
Abstract
In this manuscript, we report four series for 1,2,4-triazine derivatives as dye-sensitized solar cells (DSSCs). Density functional theory (DFT) methods via utilizing Becke's three-parameter functional and LeeeYangeParr functional (B3LYP) level with 6-31G (d, p) basis set to investigate their modeling molecular structures. Optimized molecular structures for studied molecular structures are obtained using the DFT/B3LYP/6-31G (d, p) method. In addition, the time-dependant density functional theory (TD-DFT) is used to study the optoelectronic properties and absorption spectra using DFT/CAM-B3LYP/ 6-31G + + (d, p) level in the Gaussian 09 program. The highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), energy gap (Eg), light harvest efficiency (LHE), and open-circuit voltage (Voc) of the studied molecular structures are calculated and illustrated. These properties indicate that these molecular modeling structures as good candidates for utilization in organic DSSCs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Because of the great challenge used in new research on renewable energy sources, solar cells are considered one of the most important renewable energies recently [1,2,3,4]. Photovoltaic technologies have become one of the most important topics in solar cells to convert the sun into electrical energy [5, 6]. In addition, the most important challenges are represented in capturing solar energy and converting it into electrical energy at a low cost [7]. It was taken out of the photovoltaic devices that are based on inorganic materials in their manufacture, such as crystalline and amorphous silicon, cadmium telluride (CdTe), gallium arsenide (GaAs), with the knowledge that they give their efficiency from 10 to 32% [8]. However, since these are expensive materials and scarce, in addition to their toxicity, many researchers have resorted to searching for other new, cheap, and more efficient materials. According to what will be said, solar cells based on organic compounds are considered an attractive and appropriate choice due to their flexibility and ease of processing in addition to their low cost, but their efficiency at present is considered less than those that depend on inorganic materials [9]. The efficiency obtained from these types of cells is still not marketable, as the most efficient devices are 4 to 5% [10].
Recently, new organic compounds used in solar cells have been studied and developed. Among these materials are the dyes for sensitive solar cells (DSSCs), as they receive great interest among researchers due to their low cost and high efficiency in converting solar energy into electricity [11]. Moreover, the manufacture of their devices is easy. Also, photovoltaic cells based on the DSSCs have many advantages, including their compatibility with many supporting materials and production under moderate conditions that make them less expensive compared to other DSSCs [12, 13]. The first DSSCs were based on titanium dioxide, which was discovered in 1991, and their efficiency was from 7 to 8 percent. [13].
In this manuscript, the results of quantum calculations for four organic compounds based on 1,2,4-triazine derivatives as dye-sensitized solar cells (DSSCs) are presented. In these structures, we used sulfur atom as electron donor unit (D) and the phenyl and triazine groups were used as electron acceptor groups (A) for all compounds.
Time-Dependent DFT (TD-DFT) has been widely used to discuss the properties of organic compounds in their excited state because its high accuracy is reasonable with the ab-initio method and less computational time cost. In this paper, the obtained calculated results via utilizing TD-DFT/ CAM-B3LYP/ 6–31 + + G (d, p) like the optoelectronic properties and photovoltaic properties (open-circuit voltage (Voc), oxidation potential energy, and electron injection force) of all molecular structures were investigated and presented.
Computational Investigation
The molecular modeling and photoelectronic properties of the studied compounds (DTT, BDTTB, BDTTMB, and BDTTMP) based on 1,2,4-triazine were obtained and investigated using Density functional theory (DFT) methods via utilizing Becke's three-parameter functional and LeeeYangeParr functional (B3LYP) [14] level with 6-31G(d, p) [15] basis set. All computational calculations were presented by utilizing the Gaussian 09 program [15]. Upon utilizing the DFT/B3LYP/6-31G (d, p) level, the molecular structures of neutral molecules are optimized, and their molecular electronic properties as HOMO, LUMO levels, and the energy gap (Eg) are obtained. The oscillator strengths (f) and the optical transitions have been illustrated using TD-DFT combined with CAM-B3LYP functional and 6- 31G + + (d, p) basis set. Finally, the ultraviolet–visible (UV–Vis.) absorption spectra of the studied molecules were presented utilizing Gauss View software [16].
Result and Discussions
Studied Molecular Structures
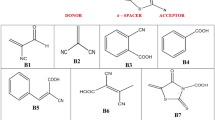
The studied molecular structures as shown in Fig. 1 are 5, 6-diphenyl-1,2,4-triazine-3(4H)-thione (DTT), 1,4-Bis((5,6-diphenyl-1,2,4-triazin-3-yl)thio)butane (BDTTB), 1,4-Bis(((5,6-diphenyl-1,2,4-triazin-3-yl)thio)methyl)benzene (BDTTMB) and 2,6-Bis(((5,6-diphenyl-1,2,4-triazin-3-yl)thio)methyl)pyridine (DTTMP). As shown in Fig. 1, the organic chemical compounds BDTTB, BDTMP and BDTMP consist of two main DTT groups, while the intermediate group differs from one compound to the other. Since in BDTTB the middle group is a butyl group, and in the BDTTMB compound, the butyl group is replaced by a p-xylenyl group, and finally in the BDTTMP compound, the middle group is 2,6-butidienyl. The four studied molecular structures under study were prepared, characterized and published in 2121 by Sakr et. al. [17].

Studied molecular structures DTT, BDTTB, BDTTMB and BDTTMP
Molecular Structure Properties
The optimization of the molecular structures under study has been obtained via using the DFT/B3LYP/6-31G (d, p) method in the gas state; the result of optimized molecular structures are presented in Fig. 2. The effect of substituents such as butyl in BDTTB, p-xylenyl in BDTTMB, and 2,6-butidienyl in BDTTMP has been studied on some important selected optimized molecular structure parameters such as (bond length in Å and dihedral angle in o) for DTT optimized molecular structure; the results are collected in Table 1. Some important comments that can be deduced are as follows; (1) The DDT molecular structure is not planar as one of the two phenyl groups rotate out of the triazine by angle 33.44° to prevent the steric hindrance. (2) all molecular structures (BDTTB, BDTTMB, and BDTTMP) are not planar as one of the phenyl groups rotates by 34.04, 15.13, and 35.49° for BDTTB, BDTTMB, and BDTTMP respectively out of the triazine moiety to decrease steric hindrance. Hence, the π-interactions among sub-systems in each molecule are small. (3) Generally, the stability of molecular structure can be judged via the length of the bond [18], where the more stable molecular structure is combined with the shorter the bond length. As shown in Table 1, the bond lengths of DDT and BDTTMB are less than that of BDTTB and BDTTMP hence, the DDT and BDTTMB are more stable molecular structures compared to BDTTB and BDTTMP. (4) The selected bond angles for all studied molecular structures refer to SP2 hybridization.

Optimized molecular structures for studied molecular structures using the DFT/B3LYP/6-31G (d, p) method
Energy levels of the studied molecular structures
The graphical presentation of the HOMO and LUMO MOs, for DDT, BDTTB, BDTTMB, and BDTTMP compounds in gas at the B3LYB/6-31G (d, p) level of theory is shown in Fig. 3. It is noticeable from Fig. 3 that the HOMO MOs of the compounds under study are localized on certain parts of these compounds (sulfur atom, triazine, and phenyl groups), but not all of them. On the other side, the LUMO MOs are localized on (triazine and phenyl groups). This indicates that the sulfur atom, triazine, and phenyl groups act as a donor, but on the other hand, triazine and phenyl groups act as an acceptor. Furthermore, the calculated energy values of HOMO, HOMO-1, HOMO-2, LUMO, LUMO + 1, and LUMO + 2 and the energy gap between the following; (HOMO and LUMO (Eg1), HOMO-1 and LUMO + 1 (Eg2) and HOMO-2 and LUMO + 2(Eg3)) of the compounds under study are written in Tables 2 and 3 The HOMO of the studied molecular structures are ordered as follows; DDT < BDTTMP < BDTTMB < BDTTB. Also, the LUMO energy MOs are arranged in the following order; BDTTMP < BDTTB < DTT < BDTTMB. The calculated Eg1 of the studied molecular structures are arranged in the following order BDTTB < BDTTMP < BDTTMB < DTT. This indicates that compound BDTTB has the highest reactivity and compound DTT has the lowest reactivity. Also, the reduction in the Eg1 value favors the red-shifted of the electronic absorption maximum peak in the UV–Vis absorption spectrum. Therefore, the electronic UV–Vis absorption spectra for BDTTB, BDTTMP, and BDTTMB are red shifted, in contrast to, the UV–Vis absorption spectrum of DTT. As a whole, the electron density of HOMO MOs is mainly located near the donor and the electron density of LUMO is mainly located near the acceptor. Consequently, the sulfur atom acts as an electron donor unit (D), and both the phenyl and triazine groups were used as electron acceptor units (A).

3 Graphical presentation of the highest occupied (HOMO), lowest unoccupied molecular (LUMO) orbitals, the energy of HOMO and LUMO, and energy gaps (Eg1) for DTT, BDTTB, BDTTMB, and BDTTMP in gas at B3LYB/6-31G(d, p) level of theory
Figure 4 exhibits the molecular orbital energy levels of the four molecular structures DTT, BDTTB, BDTTMB, and BDTTMP in gas at the DFT/B3LYB/6-31G (d, p) level of theory. For the all-molecular structures under study, the HOMO energy is lower than the energy of I−/I2 (− 4.85 eV). This indicates that all studied compounds (DTT, BDTTB, BDTTMB, and BDTTMP) can more easily recover electrons from electrolytes (I3−). In addition to, the LUMO energy of the four studied molecules (DTT, BDTTB, BDTTMB, and BDTTMP) is higher than the conduction band energy of semiconductor TiO2 (− 4.00 eV) as shown in Fig. 4. Indicating, that the electrons can be successfully transferred into TiO2 from the excited state of all studied dyes. Consequently, all studied molecular structure dyes may be good candidates for application in photovoltaic devices.

Frontier molecular orbital energies and energy gaps of TiO2, DTT, BDTTB, BDTTMB, BDTTMP, and I.−/I2
The conversion efficiency (Ƞ) of sunlight to electrical energy in solar cell devices is determined by the short-circuit current density (Jsc), the open-circuit photovoltage (Voc), and the fill factor (FF) and incident solar power (Pinc). The Ƞ can be calculated by utilizing the following Eq. (1) [19]:
From the formula, it can be seen that high VOC and JSC are the basis for producing photoelectric conversion efficiency. The maximum value for Voc is a significant photovoltaic parameter that can be obtained computationally via utilizing the difference between the HOMO of dye and the LUMO of the electron acceptor (conduction band of TiO2). The computational value of Voc has been calculated by utilizing the following (2) [20, 21]:
Whilst, in DSSCs, Voc can be approximately obtained as the different energy among LUMO of the dye and conduction band (CB) of the semiconductor
The theoretically calculated value of Voc for all studied molecular structures is presented in Table 2. The range value of Voc is 1.738 to 1.938 eV for semiconductors (TiO2). These values are positive; thus indicating that the electron transfer will be easy from all studied molecular structures (DTT, BDTTB, BDTTMB, and BDTTMP) to TiO2. Furthermore, these values are sufficient to give the best efficient electron injection. Moreover, these triazine derivatives compounds can be utilized as sensitizers of the electron injection process from the excited dye to the conduction band of TiO2.
Another parameter denoted (α) was calculated via the difference between the LUMO energy levels of the studied dyes and the LUMO energy level of PCBM (-3.2 eV) [22]. The value can be obtained by using the following Eq. (4):
The light-harvesting efficiency (LHE) is obtained using the following equation (LHE = 1-10f) [22], where f is the oscillator strength of the dye molecular structure; the calculated LHE is presented in Table 4.
As presented in Table 4, the obtained values of α were in the range of (0.938–1.138 eV). This indicates, that all LUMO levels for all studied compounds are placed higher than the LUMO level of PCBM. So, these triazine derivatives compounds can be utilized as sensitizers of the electron injection process from the excited dye to the conduction band of PCBM. Depending on the values of LHE in Table 4, the best dye that can act as DSSCs is DDT dye compared to the rest compounds under study.
UV–Vis Properties
The optical properties and the electronic transition for the all studied dyes (DTT, BDTTB, BDTTMB and BDTTMP) are investigated using TD-DFT/ CAM-B3LYP / 6-31G + + (d, p) method [23]. The calculated transition energies of studied molecular structures for the absorption wavelength, vertical excitation energy (Eex), oscillator strength (f), and the transition character of these dyes are listed in Table 4. The UV–Vis absorption spectra of triazine dye derivatives simulated at the TD-DFT/ CAM-B3LYP / 6-31G + + (d, p) method are located from 300 to 450 nm as shown in Fig. 5. The experimental electronic absorption spectra of the DDT, BDTTB, BDTTMB, and BDTTMP molecular structures were obtained [17]. The electronic experimental maximum absorption wavelengths were 400, 350, 350, and 350 for DDT, BDTTB, BDTTMB, and BDTTMP molecules respectively. The calculated electronic absorption spectrum of DTT in methanol appears as three transitions at 390, 332, and 382 nm. The first one refers to the experimental peak at 400 nm (0.005) that arises from the transition of HOMO-1—> LUMO. On the other hand, a second band (f = 0.110) arises from a transition of HOMO—> LUMO and the third one (f = 0.1210) due to electronic transition of HOMO-2—> LUMO. Also, the calculated electronic absorption of the other molecular structures (BDTTB, BDTTMB, and BDTTMP) are investigated via using the same level as presented in Fig. 5 and Table 4. The three calculated electronic transitions for BDTTB dye appear at 416 nm, 348 nm and 335 nm in methanol where in agreement with practical results. The first, second and third absorption peak arises from the electronic transition HOMO-2—> LUMO + 1, HOMO—> LUMO + 1 and HOMO-3—> LUMO + 2. The same findings were also found for both BDTTMB and BDTTMP but with red shift for DTT one.

The calculated electronic UV–Vis absorption spectra for all studied molecules (DTT, BDTTB, BDTTMB and BDTTMP) in methanol using TD-DFT/ CAM-B3LYP / 6-31G + + (d, p)
Natural Bond Orbital (NBO) Analysis
Since resonance is a fundamental phenomenon in the stability of the organic chemical structure, it was essential to study the stability of the studied molecular chemical structures (DDT, BDTTMB, BDTTB, and BDTTMP) using Natural Bond Orbital (NBO) analysis. The hyperconjugation interactions in (DDT, BDTTMB, BDTTB, and BDTTMP) molecular structures are examined via NBO analysis theory utilizing the B3LYP/6-31G(d, p)level of theory [24]. Besides, these results were obtained by utilizing second request annoyance energies (E(2)). 24] The utmost influential second-order perturbation (E(2)) delocalization energies in all studied molecular structures in their gaseous phase are presented in Table 5. These are classified as π- π* and n- π* interactions, with the latter having larger energy magnitudes. The intense interaction between C2-N4 π-bond to C1-N6 π-antibonding stabilized DDT molecular structure by 22.84 kcal/mol then from C3-N5 π-bond to C4-N38 π-antibonding via 32.31 kcal/mol. Also, the interaction between C20- C25 bond and C23-C24 π-antibonding is stabilized DDT molecule via 21.40 kcal/mol then from C20-C25 π-bond to C21-C22 π-antibonding that contributed 19.81 kcal/mol respectively as recorded in Table 5. In addition, the interaction between LP S7 to π*C3—N5 in DDT that assigned by 21.31 Kal/mol. The delocalization of MO between C1-C6 π-bond to C2-C3 and C4-C5 π-antibonding then from C4-C5 π-bond to C1-C6 π-antibonding is stabilized BDTTMB molecular structure via 21.01, 19.70 and 20.54 kcal/mol respectively. Also, the interactions among the following; (πC15—N21 and π*C17—N23, πC17—N23 and π*C16—N22, πC16—N22 and π*C15—N21, πC19—N24 and π*C18—N25, πC28—C30 and π*C29—C31 and LP S14 and π*C15—N21) are stabilized BDTTB by 21.03, 19.46, 30.86, 31.96, 19.60 and 27.17 kcal/mol respectively. Finally, the stability of BDTTMP molecular structure due to the interaction between π-bond and π-antibonding; the values of interactions are listed in Table 5.
Molecular Electrostatic Potentials Map (MEPM)
A molecular electrostatic potential map (MEPM) investigates reveals the electrophilic versus nucleophilic sites of all molecular structures under study, giving for the prediction of reaction areas [25]. The MEPMs of the studied DTT and its derivatives were calculated employing B3LYP/6-31G (d,p) in Fig. 6. The negatively, positively and neutral zones of the MEPM interfaces are represented by the colors red, blue, and green, respectively (Fig. 6). The negative site (red color) is inside terminal benzene rings, suggesting that such rings are electrophilic attacks. The positive area (blue color) encompassed the carbon and hydrogen atoms, suggesting nucleophilic attacks in these sites. The neutral areas ( green color) of the molecular structure are those that are devoid of any charge distribution. The blue area around the H-atoms of the sulfur atom was associated with electron deficiency (i.e., nucleophilic reactivity), rendering it a favorable place for intermolecular H-bonding (nucleophile attack). The electron density (red area) appeared centered within the circle, according to the ESP map. The nucleophile in the phenyl ring has had the highest negative possibility, making it an ideal subject for an electrophilic engagement [26].

Electrostatically mapped surfaces of DTT, BDTTB, BDTTMB, and BDTTMP compounds
Evaluating DTT Dye for DSSC Device
we continue our study of a prototypical dye molecule for DSSC devices from Fig. 7. In an actual device, the DTT end of the dye molecule is bound to a titanium oxide nanostructure. The original researchers [27] validated that the metal surface, in this case, can be adequately represented as a Ti(OH)3H2O moiety: the titanium atom is an octahedral complex with two positions taken up by the oxygen atoms of the acrylic acid anchor [27], three hydroxyl groups and one water molecule. Because of the high stability of the DTT compound compared to the remaining compounds under study, the DTT compound was chosen as a representative to study the DTT complexes with the Ti(OH)3H2O. The DTT/ Ti(OH)3H2O optimized structure, HOMO and LUMO MOs, and energy gap (Eg1) between HOMO and LUMO were obtained using DFT/B3LYP/LANL2DZ method and the calculated results are shown in Fig. 7(A and B). The nitrogen atom of the pyrazine ring that is present in DTT dye is linked to the titanium atom of Ti(OH)3H2O through a coordinated bond as shown in Fig. 7. The N → Ti coordination bond length is 2.016 A (see Fig. 7A), this is confirming the stability of the DTT/Ti(OH)3H2O complex. The fact that it is a charge-transfer state is again evident. Electrons are moving from the DTT benzene end to the other benzene end of the DTT molecule as shown in Fig. 7B. Interestingly. The titanium complex is also involved in HOMO and LUMO, hence it is becoming part of the donor and acceptor. The 3d orbital from the titanium atom is visible in the LUMO and HOMO, indicating that electrons in the Ti complex interfere with the electronic transition. They receive and donate electrons.

DTT/Ti(OH)3H2O optimized molecular structure (A). H and L Molecular orbitals as well as the energy gap between H and L (Eg1) (B). DFT/B3LYP/LANL2DZ method was used to obtain DTT/Ti(OH)3H2O, H, and L MOs optimized molecular structure
Conclusion
In this paper, the optimized molecular structure, and electronic and optical properties of four triazine derivative dyes DTT, BDTTB, BDTTMB, and BDTTMP were investigated via utilizing DFT/TD-DFT. According to the ground state geometry, we note that all stable conformations are not planar. The HOMO/LUMO energy gaps of DTT, BDTTB, BDTTMB and BDTTMP were calculated at DFT/B3LYP/6-31G (d, p) level are 4.168, 3.528, 3.965 and 3.921 eV respectively. So, the energy gaps differ slightly and decrease in the following order: BDTTB < BDTTMP < BDTTMB < DTT. Consequently, the calculated values of Voc/ TiO2 of our dyes are sufficient for possible efficient electron injection from the donor to the acceptor.
Data Availability
All data generated or analyzed during this study are included in this published article.
Code Availability
Not applicable.
References
Mariotti N, Bonomo M, Fagiolari L, Barbero N, Gerbaldi C, Bella F et al (2020) Recent advances in eco-friendly and cost-effective materials towards sustainable dye-sensitized solar cells. Green Chem 22:7168–7218
Vlachopoulos N, Hagfeldt A, Benesperi I, Freitag M, Hashmi G, Jia G et al (2021) New approaches in component design for dye-sensitized solar cells. Sustain Energy Fuels 5:367–383
Kokkonen M, Talebi P, Zhou J, Asgari S, Soomro SA, Elsehrawy F et al (2021) Advanced research trends in dye-sensitized solar cells. J Mater Chem A
Chalkias DA, Loizos DD, Papanicolaou GC (2020) Evaluation and prediction of dye-sensitized solar cells stability under different accelerated ageing conditions. Sol Energy 207:841–850
Ansari AA, Nazeeruddin MK, Tavakoli MM (2021) Organic-inorganic upconversion nanoparticles hybrid in dye-sensitized solar cells. Coord Chem Rev 436:213805
Liao C, Zeng K, Wu H, Zeng Q, Tang H, Wang L et al (2021) Conjugating pillararene dye in dye-sensitized solar cells. Cell Reports Phys Sci 2:100326
Lee M-W, Kim J-Y, Lee H-G, Cha HG, Lee D-H, Ko MJ (2021) Effects of structure and electronic properties of D-π-A organic dyes on photovoltaic performance of dye-sensitized solar cells. J Energy Chem 54:208–216
Zakutayev A, Major JD, Hao X, Walsh A, Tang J, Todorov TK et al (2021) Emerging inorganic solar cell efficiency tables (version 2). J Phys Energy 3:32003
Grätzel M (2009) Recent advances in sensitized mesoscopic solar cells. Acc Chem Res 42:1788–1798
Li G, Shrotriya V, Huang J, Yao Y, Moriarty T, Emery K et al (2011) High-efficiency solution processable polymer photovoltaic cells by self-organization of polymer blends. Mater Sustain Energy A Collect Peer-Reviewed Res Rev Artic from Nat Publ Gr, World Scientific 80–4
Yang W, Cao D, Zhang H, Yin X, Liao X, Huang J et al (2018) Dye-sensitized solar cells based on (DĀpĀA) 3L2 phenothiazine dyes containing auxiliary donors and flexible linkers with different length of carbon chain. Electrochim Acta 1732;283
Ardo S, Meyer GJ (2009) Photodriven heterogeneous charge transfer with transition-metal compounds anchored to TiO 2 semiconductor surfaces. Chem Soc Rev 38:115–164
O’regan B, Grätzel M (1991) A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO 2 films. Nature 353:737–740
Horiuchi T, Miura H, Sumioka K, Uchida S (2004) High efficiency of dye-sensitized solar cells based on metal-free indoline dyes. J Am Chem Soc 126:12218–12219
Jia C, Wan Z, Zhang J, Li Z, Yao X, Shi Y (2012) Theoretical study of carbazole–triphenylamine-based dyes for dye-sensitized solar cells. Spectrochim Acta Part A Mol Biomol Spectrosc 86:387–391
Lin Y, Fan H, Li Y, Zhan X (2012) Thiazole-based organic semiconductors for organic electronics. Adv Mater 24:3087–3106
Sakr MAS, Mohamed AA, Abou Kana MTH, Elwahy AHM, El-Daly SA, Ebeid EZM (2021) Synthesis, characterization, DFT and TD-DFT study of novel bis (5, 6-diphenyl-1, 2, 4-triazines). J Mol Struct 1226:129345
Luo-Xin W, Yong L, Xin-Lin T, Song-Nian L, Mao-Gong W (2007) Effect of H+ and NH4+ on the N-NO2 bond dissociation energy of HMX. Acta Physico-Chimica Sin 23:1560–1564
Ghosh NN, Habib M, Pramanik A, Sarkar P, Pal S (2019) Molecular engineering of anchoring groups for designing efficient triazatruxene-based organic dye-sensitized solar cells. New J Chem 43:6480–6491
Bourass M, Benjelloun AT, Benzakour M, Mcharfi M, Jhilal F, Serein-Spirau F et al (2017) DFT/TD-DFT characterization of conjugational electronic structures and spectral properties of materials based on thieno [3, 2-b][1] benzothiophene for organic photovoltaic and solar cell applications. J Saudi Chem Soc 21:563–574
Bourass M, Benjelloun AT, Benzakour M, Mcharfi M, Hamidi M, Bouzzine SM et al (2016) DFT and TD-DFT calculation of new thienopyrazine-based small molecules for organic solar cells. Chem Cent J 10:1–11
Bouachrine M (n.d.) New organic dyes based on phenylenevinylene for solar cells: DFT and TD-DFT investigation
Zhao Y, Tishchenko O, Truhlar DG (2005) How well can density functional methods describe hydrogen bonds to π acceptors? J Phys Chem B 109:19046–19051
Reed AE, Weinhold F (1983) Natural bond orbital analysis of near-Hartree–Fock water dimer. J Chem Phys 78:4066–4073
Aravindan P, Sivaraj K, Kamal C, Vennila P, Venkatesh G (2021) Synthesis, Molecular structure, Spectral Characterization, Molecular docking and biological activities of (E)-N-(2-methoxy benzylidene) anthracene-2-amine and Co(II), Cu(II) and Zn(II) complexes. J Mol Struct 1229:129488. https://doi.org/10.1016/j.molstruc.2020.129488
Fetoh A, Mohammed MA, Youssef MM, Abu El-Reash GM (2019) Characterization, cyclic voltammetry and biological studies of divalent Co, Ni and Cu complexes of water-soluble, bioactive and photoactive thiosemicarbazone salt. J Mol Liq 287:110958. https://doi.org/10.1016/j.molliq.2019.110958
Forseman JB, Frisch MJ (1996) Exploring Chemistry with Electronic Structure Methods. Gaussian. Inc, Pittsburgh, PA
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). This work was supported by Misr University for Science and Technology and Cairo University (Laser Systems Department, National Institute of Laser- Enhanced Science (NILES).
Author information
Authors and Affiliations
Contributions
Mahmoud A. S. Sakr, Data Curation, Writing-original draft, and Software. Maram T. H. Abou Kana, Visualization and Investigation.
Corresponding author
Ethics declarations
Ethical Approval
This article does not contain any studies involving animals performed by any of the authors.
Consent to Participate
This article does not contain any studies involving animals performed by any of the authors.
Consent to Publish
All authors mentioned in the manuscript have given consent for submission and subsequent publication of the manuscript.
Conflict of Interest
The authors have declared no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sakr, M.A.S., Kana, M.T.H.A. 1,2,4-Triazine-based Materials: Spectroscopic Investigation, DFT, NBO, and TD-DFT Calculations as Well As Dye-sensitized Solar Cells Applications. J Fluoresc 32, 2053–2063 (2022). https://doi.org/10.1007/s10895-022-03005-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10895-022-03005-1