
Abstract
Hereditary angioedema (HAE) due to C1-inhibitor deficiency is a rare, debilitating, genetic disorder characterized by recurrent, unpredictable, attacks of edema. The clinical symptoms of HAE arise from excess bradykinin generation due to dysregulation of the plasma kallikrein-kinin system (KKS). A quantitative systems pharmacology (QSP) model that mechanistically describes the KKS and its role in HAE pathophysiology was developed based on HAE attacks being triggered by autoactivation of factor XII (FXII) to activated FXII (FXIIa), resulting in kallikrein production from prekallikrein. A base pharmacodynamic model was constructed and parameterized from literature data and ex vivo assays measuring inhibition of kallikrein activity in plasma of HAE patients or healthy volunteers who received lanadelumab. HAE attacks were simulated using a virtual patient population, with attacks recorded when systemic bradykinin levels exceeded 20 pM. The model was validated by comparing the simulations to observations from lanadelumab and plasma-derived C1-inhibitor clinical trials. The model was then applied to analyze the impact of nonadherence to a daily oral preventive therapy; simulations showed a correlation between the number of missed doses per month and reduced drug effectiveness. The impact of reducing lanadelumab dosing frequency from 300 mg every 2 weeks (Q2W) to every 4 weeks (Q4W) was also examined and showed that while attack rates with Q4W dosing were substantially reduced, the extent of reduction was greater with Q2W dosing. Overall, the QSP model showed good agreement with clinical data and could be used for hypothesis testing and outcome predictions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hereditary angioedema (HAE) is a rare, autosomal dominant genetic disorder caused by a deficiency in functional C1 esterase inhibitor (C1-INH) with an estimated prevalence of 1 in 50,000 people worldwide [1]. HAE is potentially life-threatening and is characterized by unpredictable recurrent attacks of swelling in the extremities, face, larynx, and mucosal tissues. Attacks are highly variable in their time of onset, frequency, severity, and duration [2].
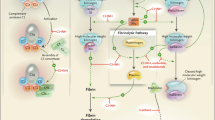
The underlying cause of HAE is a mutation in the gene encoding C1-INH, SERPING-1, resulting in reduced levels (HAE type 1) or function (HAE type 2) of the C1-INH protein [3], a negative regulator of plasma kallikrein. The kallikrein-kinin system (KKS) may become aberrantly activated in response to a variety of triggers including stress, hormonal changes, injury, exertion, sun exposure, and infection [4]. These triggers, the exact biochemical identification of which remains to be elucidated, may cause autoactivation of factor XII (FXII) to its activated form (FXIIa), which in turn converts prekallikrein to plasma kallikrein (referred to herein as kallikrein). In the absence of C1-INH, kallikrein cleaves high molecular weight kininogen (HMWK) to produce bradykinin and cleaved HMWK (cHMWK). Bradykinin activates bradykinin receptors on the surface of endothelial cells to cause vasodilation and increased blood vessel permeability, leading to subcutaneous or submucosal swelling and pain [5, 6].
Various pharmacological therapies are approved for the treatment of HAE. Icatibant is a competitive antagonist of the bradykinin B2 receptor that is indicated for the on-demand treatment of attacks [7]. In adults, a single subcutaneous injection of 30 mg icatibant provided symptom relief regardless of HAE attack location [8]. Treatments for the prevention of attacks include the following: Cinryze®, a plasma-derived C1-INH replacement therapy administered intravenously at a fixed dose of 1000 U every 3 or 4 days [9]; Haegarda®, another form of plasma-derived C1-INH, administered subcutaneously as weight-adjusted doses of 60 U/kg every 3 or 4 days [10]; berotralstat, a kallikrein inhibitor that is administered once daily as an oral 150 mg capsule [11]; and lanadelumab, a monoclonal antibody inhibitor of activated kallikrein, indicated for prophylaxis with a 300 mg dose administered every 2 weeks (Q2W), although this can be reduced to every 4 weeks (Q4W) if patients are attack free for more than 6 months [12].
We developed a mechanistic quantitative systems pharmacology (QSP) model that integrates current understanding of the pathophysiology of HAE with molecular, preclinical, and clinical data on the pharmacokinetics (PK), pharmacodynamics (PD), and efficacy of HAE treatments that target the KKS. QSP has been applied across many diseases as a strategy to aid in clinical drug development and decision making, such as patient and dose selection [13]. The QSP model described herein was developed to understand the relative effectiveness of various HAE treatments in suppressing HAE attacks, and to explore the optimal dosing regimen of these treatments and the impact of nonadherence on treatment outcomes.
Methods
Model description
An overview of the process that was followed for development of the model is shown in Fig. S1; the numbered sections described below refer to the steps in the figure. Bradykinin levels were linked to the onset of HAE attacks by modeling attack events observed in patients with HAE. The QSP model assumes that all attack triggers lead to a systematic perturbation that activates the KKS (i.e. increases the circulating plasma concentration of FXIIa), which generates bradykinin above a previously reported threshold plasma concentration of 20 pM [14], resulting in an HAE attack.
The overall QSP model for HAE incorporates both pharmacological properties of the drug such as the PK characteristics of the drug of interest and the pharmacodynamic (inhibitory) properties of the drug, as well as the disease specific biology associated with HAE such as the bradykinin threshold level to induce an HAE attack (Fig. S1). The model was developed initially alongside data for lanadelumab and was then modified to incorporate data for fixed-dose C1-INH. Clinical data from patients with HAE who received these therapies were used to calibrate and validate the model.
To test the model for the untreated state, a comparison of steady-state protein levels predicted by the KKS model versus reported levels in healthy individuals and in patients with HAE during remission was performed.
Model structure and parameterization
As illustrated by the model diagram in Fig. 1 and model parameterization step in Fig. S1, a multistep process was used to parameterize and validate the PK, PD, and HAE disease clinical endpoint of the model. Clinical studies from which data were used to develop the model are listed in Table S1. The process is as follows:
-
(1)
Pharmacokinetics. The concentration–time profile of lanadelumab was simulated using a one-compartment model with values for first-order rate of absorption, volume of distribution, linear clearance, and interindividual variability obtained from a population PK analysis [15]. The pharmacokinetic parameters of fixed-dose C1-INH were obtained from the literature [16, 17].
-
(2)
Ex vivo QSP model of the KKS system. A two-step approach was used to establish the PD parameters of the key proteins in the KKS and their interactions. First, kallikrein activity from an ex vivo prekallikrein activation assay (Fig. S2) was modeled to verify the inhibitory effect of lanadelumab against kallikrein activity in plasma from healthy volunteers and patients with HAE who received lanadelumab in phase 1 studies [18, 19]. In this assay, the addition of FXIIa to plasma simulates activation of the KKS and is described mathematically by enzymatic reactions that form kallikrein in the presence of inhibitors (e.g. lanadelumab) (Table S2). Initial parameter values for the model were obtained from the literature (Table S3). The inhibition of kallikrein activity was calculated as:
$$\%\;\mathrm{inhibition}=\left(1-\frac{{\text{KAL}}}{{{\text{KAL}}}_{{\text{control}}}}\right)*100{\%}$$where KALcontrol was the simulated kallikrein activity in control samples after 2 min of FXIIa activation. Second, parameters developed from the ex vivo modeling described above were calibrated using plasma concentrations of KKS components from the literature, as described below.
-
(3)
In vivo QSP model. A biological map of the KKS and pharmacodynamic interactions assembled from literature is presented in Fig. S3, and details on the components and interactions are presented in the Supplement methods. The QSP model was constructed using the components and mathematical equations listed in Table S4 and Table S5, respectively, and the model parameters and model assumptions listed in Tables S6 and S7, respectively. The rate of change of each species was governed by its steady-state level, binding affinity, synthesis rate, and degradation rate. An additional parameter for the catalytic conversion rate was included for enzymatic conversion reactions. Basal levels of all species were obtained from the literature and were used to guide the calibration of the synthesis rate and catalytic rate for each species. Binding affinity of lanadelumab to kallikrein was obtained from the literature and verified against data from the fluorogenic assay. Binding affinities and degradation rates for other species were obtained from the literature, except for the degradation rate of cHMWK, which was calibrated to match reported levels in healthy subjects and patients with HAE. To be consistent with C1-INH deficiency in HAE, the synthesis rate of C1-INH for HAE patients was set to be lower than that of healthy individuals. An estimate of the kallikrein concentration generated during an HAE attack was obtained from the measurement of α-2-macroglobulin complexed with kallikrein [20]. Based on published physiological parameters [21], the vascular volume circulation time is approximately 100 s; therefore, for all soluble KKS components except bradykinin, the distribution clearance between proximal and vascular space was assumed to be the product of proximal volume and the inverse of circulation time. Bradykinin was assumed to have a faster exchange rate due to its smaller size compared to other species; thus, the distribution clearance was calibrated to capture the basal level in plasma. All other model parameters were the same for healthy subjects, HAE patients during remission, and HAE patients during an attack.
-
(4)
Modelling HAE efficacy endpoint. The link between PD biomarker (cHMWK level) and clinical endpoint (number of attacks) was modeled using attack events for a virtual cohort of patients with HAE (virtual patients differ in their PK profile, attack frequency, timing of triggering event, and attack severity). The propensity of a virtual patient to experience an attack was modeled by a distribution of varying attack frequency and severity. The trigger for an attack leading to autoactivation of the KKS was modeled as a random increase in the FXII autoactivation rate; the increase occurred according to simulated timing based on a Poisson process [22], with an assigned increase in autoactivation rate following a normal distribution, and based on input of the average attack frequency in patients. Attack occurrence and monthly attack rate were recorded when the bradykinin level exceeded 20 pM [14]. The baseline attack rate for each virtual patient was randomly sampled from a normal distribution with a mean of 3.5 attacks/month obtained from the HELP study (NCT02586805) [15]. An individual HAE attack can occur at any time and is independent of the time since the last attack, but attacks overall tend to occur at a constant rate [23]. Furthermore, the time between events is exponentially distributed with parameter λ (average number of events in a given time period); therefore, the time interval between events was sampled using the inverse cumulative density function method with λ = 3.5:
$$\mathrm{Time\;interval}= \frac{-\mathrm{ ln}(1-{\text{p}})}{\uplambda } (0<{\text{p}}<1)$$

Overall scope of proposed QSP model. The model mechanistically links drug exposure to PD biomarkers and clinical outcome via the key biological pathway in HAE pathophysiology. CL clearance, HAE hereditary angioedema, f HAE attack frequency, HMWK high molecular weight kininogen, ka rate of absorption after subcutaneous administration, PD pharmacodynamics, PK pharmacokinetics, QSP quantitative systems pharmacology, Vss volume of disease at steady state. *HAE attack simulations were initiated by factor XIIa generation
A duration of 12 h was used for all attacks in this study; this was based on the different phases of the pain score [24] for a patient with HAE experiencing an untreated attack. The pain stemming from swelling typically increases during the first 8–24-h and then gradually subsides over the next 24–72 h [24,25,26]. It was assumed that the onset of pain is triggered by an attack and the presence of pain represents the duration of the attack (i.e. the time during which the level of FXII autoactivation remained elevated). HAE attacks are assessed in clinical studies using patient-reported tools such as the visual analog score or with quality of life instruments [27], which are difficult to apply in modeling without a direct link to KKS components. HAE attacks are understood to be related to the level of bradykinin, and the increase in bradykinin during an attack is caused by an increase in KKS activation. The model thus correlates attack severity with the magnitude of increase in FXII autoactivation. We estimated the extent of the increase in FXII autoactivation rate in a population by calibrating the model so that levels of bradykinin and cHMWK from virtual HAE patients (n = 1000 in each case) during an attack match disease activity in the absence of prophylactic therapy.
-
(5)
Model validation
After parameterization, the model was validated using clinical data from patients treated with lanadelumab. First, the KKS model excluding attack events was run to verify that lanadelumab inhibits plasma kallikrein (i.e. suppresses the formation of cHMWK) by comparing the simulation output to results from a phase 1b study [19] (Fig. S4). Next, the KKS model including attack events was run for 1000 virtual patients to simulate variabilities in lanadelumab PK [15], attack frequency, and attack severity. Results from the simulations were compared with cHMWK levels and monthly attack rates observed in the HELP study [28].
-
(6)
Inclusion of the impact of C1-INH in the model
To evaluate the KKS model was updated to account for treatment with C1-INH concentrates using data for fixed-dose C1-INH. Reactions for the inhibition of kallikrein and FXIIa following the administration of C1-INH were added to the model, and system parameters for endogenous C1-INH were also applied. A one-compartment model with reported PK parameters [17] was used to describe the disposition of fixed-dose C1-INH. A new variable, “C1-INH activity”, was calculated as the ratio of total endogenous and administered C1-INH versus C1-INH in healthy controls. Baseline C1-INH activity was fixed at 31% of the healthy controls for all virtual patients, reflecting their measured baseline levels [9]. A baseline monthly attack rate of 4.23 was applied for virtual patients to replicate the baseline characteristics of patients from a clinical trial [29]. Other system parameters were the same as those for simulations of lanadelumab treatment. To validate the updated model, simulations were run for HAE patients treated with a single intravenous (IV) dose of 1000 U C1-INH or 1000 U C1-INH twice/week for 12 weeks to compare the predicted PK characteristics to those observed in clinical studies. In addition, attack rate predictions for patients treated with 1000 U C1-INH twice/week for 12 weeks were compared with reported attack suppression [29].
-
(7)
Application of the model
The validated model was used to evaluate the impact of treatment nonadherence and changes in dosing regimen on attack rates during treatment. Simulations for 1000 virtual patients with HAE were performed for the switch from 6 months of treatment with lanadelumab 300 mg Q2W to 6 months of treatment with 300 mg Q4W.
Software
The QSP model was initially developed and calibrated using J2 Dynamic Modeling and Optimization Software (RES Group, Needham, MA, USA). J2 provides a high level, declarative input language suitable for describing complex mechanistic models. The algorithm implemented in J2 has been described previously [30,31,32,33]. The model was implemented using R software (version 3.5; R Foundation for Statistical Computing, Vienna, Austria). The R package “deSolve” (version 1.21) was used to run simulations. The R model code can be found in the Supplementary Material.
Results
Establishing the base system pharmacology model of the KKS in patients with HAE during remission
The ex vivo QSP model of the KKS was established using parameters from modeling of ex vivo plasma activation and fitted against published data. Data from the ex vivo fluorogenic assay of kallikrein activity using samples from healthy volunteers and HAE patients who received lanadelumab were compared with kallikrein activity results from clinical studies of lanadelumab [18, 19] (Fig. 2). Observed kallikrein inhibition data were well within the simulated results.

Ex vivo plasma prekallikrein activation assay. Citrated plasma was activated by the addition of FXIIa. The simulation output (shaded band) was compared to experimentally observed prekallikrein activity data from (a) healthy volunteers in phase 1a [18] and (b) HAE patients in phase 1b [19] studies at a dose of 3 mg/kg lanadelumab and 300 mg lanadelumab, respectively. The basal prekallikrein range corresponds to a previously reported range between 250 and 650 nM [36]. FXIIa activated factor XII, HAE hereditary angioedema, PKa prekallikrein
Using reference levels of all model species (Table 1), synthesis rates and catalytic rates in the KKS model were calibrated for agreement with reported values. A comparison of steady-state protein levels predicted by the KKS model versus reported levels in healthy subjects and in patients with HAE during remission is shown in Fig. S5.
Linking the KKS model to HAE attacks
Once the ex vivo QSP model of the KKS was established, the QSP model was developed further by linking biochemical changes in the KKS to HAE attacks. To trigger an attack, FXII autoactivation was increased. Different rates of FXII autoactivation were sampled from a normal distribution so that plasma bradykinin levels during an attack were in line with the reported range of 15–90 pM [34]. A mean (SD) 4.2-fold (1.2-fold normally distributed) increase in FXII autoactivation rate (Fig. 3a) was estimated to be required to match bradykinin levels during an attack.

Illustration of acute attack event modeling. a Distribution of fold increase in autoactivation rate of FXII fitted to published bradykinin data [14]. b Comparison of % cHMWK obtained from the simulation of 1000 virtual patients to median levels observed in the HELP study (58%) [35], where % cHMWK is the percentage of cHMWK relative to the total of cHMWK and HMWK. c Calculated bradykinin level from the virtual population simulation. d The simulated time interval between attacks compared to the data from the same dose group. The attack occurrence events sampled from a Poisson distribution were consistent with the observed data. e A sample output of attack frequency and severity. The sample used 3.2 attacks per month [19]. The value within each attack event represents the fold increase in the autoactivation rate of FXII; nonzero values are highlighted. The X represents mean value and the box plots are min, Q1, median, Q3, and max. cHMWK cleaved high molecular weight kininogen, conc concentration, HAE hereditary angioedema, HMWK high molecular weight kininogen, FXII factor XII, max maximum, min minimum, Q quartile
Autoactivation of FXII and subsequent activation of plasma kallikrein results in a spike in bradykinin and cHMWK levels. Using the estimated extent of FXII autoactivation required for an attack as determined above, simulations were run for 1000 virtual patients over a 3-month baseline period. Based on the mean value of % cHMWK (difference between the peak and base) from each spike, a mean % cHMWK of 59% was derived during HAE attacks, similar to the reported level of 58% [35] (Fig. 3b).
The maximum plasma bradykinin [19] concentration during an attack as derived from simulations of virtual patient populations is shown in Fig. 3c; the mean and 25th/75th percentiles are well within the range of measured data [34]. In these simulations, a minimum threshold of 20 pM bradykinin in plasma was used as the criterion for an attack, representing the low end of the range of measured bradykinin levels for HAE patients during an attack.
The time intervals between successive attacks for 1000 virtual patients and the time intervals observed for the placebo group in a phase 1b study [19], both with an attack rate of 2.1 attacks/month, were well-aligned, indicating that the attack occurrences sampled from a Poisson distribution were consistent with the observed data (Fig. 3d). A sample output of attack frequency and severity for 1000 virtual patients over 30 days is shown in Fig. 3e. For each virtual patient, an attack rate of 3.2 attacks/month was used as input, and an attack event was generated via random sampling. The severity of each attack event represents the fold increase in the autoactivation rate of FXII.
Model verification using data from a lanadelumab phase 3 study
Next, we investigated the ability of the model to predict biochemical changes in the KKS when a pharmacological intervention, lanadelumab, is administered. PK parameters for lanadelumab were obtained from the fitting of data from the lanadelumab clinical studies [19]. Figure 4 shows the relationship between lanadelumab dose and cHMWK levels from clinical data and the corresponding simulated profiles for virtual patients treated with 30, 100, 300, and 400 mg of lanadelumab. Results from the simulations agreed with clinical data showing an inverse correlation between drug concentration and cHMWK, and a notable reduction in cHMWK levels during the time of peak drug exposure (up to 8 weeks for the 300 mg and 400 mg dose groups).

Comparison of cHMWK levels from a clinical study [19] to the simulation output from the QSP model for HAE patients treated with (a) 30 mg, (b) 100 mg, (c) 300 mg, and (d) 400 mg lanadelumab. The predicted PK profile is based on a one-compartment model. % cHMWK is the percentage of cHMWK relative to the sum of cHMWK + HMWK. cHMWK cleaved high molecular weight kininogen, conc concentration, HAE hereditary angioedema, HMWK high molecular weight kininogen, Lana lanadelumab, mAb monoclonal antibody, PK pharmacokinetics, QSP quantitative systems pharmacology, w week
Furthermore, cHMWK levels from the HELP study [28] were compared with outputs from simulations of patients treated with lanadelumab 150 mg Q4W, 300 mg Q4W, and 300 mg Q2W (Fig. 5). In agreement with the clinical findings, the simulations showed a dose–response relationship in that cHMWK levels were reduced to a greater extent with higher dose (150 mg Q4W versus 300 mg Q4W) and more frequent dosing (300 mg Q4W versus 300 mg Q2W). Of note, the cHMWK level measured for some individual patients exceeded the 95th percentile envelope of the simulation; this may be attributed to the comparison data from individuals against a statistical output (percentile) from the simulation. The simulations also showed that all dose regimens are effective in suppressing the frequency of HAE attacks, in agreement with clinical observations (Fig. 6).

Comparison of cHMWK levels observed from clinical data to predicted levels from the QSP model simulation for virtual HAE patients treated with (a) 150 mg Q4W, (b) 300 mg Q4W, and (c) 300 mg Q2W lanadelumab. Symbols are data lines from simulation. The model prediction was made using 1000 virtual patients for each regimen group and the lanadelumab concentration was provided from the population PK model [15]. % cHMWK is the percentage of cHMWK relative to the total of cHMWK + HMWK. cHMWK cleaved high molecular weight kininogen, HAE hereditary angioedema, HMWK high molecular weight kininogen, PK pharmacokinetics, Q2W every 2 weeks, Q4W every 4 weeks, QSP quantitative systems pharmacology

Comparison of HAE attack rates from observed clinical data and simulation output from the QSP model for a virtual cohort of 1000 HAE patients treated with (a) 150 mg Q4W, (b) 300 mg Q4W, and (c) 300 mg Q2W lanadelumab. The error bars represent 1 standard deviation. The clinical study had a duration of 28 weeks and no observed clinical data are available for weeks 32 and 36. HAE hereditary angioedema, Q2W every 2 weeks, Q4W every 4 weeks, QSP quantitative systems pharmacology
Model validation with clinical data for fixed-dose C1-INH
To generate further confidence in the KKS QSP model, the impact of fixed-dose C1-INH was investigated and compared to observed data. Without modifying or recalibrating the parameters of the above described KKS QSP model, simulated PK profiles for fixed-dose C1-INH were in agreement with mean concentrations of functional C1-INH protein in patients with HAE (Fig. S6) [29]. The same model was then used to run simulations for 1000 virtual patients following twice/week dosing of 1000 U C1-INH for 12 weeks. Attack rates from the model were consistent with attack rates observed in a clinical study (Fig. 7).

Simulation of attack frequency for fixed-dose C1-INH in comparison with clinical data [29]. C1-INH C1 esterase inhibitor, HAE hereditary angioedema
Application of the model: effect of a missed dose
Nonadherence to preventive therapy was studied for a hypothetical oral drug with a shorter half-life (“drug X”) using simulations of virtual populations with an assumed average occurrence of nonadherence (random missing dose) of 20% (i.e. six missed doses in a 30-day month). Drug X was modeled as a once daily oral KAL inhibitor with a Kd of 0.44 nM and T1/2 of 19 h, which would result in an attack suppression of 54% during the first month after treatment. The model suggests that nonadherence for daily dosing of “drug X” could negatively impact the suppression of HAE attacks, as each missed dose would reduce drug levels, resulting in bradykinin peaks that exceed the 20 pM threshold for attacks (Fig. 8a). The monthly attack profiles showed that more missed doses resulted in increased occurrence of HAE attacks (Fig. 8b). Simulations showed that 6–8 days of randomly missed doses out of 4 weeks of daily dosing could translate into an approximately two-fold decline in efficacy. If ≥ 50% of doses are missed, efficacy becomes marginal.

a Example pharmacokinetics of “drug X” 150 mg QD and bradykinin profiles with a nonadherence rate of 20%. b Simulated numbers of monthly attacks during treatment of “drug X” 150 mg QD with different nonadherence rates. Percentages represent reduction of attack rates compared with placebo. QD once daily
Application of the model: effect of lanadelumab down-titration
In simulations of virtual patients with a baseline attack rate of 3.5 attacks/month (similar to the mean attack rate among patients enrolled in the HELP study [28]), lanadelumab Q2W dosing resulted in mean minimum and maximum concentrations of lanadelumab above the 90% inhibitory concentration (IC90) of 18.8 µg/mL [15], mean cHMWK levels below 51%, and bradykinin levels below the 0.02 nM threshold for attacks (Fig. 9). After 6 months of Q2W treatment, the dosing frequency was reduced to Q4W, resulting in mean lanadelumab maximum concentrations, but not minimum concentrations, above IC90. However mean cHMWK levels remained below 51%, and bradykinin levels were less than 0.02 nM. Attack-free status, particularly in patients with high baseline attack rates (Fig. S7), was best achieved with Q2W dosing.

Simulations of lanadelumab 300 mg Q2W dosing for 6 months followed by Q4W dosing for 6 months. HAE hereditary angioedema, Q2W every 2 weeks, Q4W every 4 weeks
Discussion
Model-informed drug development has been used to integrate information from diverse data sources to inform and support decision making during the drug development process. QSP is one approach that leverages thorough understandings of the disease pathology and the drug. A QSP model of the KKS was developed to quantitatively describe the dynamic interactions of the key components and biological pathways of the KKS and therapeutics that target the system. The model was linked to the frequency of attacks in HAE and provides a quantitative understanding of outcomes in clinical investigations of HAE therapeutics. Clinical drug development for rare diseases such as HAE is particularly challenging due to the small pool of patients available to participate in trials. QSP modeling is advantageous because it allows for outcome predictions in the absence of an additional clinical trial, thereby providing answers to questions quickly and accurately.
The workflow of the QSP model development began with a subset of the KKS using ex vivo data to explore and define the optimal scope of a model that can be reliably parameterized and verified. With all parameters initially estimated from literature data, the model for the ex vivo fluorescent assay was able to estimate observed inhibition of kallikrein activity [36]. This model was then expanded to include all key molecular elements in the KKS that are involved in HAE. KKS activation is localized on physiological surfaces and kallikrein is largely tethered to the endothelial cell surface through the prekallikrein–HMWK complex [37]. Consequently, the local (proximal space) concentration of kallikrein is simulated to approach the level of its precursor, prekallikrein (~ 500 nM) [38], with a simulated range between 40–400 nM during attacks depending on factors such as the volume of the local environment. While literature estimates for free, circulating kallikrein levels were unavailable, the model predicted a range from 0.2 to 1.5 nM. This low concentration of free kallikrein in the circulation could be due to the short half-life of kallikrein (~ 5 min) [39] and may contribute to attack localization, as opposed to systemic angioedema in HAE.
Alongside the modeling of HAE attack events, the QSP model was applied to verify the inhibitory effect of various therapeutics in HAE patients by comparing results from the simulations to biomarker data and attack rates observed in clinical trials. Reduction of cHMWK formation owing to kallikrein inhibition was sustained throughout the lanadelumab treatment period. The model demonstrated that the 300 mg Q2W regimen is optimal for achievement of therapeutic efficacy as indicated by the fastest and highest extent of cHMWK suppression over time, as well as the lowest average monthly attack rate compared with the other dosing regimens. Furthermore, the model showed that down-titration to Q4W dosing may still provide significant protection against HAE attacks, depending on the goals of treatment. The model supports the 300 mg Q2W regimen to achieve the therapeutic goal of being attack free; however, 300 mg Q4W dosing may be an appropriate option for some patients. Notably, a new steady state is reached approximately 70 days after the switch to Q4W dosing; thus, patients and their physicians should be mindful of this when monitoring the effectiveness of the new dose over time.
The key advantage of this QSP model is that it was constructed to cover the essential biological network of the KKS in HAE. Thus, the model allows for testing of its predictive capability against different treatment options. For example, the model was applied to analyze the impact of fixed-dose C1-INH using the same system parameters as the lanadelumab model, with only the PK component being updated for C1-INH; the primary endpoint (number of attacks) obtained from the model was in good agreement with reported results from clinical trials.
The model was also applied to investigate the potential impact of nonadherence to treatment with a theoretical orally administered daily inhibitor of plasma kallikrein, as a mean adherence rate of 80% (range ~ 65–90%) in the real world has been reported for daily oral therapy [40]. The model suggested that a nonadherence rate of 20% (i.e. five or six doses missed randomly in a month) results in a 35% increase in attack rate. Thus, with the short half-life of medications that must be administered daily, the apparent convenience of an oral dosing regimen is countered by a need to ensure strict adherence to treatment as a way of maintaining efficacy and cost effectiveness. On the other hand, the long half-life of lanadelumab results in maintenance of trough plasma concentrations above IC90 even with Q2W dosing, and less frequent dosing (13–26 doses/year) provides fewer opportunities to miss doses and experience breakthrough attacks.
QSP models mechanistically link different components in biological pathways to mathematically represent complex physiology and drug interactions. Application of QSP models can be challenging due to biological uncertainty and variability. To develop a model with a high level of validity, the goal was to improve understanding of the connection between mechanisms and outcomes. A virtual cohort of patients was generated to explore the impact of biological uncertainty and variability on response to therapies. Lastly, the model was challenged with approved medications targeting different elements of the KKS. The model may serve as a predictive platform for new drug development in the area of disease pathophysiology involving the KKS.
Conclusions
The QSP model of KKS dysfunction in HAE shows promise as a useful predictive platform for new drug development in HAE. With rare diseases such as HAE, prospective trials that are meant to assess the impact of PK/PD changes on patient outcomes can be challenging to conduct. This model allows for hypothesis testing in the absence of an additional clinical trial, thereby facilitating drug discovery and development in HAE.
Data availability
The datasets, including the redacted study protocol, redacted statistical analysis plan, and individual participants’ data supporting the results reported in this article, will be made available within 3 months from initial request to researchers who provide a methodologically sound proposal. The data will be provided after its de-identification, in compliance with applicable privacy laws, data protection, and requirements for consent and anonymization.
References
Maurer M, Magerl M, Ansotegui I, Aygören-Pürsün E, Betschel S, Bork K, Bowen T, Balle Boysen H, Farkas H, Grumach AS, Hide M, Katelaris C, Lockey R, Longhurst H, Lumry WR, Martinez-Saguer I, Moldovan D, Nast A, Pawankar R, Potter P, Riedl M, Ritchie B, Rosenwasser L, Sánchez-Borges M, Zhi Y, Zuraw B, Craig T (2018) The international WAO/EAACI guideline for the management of hereditary angioedema-The 2017 revision and update. Allergy 73:1575–1596. https://doi.org/10.1111/all.13384
Weis M (2009) Clinical review of hereditary angioedema: diagnosis and management. Postgrad Med 121:113–120. https://doi.org/10.3810/pgm.2009.11.2071
Lumry WR (2013) Overview of epidemiology, pathophysiology, and disease progression in hereditary angioedema. Am J Manag Care 19:s103–s110
Zotter Z, Csuka D, Szabó E, Czaller I, Nébenführer Z, Temesszentandrási G, Fust G, Varga L, Farkas H (2014) The influence of trigger factors on hereditary angioedema due to C1-inhibitor deficiency. Orphanet J Rare Dis 9:44. https://doi.org/10.1186/1750-1172-9-44
Walker K, Perkins M, Dray A (1995) Kinins and kinin receptors in the nervous system. Neurochem Int 26:1–16. https://doi.org/10.1016/0197-0186(94)00114-a. (discussion 17–26)
Choi SI, Hwang SW (2018) Depolarizing effectors of bradykinin signaling in nociceptor excitation in pain perception. Biomol Ther (Seoul) 26:255–267. https://doi.org/10.4062/biomolther.2017.127
Dubois EA, Cohen AF (2010) Icatibant. Br J Clin Pharmacol 69:425–426. https://doi.org/10.1111/j.1365-2125.2010.03642.x
Lumry WR, Farkas H, Moldovan D, Toubi E, Baptista J, Craig T, Riedl M (2015) Icatibant for multiple hereditary angioedema attacks across the controlled and open-label extension phases of FAST-3. Int Arch Allergy Immunol 168:44–55. https://doi.org/10.1159/000441060
Takeda (2021) Cinryze® prescribing information. http://pi.shirecontent.com/PI/PDFs/Cinryze_USA_ENG.pdf. Accessed 11 October 2021
CSL Behring (2022) Haegarda® prescribing information. https://labeling.cslbehring.com/PI/US/HAEGARDA/EN/HAEGARDA-Prescribing-Information.pdf. Accessed 18 August 2023
Biocryst Pharmaceuticals, Inc. (2020) ORLADEYO™ (berotralstat) capsules, for oral use, prescribing information. https://www.biocryst.com/wp-content/uploads/2020/12/ORLADEYO_PI_V1_2020.pdf. Accessed 18 August 2023
Shire plc (2018) TAKHZYRO® prescribing information. https://www.shirecontent.com/PI/PDFs/TAKHZYRO_USA_ENG.pdf. Accessed 11 October 2021
Aghamiri SS, Amin R, Helikar T (2021) Recent applications of quantitative systems pharmacology and machine learning models across diseases. J Pharmacokinet Pharmacodyn 49:1–19. https://doi.org/10.1007/s10928-021-09790-9
Nussberger J, Cugno M, Amstutz C, Cicardi M, Pellacani A, Agostoni A (1998) Plasma bradykinin in angio-oedema. The Lancet 351:1693–1697. https://doi.org/10.1016/S0140-6736(97)09137-X
Wang Y, Marier J-F, Kassir N, Chang C, Martin P (2020) Pharmacokinetics, pharmacodynamics, and exposure-response of lanadelumab for hereditary angioedema. Clin Transl Sci 13:1208–1216. https://doi.org/10.1111/cts.12806
Wang Y, Jomphe C, Marier JF, Martin P (2021) Population pharmacokinetics and exposure-response analyses to guide dosing of icatibant in pediatric patients with hereditary angioedema. J Clin Pharmacol 61:555–564. https://doi.org/10.1002/jcph.1768
Shire (2017) Cinryze(R) assessment report. https://www.ema.europa.eu/en/documents/assessment-report/cinryze-epar-public-assessment-report_en.pdf. Accessed October 11, 2021
Chyung Y, Vince B, Iarrobino R, Sexton D, Kenniston J, Faucette R, TenHoor C, Stolz LE, Stevens C, Biedenkapp J (2014) A phase 1 study investigating DX-2930 in healthy subjects. Ann Allergy Asthma Immunol 113:460-466.e462
Banerji A, Busse P, Shennak M, Lumry W, Davis-Lorton M, Wedner HJ, Jacobs J, Baker J, Bernstein JA, Lockey R (2017) Inhibiting plasma kallikrein for hereditary angioedema prophylaxis. N Engl J Med 376:717–728. https://doi.org/10.1056/NEJMoa1605767
Kaufman N, Page JD, Pixley RA, Schein R, Schmaier AH, Colman RW (1991) Alpha 2-macroglobulin-kallikrein complexes detect contact system activation in hereditary angioedema and human sepsis. Blood 77:2660–2667
Shah DK, Betts AM (2012) Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J Pharmacokinet Pharmacodyn 39:67–86
Navidi WC (2008) Statistics for engineers and scientists, 2nd end. McGraw-Hill Higher Education, New York, NY, p 901. https://books.google.com/books/about/Statistics_for_Engineers_and_Scientists.html?id=I9FFAQAAIAAJ
Lumry WR, Castaldo AJ, Vernon MK, Blaustein MB, Wilson DA, Horn PT (2010) The humanistic burden of hereditary angioedema: Impact on health-related quality of life, productivity, and depression. Allergy Asthma Proc 31:407–414
Bork K, Staubach P, Eckardt AJ, Hardt J (2006) Symptoms, course, and complications of abdominal attacks in hereditary angioedema due to C1 inhibitor deficiency. Am J Gastroenterol 101:619–627. https://doi.org/10.1111/j.1572-0241.2006.00492.x
Gompels M, Lock R, Abinun M, Bethune C, Davies G, Grattan C, Fay A, Longhurst H, Morrison L, Price A (2005) C1 inhibitor deficiency: consensus document. Clin Exp Immunol 139:379–394. https://doi.org/10.1111/j.1365-2249.2005.02726.x
Bork K, Hardt J, Schicketanz K-H, Ressel N (2003) Clinical studies of sudden upper airway obstruction in patients with hereditary angioedema due to C1 esterase inhibitor deficiency. Arch Intern Med 163:1229–1235. https://doi.org/10.1001/archinte.163.10.1229
Bygum A, Busse P, Caballero T, Maurer M (2017) Disease severity, activity, impact, and control and how to assess them in patients with hereditary angioedema. Front Med (Lausanne) 4:212. https://doi.org/10.3389/fmed.2017.00212
Banerji A, Riedl MA, Bernstein JA, Cicardi M, Longhurst HJ, Zuraw BL, Busse PJ, Anderson J, Magerl M, Martinez-Saguer I (2018) Effect of lanadelumab compared with placebo on prevention of hereditary angioedema attacks: a randomized clinical trial. JAMA 320:2108–2121. https://doi.org/10.1001/jama.2018.16773
Zuraw BL, Busse PJ, White M, Jacobs J, Lumry W, Baker J, Craig T, Grant JA, Hurewitz D, Bielory L (2010) Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med 363:513–522. https://doi.org/10.1056/NEJMoa0805538
Brenan KE, Campbell SL, Petzold LR (1995) Numerical Solution of Initial-Value Problems in Differential-Algebraic Equations. Soc Industrial Appl Math [SIAM], https://doi.org/10.1137/1.9781611971224
Feehery WH, Tolsma JE, Barton PI (1997) Efficient sensitivity analysis of large-scale differential-algebraic systems. Appl Numer Math 25:41–54
Goh CJ, Teo KL (1988) Control parametrization: A unified approach to optimal control problems with general constraints. Automatica 24:3–18
Zhu C, Byrd RH, Lu P, Nocedal J (1997) Algorithm 778: L-BFGS-B: Fortran subroutines for large-scale bound-constrained optimization. ACM Trans Math Software 23:550–560
Nussberger J, Cugno M, Cicardi M (2002) Bradykinin-mediated angioedema. N Engl J Med 347:621–622. https://doi.org/10.1056/NEJM200208223470820
Suffritti C, Zanichelli A, Maggioni L, Bonanni E, Cugno M, Cicardi M (2014) High-molecular-weight kininogen cleavage correlates with disease states in the bradykinin-mediated angioedema due to hereditary C 1-inhibitor deficiency. Clin Exp Allergy 44:1503–1514. https://doi.org/10.1111/cea.12293
Madsen DE, Sidelmann JJ, Biltoft D, Gram J, Hansen S (2015) C1-inhibitor polymers activate the FXII-dependent kallikrein–kinin system: Implication for a role in hereditary angioedema. Biochim Biophys Acta 1850:1336–1342. https://doi.org/10.1016/j.bbagen.2015.03.005
Colman RW, Schmaier AH (1997) Contact system: a vascular biology modulator with anticoagulant, profibrinolytic, antiadhesive, and proinflammatory attributes. Blood 90:3819–3843
Kenniston JA, Faucette RR, Martik D, Comeau SR, Lindberg AP, Kopacz KJ, Conley GP, Chen J, Viswanathan M, Kastrapeli N (2014) Inhibition of plasma kallikrein by a highly specific active site blocking antibody. J Biol Chem 289:23596–23608. https://doi.org/10.1074/jbc.M114.569061
Cumming A, Robertson C, Jeffrey S, Robson J, Ledingham I (1984) The plasma kallikrein kinin system in severely ill and traumatised patients. Arch Emerg Med 1:135–142. https://doi.org/10.1136/emj.1.3.135
Osterberg L, Blaschke T (2005) Adherence to medication. N Engl J Med 353:487–497. https://doi.org/10.1056/NEJMra050100
Weidmann H, Heikaus L, Long AT, Naudin C, Schlüter H, Renné T (2017) The plasma contact system, a protease cascade at the nexus of inflammation, coagulation and immunity. Biochim Biophys Acta Mol Cell Res 1864:2118–2127. https://doi.org/10.1016/j.bbamcr.2017.07.009
Madsen DE, Sidelmann JJ, Overgaard K, Koch C, Gram JB (2013) ELISA for determination of total coagulation factor XII concentration in human plasma. J Immunol Methods 394:32–39. https://doi.org/10.1016/j.jim.2013.04.012
Schapira M, Silver LD, Scott CF, Schmaier AH, Prograis LJ Jr, Curd JG, Colman RW (1983) Prekallikrein activation and high-molecular-weight kininogen consumption in hereditary angioedema. N Engl J Med 308:1050–1053. https://doi.org/10.1056/nejm198305053081802
Cardigan RA, Crook M, Mackie IJ, Machin SJ (2001) Plasma levels of factor XIIa and factor VIIa are increased but not related in primary hyperlipidaemia. Blood Coagul Fibrinolysis 12:187–192. https://doi.org/10.1097/00001721-200104000-00004
Cugno M, Cicardi M, Bottasso B, Coppola R, Paonessa R, Mannucci PM, Agostoni A (1997) Activation of the coagulation cascade in C1-inhibitor deficiencies. Blood 89:3213–3218
Zhang G, Sexton DJ, Faucette RR, Qiu Y, Wu J (2017) 2D-LC-MS/MS to measure cleaved high-molecular-weight kininogen in human plasma as a biomarker for C1-INH-HAE. Bioanalysis 9:1477–1491
Kaplan AP, Ghebrehiwet B (2010) The plasma bradykinin-forming pathways and its interrelationships with complement. Mol Immunol 47:2161–2169. https://doi.org/10.1016/j.molimm.2010.05.010
Wu Y (2015) Contact pathway of coagulation and inflammation. Thromb J 13:17
Drouet C, Ponard D, Ghannam A (2018) Chapter 23: C1 inhibitor. The Complement FactsBook, 2nd edn. Academic Press, Cambridge, Massachusetts, pp 241–249
Cicardi M, Igarashi T, Rosen FS, Davis AE 3rd (1987) Molecular basis for the deficiency of complement 1 inhibitor in type I hereditary angioneurotic edema. J Clin Invest 79:698–702. https://doi.org/10.1172/jci112873
Konings J, Cugno M, Suffritti C, ten Cate H, Cicardi M, Govers-Riemslag JWP (2013) Ongoing contact activation in patients with hereditary angioedema. PLoS ONE 8:e74043. https://doi.org/10.1371/journal.pone.0074043
Nussberger J, Cugno M, Cicardi M, Agostoni A (1999) Local bradykinin generation in hereditary angioedema. J Allergy Clin Immunol 104:1321–1322. https://doi.org/10.1016/S0091-6749(99)70030-8
Acknowledgements
Under direction of the authors, Shirley Teng, PhD, CMPP, of Excel Medical Affairs provided writing assistance for this manuscript. Editorial assistance in formatting, proofreading, and copyediting also was provided by Excel Medical Affairs. Takeda Pharmaceuticals USA, Inc., provided funding to Excel Medical Affairs for support in writing and editing this manuscript.
Funding
The study was sponsored by Takeda Development Center Americas, Inc.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design, data analysis, critically reviewed the manuscript, and approved the final version of the manuscript for submission.
Corresponding author
Ethics declarations
Competing interests
S. Juethner and A. Z. X. Zhu are full-time employees of Takeda and hold stocks/options in Takeda. D. Sexton and H. Q. Nguyen were employees of Takeda at the time of this analysis. H. Luo, Z. Zhang, and P. Jasper are full-time employees of the RES Group, which was contracted by Takeda to develop the QSP HAE model.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Hoa Q. Nguyen was an employee of Takeda Development Center of Americas at the time of this study.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sexton, D., Nguyen, H.Q., Juethner, S. et al. A quantitative systems pharmacology model of plasma kallikrein-kinin system dysregulation in hereditary angioedema. J Pharmacokinet Pharmacodyn (2024). https://doi.org/10.1007/s10928-024-09919-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10928-024-09919-6