be formed by the Ti-Si material and middle size pore structure might be formed by the SAT material. The large pore-structure at the size between 10 and 20 nm might be formed by the secondary pores structures which were piled on the initial materials. For the NiMo/SAT catalyst, almost all the pore structures between 10 and 20 nm were diminished and most of the small pores were reduced, which was because they were occupied by the NiMo covering or blocking on the surface or inside the pore structures. For the NiMo/AZ catalyst, the pore size is distributed between 3 and 20 nm.

a Pore size distribution calculated by BJH Adsorption dV/dlog(D) Pore Volume method NM/SAT-NiMo/SAT; NM/AZ-NiMo/AZ. b and c The Original N2-physisorption Isotherm Linear Plots
Table 1 lists the surface properties of the SAT, NiMo/SAT, AZ, and NiMo/AZ. For the SAT, the BET surface area and pore volume were very high. This is because Si was coated on the outer surface of the Ti and Al was coated on the outer surface of the Si–Ti. For the NiMo/SAT catalyst, more than half of the surface area, pore volume, and micropore volume was diminished, which was caused by the NiMo supported on the surface or inside the pore structures.
Figure 3 presents the TEM results of the NiMo/AZ and NiMo/SAT. After sulfidation, one molybdenum atom and two sulfur atom layers produce a S–Mo–S sandwich-like structure and Ni atoms are positioned on the above structure’s edge. The short lines in Fig. 3 were the MoS2 crystalline layer. The number of short lines illustrates the number of MoS2 crystalline layers. The length and layer number of the MoS2 crystalline are statistic through over 300 crystallites in different images which are listed in Table 1 [25, 26]. The average length and average slabs number is 6.46 nm and 4.30 for the NiMo/SAT and 5.10 nm and 1.74 for the NiMo/AZ. Because zeolite supports may have some strong acid sites, which might have a stronger interaction between metal and the support than that of the NiMo/SAT, they attract more NiMo precursors along the surface of the support forming thinner NiMoS slabs.

TEM images of the sulfided catalysts
Figure 4 shows the Pyridine-FTIR results of the SAT, NiMo/SAT, AZ, and NiMo/AZ. The acidities of the NiMo/SAT and NiMo/AZ were calculated when the desorption of Pyridine at 100 °C, 200 °C, and 300 °C. Lewis or Brønsted acids are defined according to the wavenumbers of the peaks [27]. For the SAT and NiMo/SAT, Lewis acids are the most dominant acid types. As our previous publication [22], only Lewis acids on the SAT catalyst, and the Bronsted acids produce when MoO3 loads on SAT. Bronsted acids is the most dominant acid type for the AZ catalyst and the total amounts of Brønsted acid decrease significantly when AZ is loaded by Ni and Mo. Ni and Mo may occupy the Brønsted acids. The total amounts of acids decreased a little for SAT, but significantly for AZ when NiMo was loaded. NiMo/AZ had more Brønsted and similar Lewis acids compare to that of the NiMo/SAT. Furthermore, more strong acid sites were also found in NiMo/AZ, which favor both the cleavage of the C–S and isomerization of methyl-substituted DBTs [28].

Pyridine-IR results of the SAT, NiMo/SAT, AZ, and NiMo/AZ
3.2 Reaction performances
3.2.1 Aromatics hydrogenation (HYD)
Figure 5 shows the non-aromatics (NAHs) and aromatics (including one-ring aromatics (MAHs), two-ring aromatics (DAHs), and three and more ring aromatics (PAHs)) hydrocarbon distributions in the final products. HDA activity (according to the increase of the non-aromatics (NAHS)) decreased in the order NiMo/AZ > NiMo/SAT no matter what the reaction conditions. Most of the diaromatics (67–75%) were converted into monoaromatics and non-aromatics at 375 °C, 1100 psig, over NiMo/AZ and NiMo/SAT. The amounts of the non-aromatics increased twofold. The results suggest that NiMo/SAT has a relatively high HYD capability—close to that of the NiMo/AZ catalyst.

Saturated and aromatics hydrocarbon distributions in the final products
The morphology of NiMoS affects the HYD ability of the catalyst. It is easy for HYD to react on the top edges of MoS2 clusters because of less steric hindrance [29, 30]. In the HYD reaction, it has been found that, compared with basal-bonded MoS2 clusters, edge-bonded MoS2 clusters have superior intrinsic activity [31]. For Al2O3 supported by catalyst, MoS2 clusters with low stacking are of HYD-orientation, which is in accordance with the rim-edge model [29]. For TiO2 supported by catalyst, multi-layered MoS2 clusters possess higher intrinsic activities than single-layered MoS2 clusters, which is in accordance with the comparison between Co–Mo–S(I) and Co–Mo–S(II) [31]. The analysis results suggest that multilayered MoS2 clusters had more multi-vacancies than single-layered ones did, facilitating the adsorption of heavy molecular compounds and their HYD [31]. In addition, multi-layered Co–Mo–S(II) has rare electronic interaction with the catalyst support, showing higher intrinsic activity than single-layered Co–Mo–S(I) [31]. As shown in Table 1 and Fig. 5, NiMo/SAT catalyst has more rims sites, less aspect ratio [31], and more multi-layered MoS2, and shows a comparable HYD ability than that of NiMo/AZ.
3.2.2 Hydrodesulfurization
Figure 6 shows the hydrodesulfurization results of NiMo/SAT on a microreactor. The reaction products were detected by GC-FID. As shown in Fig. 6, For the LCO feed with 14,930 ppm S, the main sulfur species were: benzene-thiophene; methyl-(Me-), ethyl-(Et), propyl-(Pr), and butyl-(Bu) substituted benzene-thiophene; dibenzenethiophene; and Me-, Et-, Pr-, and Bu-substituted dibenzenethiophene. After reacting under the condition of 300 °C and 1100 psig, about 75% sulfur was removed. However, dibenzenethiophene and alkyl-substituted dibenzenethiophene were still unconverted under this condition. When the reaction occurred under the condition of 375 °C and 550 psig, most of dibenzenethiophene and partial alkyl-substituted dibenzenethiophene were converted. The sulfur content dropped significantly from 3470 ppmw sulfur to 1079 ppmw. When the reaction occurred under the condition of 375 °C and 1100 psig, the sulfur content was only 470 ppmw. Most refractory sulfur compounds, such as 4-methyl and some dimethyl-dibenzenethiophene, were left in the final products.

Sulfur compounds in the products under different reaction conditions
Figure 7 shows the detailed results of the refractory sulfur compounds, such as DBT, C1-DBT, C2-DBT, and C3-DBT, in the final products over the NiMo/SAT catalyst. It is seen that C1-DBT was the most plentiful sulfur compound in the feed. When the reaction was performed under the condition of 375 °C and 1100 psig, the selectivity for the DBTs was C3-DBT ≈ C2-DBT ≈ C1-DBT > DBT. Alkyl-substituted dibenzenethiophene might have more steric hindrance than that of the non-substituted dibenzenethiophene. When the reaction was performed under other conditions, the selectivity for the DBTs is C3-DBT > C2-DBT ≈C1DBT > DBT. C3-DMDBT shows slightly higher steric hindrance than that of the C1 and C2-dibenzenethiophene.

The refractory Sulfur compounds in the final products over the NiMo/SAT
Table 2 compares the HDS and HDN performances of LCO over NiMo/AZ and NiMo/SAT under different reaction conditions. Normally, HDS reaction is carried out through two parallel reactions. (a) Direct desulfurization (DDS), and (b) desulfurization through HYD [32, 33]. For the DDS pathway, one of the double bonds in the vicinity of the sulfur atom undergoes partial HYD to form a dihydro-intermediate. Then, a C–S bond is cleaved by an elimination process to form an SH group and the second C–S bond is broken to produce the final desulfurized products. For the HYD pathway, one aromatic ring of 4,6-DMDBT was hydrogenated to form a tetrahydromethyl-substituted dibenzothiophene. The later one eliminates the C–S bond to form final desulfurized products [34].
Under either low temperature or low pressure, NiMo/AZ showed significantly higher HDS activity than the NiMo/SAT. However, under high temperature and high pressure, NiMo/SAT shows a slightly higher HDS activity than the NiMo/AZ. When the H2 pressure increased from 550 to 1100 psig, the residual sulfur in the products decreased from 1079 to 470 ppm for the NiMo/SAT and from 612 ppm to the 521 ppm for the NiMo/AZ. NiMo/SAT and NiMo/AZ have similar HYD ability. When acidic compounds, such as zeolite, are introduced into the NiMo/AZ, they shift the methyl groups from 4,6-position to the 3, 6 or other positions, which reduce the steric hindrance between 4,6-DMDBT molecular and catalyst active sites and thereafter increasing the HDS reactivity. Therefore, for the NiMo/AZ catalyst under the condition of 375 °C and 550 psig, the refractory sulfur compounds were removed not only by the HYD and DDS process, but also by the isomerization firstly, and then DDS and HYD pathways.
NiMo/SAT is sensitive to temperature and pressure than NiMo/AZ. The total Bronsted acids or the weak and medium Bronsted acids may be the key impact factor for both HDS and HDN under the relatively low temperature or low pressure, as is the strong Bronsted or the Lewis acids under both relatively high temperature and pressure. Moreover, the layer numbers of MoS2 may contribute to this phenomenon too.
When the reaction was carried out under the condition of 300 °C and 1100 psig, the residual sulfur in the products was much higher than that of 375 °C and 1100 psig, either with NiMo/SAT or NiMo/AZ catalyst. At lower temperature, DDS may be the prominent reaction pathway in the HDS process. Thiophenes, benzene-thiophenes, and alkyl-substituted thiophene were removed during the reaction. However, some DBT and methyl-substituted dibenzothiophene were still left in the products. The acidic sites prefer the cleavage of C–S bond and improve the DDS pathway, which in turn increases the activity of the HDS. It is concluded that to HDS, both the temperature and the pressure were significant factors when over the NiMo/SAT; however, the temperature was a much more important factor than pressure when over the NiMo/AZ catalyst.
3.2.3 Hydrodenitrogentation
The HDN activity is also listed in Table 2. HDN frequently occurs by the HYD of nitrogen containing rings, followed by the cleavage of C–N bond [35]. Higher stacked MoS2 slabs favor the N-rings HYD and strong Bronsted acid sites favor the C–N bonds cleavage. Under the condition of 375 °C and 550 psig, the HDN activity (according to the reduction of the nitrogen in the products) of NiMo/SAT is slightly lower than that of the NiMo/AZ, however, under the condition of 375 °C and 1100 psig. NiMo/SAT has a relatively better HDN performance than NiMo/AZ does under the condition of 375 °C and 1100 psig, indicating that NiMo/SAT has a higher HYD ability under the high H2 pressure. When the reaction was carried out at the low temperature, NiMo/SAT has a worse HDN performance than NiMo/AZ does. Zeolite in the catalyst may favor the cleavage of the C–N bond.
4 Conclusion
A trinary porous SiO2–Al2O3–TiO2 support was synthesized in this work, which has the advantaged of simple synthesis procedures, low input cost, and comparable hydrotreating performances under high temperature and high pressure to zeolite addition catalyst.
The synthesized SAT material has a high surface area and a high pore volume. The synthesis method forms Ti–Si and Ti–Si–Al, which forms a different mesopores and secondary pores structure. Furthermore, the synthesized NiMo/SAT favors the cleavage of the C–N and C–S more than the NiMo/AZ does under a higher temperature at 375 °C and a high pressure at 1100 psi. The hydrotreating of the NiMo/SAT was exhibited a comparably even better HDS, HDN, and HDA performances than those of NiMo/AZ when the reaction was performed under the condition of 375 °C and 1100 psi. To HDS, both the temperature and the pressure were significant factors when over the NiMo/SAT; however, the temperature was a much more important factor than the pressure when over the NiMo/AZ catalyst. The selectivity of the converted DBTs species decreased in the order C3-DBT > C2-DBT ≈C1DBT > DBT under the condition of 375 °C and 1100 psi. C3-DMDBT has a relatively higher steric hindrance than that of the C1 and C2-dibenzenethiophenes.
References
Tang, Oil industry data analysis: It is predicted that the global demand for diesel oil in 2020 will be 30.8 million b/d, iiMedia, 2020.
T. Fitzgibbon, C. Ding, P. Szabat, Diesel demand: still growing globally despite Dieselgate Mckinsey&Company, 2018.
J.-I. Park, J.-K. Lee, J. Miyawaki, Y.-K. Kim, S.-H. Yoon, I. Mochida, Hydro-conversion of 1-methyl naphthalene into (alkyl)benzenes over alumina-coated USY zeolite-supported NiMoS catalysts. Fuel 90, 182–189 (2011)
K.G. Knudsen, B.H. Cooper, H. Topsoe, Catalyst and process technologies for ultra low sulfur diesel. Appl. Catal. A General 189, 205–215 (1999)
L. Ding, Y. Zheng, Z. Zhang, Z. Ring, J. Chen, Hydrotreating of light cycled oil using WNi/Al2O3 catalysts containing zeolite beta and/or chemically treated zeolite Y. J. Catal. 241, 435–445 (2006)
L. Ding, Y. Zheng, Z. Zhang, Z. Ring, J. Chen, Hydrotreating of light cycle oil using WNi catalysts containing hydrothermally and chemically treated zeolite Y. Catal. Today 125, 229–238 (2007)
L. Ding, Y. Zheng, H. Yang, R. Parviz, LCO hydrotreating with Mo-Ni and W-Ni supported on nano- and micro-sized zeolite beta. Appl. Catal. A General 353, 17–23 (2009)
L. Ding, M. Zheng, A. Wang, T. Zhang, A novel route to the preparation of carbon supported nickel phosphide catalysts by a microwave heating process. Catal. Lett. 135, 305–311 (2010)
S. Yao, Y. Zheng, L. Ding, S. Ng, H. Yang, Co-promotion of fluorine and boron on NiMo/Al2O3 for hydrotreating light cycle oil. Catal. Sci. Technol. 2, 1925–1932 (2012)
S. Yao, Y. Zheng, S. Ng, L. Ding, H. Yang, The role of nanobeta zeolite in NiMo hydrotreating catalysts. Appl. Catal. A General 435, 61–67 (2012)
M. Breysse, J.L. Portefaix, M. Vrinat, Support effects on hydrotreating catalysts. Catal. Today 10, 489–505 (1991)
Y. Okamoto, A. Maezawa, T. Imanaka, Active sites of molybdenum sulfide catalysts supported on Al2O3 and TiO2 for hydrodesulfurization and hydrogenation. J. Catal. 120, 29–45 (1989)
F. Sanchez-Minero, J. Ramirez, R. Cuevas-Garcia, A. Gutierrez-Alejandre, C. Fernandez-Vargas, Kinetic study of the HDS of 4,6-DMDBT over NiMo/Al2O3−SiO2(x) catalysts. Ind. Eng. Chem. Res. 48, 1178–1185 (2009)
J. Escobar, S. Nunez, A. Montesinos-Castellanos, J. Antonio de los Reyes, Y. Rodriguez, O.A. Gonzalez, Dibenzothiophene hydrodesulfurization over PdPt/Al2O3-TiO2 center dot Influence of Ti-addition on hydrogenating properties. Mater. Chem. Phys. 171, 185–194 (2016)
V. Tajer-Kajinebaf, H. Sarpoolaky, T. Mohammadi, Sol-gel synthesis of nanostructured titania-silica mesoporous membranes with photo-degradation and physical separation capacities for water purification. Ceram. Int. 40, 1747–1757 (2014)
T.M. Lima, W.N. Castelblanco, A.D. Rodrigues, R.E. Roncolatto, L. Martins, E.A. Urquieta-Gonzalez, CO oxidation over Co-catalysts supported on silica-titania—the effects of the catalyst preparation method and the amount of incorporated Ti on the formation of more active Co3+ species. Appl. Catal. A General 565, 152–162 (2018)
G. Perez Lopez, R. Ramirez Lopez, T. Viveros, Dehydrocyclization of n-heptane over Pt catalysts supported on Al- and Si-promoted TiO2. Catal. Today 220, 61–65 (2014)
G. Wan, A. Duan, Z. Zhao, G. Jiang, D. Zhang, R. Li, T. Dou, K.H. Chung, Al2O3-TiO2/Al2O3-TiO2-SiO2 composite-supported bimetallic Pt-Pd catalysts for the hydrodearomatization and hydrodesulfurization of diesel fuel. Energy Fuels 23, 81–85 (2009)
D.P. Debecker, K. Bouchmella, M. Stoyanova, U. Rodemerck, E.M. Gaigneaux, P.H. Mutin, A non-hydrolytic sol-gel route to highly active MoO3-SiO2-Al2O3 metathesis catalysts. Catal. Sci. Technol. 2, 1157–1164 (2012)
D. Shee, G. Deo, In situ DRIFT studies of alkane adsorption on vanadia supported titania-doped catalysts. Catal. Today 325, 25–32 (2019)
H. Wang, L. Zhang, G. Li, K. Rogers, H. Lin, P. Seers, T. Ledan, S. Ng, Y. Zheng, Application of uniform design experimental method in waste cooking oil (WCO) co-hydroprocessing parameter optimization and reaction route investigation. Fuel 210, 390–397 (2017)
H. Wang, G. Li, K. Rogers, H. Lin, Y. Zheng, S. Ng, Hydrotreating of waste cooking oil over supported CoMoS catalyst Catalyst—deactivation mechanism study. Mol. Catal. 443, 228–240 (2017)
H. Wang, K. Rogers, H. Zhang, G. Li, J. Pu, H. Zheng, H. Lin, Y. Zheng, S. Ng, The effects of catalyst support and temperature on the hydrotreating of waste cooking oil (WCO) over CoMo sulfided catalysts. Catalysts 9, 689 (2019)
H. Wang, H. Lin, P. Feng, X. Han, Y. Zheng, Integration of catalytic cracking and hydrotreating technology for triglyceride deoxygenation. Catal. Today 291, 172–179 (2017)
M.F. Li, H.F. Li, F. Jiang, Y. Chu, H. Nie, The relation between morphology of (Co)MoS2 phases and selective hydrodesulfurization for CoMo catalysts. Catal. Today 149, 35–39 (2010)
D. Ferdous, A.K. Dalai, J. Adjaye, L. Kotlyar, Surface morphology of NiMo/Al2O3 catalysts incorporated with boron and phosphorus: experimental and simulation. Appl. Catal. A General 294, 80–91 (2005)
T.R. Hughes, H.M. White, A study of the surface structure of decationized Y zeolite by quantitative infrared spectroscopy. J. Phys. Chem. 71, 2192–2201 (1967)
P. Michaud, J.L. Lemberton, G. Perot, Hydrodesulfurization of dibenzothiophene and 4,6-dimethyldibenzothiophene: effect of an acid component on the activity of a sulfided NiMo on alumina catalyst. Appl. Catal. A General 169, 343–353 (1998)
M. Daage, R.R. Chianelli, Structure-function relations in molybdenum sulfide catalysts: the “rim-edge” model. J. Catal. 149, 414–427 (1994)
X. Rozanska, X. Saintigny, R.A. van Santen, S. Clemendot, F. Hutschka, A theoretical study of hydrodesulfurization and hydrogenation of dibenzothiophene catalyzed by small zeolitic cluster. J. Catal. 208, 89–99 (2002)
H. Shimada, Morphology and orientation of MoS2 clusters on Al2O3 and TiO2 supports and their effect on catalytic performance. Catal. Today 86, 17–29 (2003)
V. Meille, E. Schulz, M. Lemaire, M. Vrinat, Hydrodesulfurization of alkyldibenzothiophenes over a NiMo/Al2O3 catalyst: kinetics and mechanism. J. Catal. 170, 29–36 (1997)
M.L. Vrinat, The kinetics of the hydrodesulfurization process—a review. Appl. Catal. 6, 137–158 (1983)
F. Bataille, J.L. Lemberton, P. Michaud, G. Perot, M. Vrinat, M. Lemaire, E. Schulz, M. Breysse, S. Kasztelan, Alkyldibenzothiophenes hydrodesulfurization-promoter effect. Reactivity, and reaction mechanism. J. Catal. 191, 409–422 (2000)
T.C. Ho, Hydrodenitrogenation catalysis. Catal. Rev. Sci. Eng. 30, 117–160 (1988)
Funding
(Jiaxing Science and Technology Bureau, Grant Number 2020AZ30001; and Jiaxing University, Grant Numbers 70518034 and CD70518034.)
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hu, W., Zhang, H., Wang, M. et al. Hydro‐upgrading of light cycle oil‐synthesis of NiMo/SiO2-Al2O3-TiO2 porous catalyst. J Porous Mater 28, 867–874 (2021). https://doi.org/10.1007/s10934-021-01040-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10934-021-01040-0