
Abstract
Human herpesvirus-6 (HHV-6) infection can rarely cause life-threatening conditions, such as encephalitis, in otherwise healthy children, with unclear pathogenesis. We studied a child who presented with acute HHV-6 encephalitis at the age of 10 months and who was homozygous for a novel missense mutation in IRAK4, encoding interleukin-1 receptor-associated kinase 4, identified by whole-exome sequencing. We tested the damaging impact of this mutation in silico by molecular dynamics simulations and in vitro by biochemical and functional experiments utilizing cell lines and patient’s cells. We found that the mutation is severely hypomorphic, impairing both the expression and function of IRAK-4. Patient’s leukocytes had barely detectable levels of IRAK-4 and diminished anti-viral immune responses to various stimuli inducing different Toll-like receptors and cytosolic nucleic acid sensors. Overall, these findings suggest that acute HHV-6 encephalitis can result from inborn errors of immunity to virus. This study represents the first report of isolated acute HHV-6 infection causing encephalitis in an inherited primary immunodeficiency, notably autosomal recessive (AR) partial IRAK-4 deficiency, and the first report of AR IRAK-4 deficiency presenting with a severe viral disease, notably HHV-6 encephalitis upon an acute infection, thereby expanding the clinical spectrum of IRAK-4 deficiency.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Human herpesvirus-6 (HHV-6) is a ubiquitously found virus in the human population worldwide, with a seropositivity ranging from 70 to 95% among people older than 2 years [1,2,3,4]. Although HHV-6 is a lymphotropic double-stranded DNA (dsDNA) virus, predominantly targeting CD4+ T cells, it can infect a wide variety of cells in the body, such as monocytes/macrophages, hepatocytes, and neuronal cells [5, 6]. Primary infection with HHV-6 mostly occurs within very early years of life, culminating in a life-long latency, as do all other herpesviruses. HHV-6 is a common cause of acute febrile illness, with or without a rash, and roseola infantum among young children, yet majority of infected children remain asymptomatic [5]. In very rare cases, HHV-6 can cause neurological manifestations, such as febrile seizures and encephalitis, during primary infection [5, 6]. On the contrary, HHV-6-associated encephalitis or other severe manifestations can be common in immunocompromised individuals, including cancer patients, hematopoietic stem cell or solid organ transplant recipients [5,6,7,8]. However, establishing HHV-6 as the culprit in such individuals is dubious as they are also susceptible to other opportunistic infections. Interestingly, there is not yet any known inherited immunodeficiency associated with encephalitis upon infection with HHV-6 [9]. Recently, one IRAK-4 (interleukin-1 receptor (IL-1R)-associated kinase 4)-deficient patient presenting with anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis and HHV-6 reactivation was reported [10]. Human IRAK-4 deficiency impairs the Toll-like receptor (TLR)/IL-1R-mediated immunity, rendering affected individuals predominantly susceptible to recurrent life-threatening bacterial infections [11,12,13]. However, there was no increased predisposition to severe viral diseases reported in known IRAK-4-deficient patients [11,12,13]. In this study, we report a rare case of acute HHV-6 encephalitis in a male infant carrying a novel homozygous mutation in IRAK4. We experimentally validated the IRAK-4 deficiency and revealed impaired anti-viral immune responses in the patient.
Materials and Methods
Case Report
The patient was born in Turkey in 2020 at term with a normal birth weight after an uneventful pregnancy. The umbilical cord separation time was the postnatal 8th day. His parents were second-degree cousins and he had a 2 years older healthy sister. The patient, at the age of 10 months, presented with fever for 3 days, lethargy, vomiting, and focal seizure. On physical examination, his body weight was 9 kg (50th percentile), height was 71 cm (25th percentile), and head circumference was 45.5 cm (50th percentile). The patient’s temperature was 39 °C. He was lethargic, but his vital signs were stable. The other physical examination was normal. According to the national immunization schedule, he was fully vaccinated with the Bacillus Calmette-Guérin vaccine, hepatitis B vaccine, diphtheria, tetanus, acellular pertussis, inactive polio and Haemophilus influenzae type B vaccine, 13-valent pneumococcal conjugate vaccine, and oral polio vaccine. He had only recurrent upper respiratory tract infections in his past medical history. There was no family history of primary immunodeficiency or other disorders.
Patchy hyperdense density changes were observed in both frontal periventricular white matter in the brain computerized tomography (CT) (Fig. 1a and b). In the brain magnetic resonance imaging (MRI), the axial T2-weighted images showed periventricular white matter hyperintensities in both frontal horn and centrum semiovale (Fig. 1c and d). Postcontrast axial T1-weighted images showed contrast enhancement in the frontal horn neighborhood (Fig. 1e). Diffusion-weighted images and apparent diffusion coefficient maps showed restriction of diffusion in the frontal horn neighborhood (Fig. 1f and g). With these findings, meningoencephalitis was considered, and lumbar puncture was performed. Cerebrospinal fluid (CSF) analysis revealed no white or red blood cells, increased protein level (123 mg/dL; normal: < 40 mg/dL) and decreased CSF/plasma glucose ratio (0.4; normal: > 0.5). No cells or microorganisms were seen in the Gram staining of the CSF. A CSF sample was taken for the viral encephalitis panel including adenovirus, cytomegalovirus (CMV), Epstein-Barr virus (EBV), herpes simplex virus (HSV)-1, HSV-2, varicella zoster virus (VZV), human herpesvirus-6 (HHV-6), HHV-7, Enterovirus, Parechovirus, and Parvovirus-B19. Levetiracetam, empirical vancomycin, and acyclovir were started. During the follow-up, his fever continued and an erythematous maculopapular rash appeared on the 5th day of fever. The rash started in the trunk and spread to the face and limbs, and then the fever subsided. The CSF PCR was reported to be positive for HHV-6 DNA. The SARS-CoV-2 PCR test on the nasopharyngeal swab was negative. The patient was diagnosed with acute HHV-6 encephalitis.

Computerized tomography (CT) scan images and magnetic resonance imaging (MRI) of the patient. a, b Brain CT scan: axial a, and sagittal b, images showing high-density regions in both frontal periventricular white matter lesions (black arrows). c, d, e Brain MRI: Axial T2-weighted images showing periventricular white matter hyperintensities in both frontal c, and centrum semiovale d (black arrows). Postcontrast axial T1 weighted images showing contrast enhancement in the frontal horn neighborhood, e (white arrows). f, g Brain MRI: diffusion-weighted images f, (black arrows), and apparent diffusion coefficient maps g, (white arrows), showing restriction of diffusion in the frontal horn neighborhood
As the patient did not have any previous history of a disease with skin rash after fever, a primary infection with HHV-6 was strongly considered to cause acute encephalitis. Erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), and interleukin-6 (IL-6) levels were elevated. Immunological evaluation revealed normal neutrophil and lymphocyte counts; slightly low IgG level; normal IgA, IgM, and IgE levels; normal peripheral blood lymphocyte subsets; normal recent thymic emigrant cells (RTEs); normal activation response to PHA; and normal dihydrorhodamine (DHR) test (Table 1). The general condition of the patient improved, and he was discharged from the hospital on the 11th day. During the follow-up, he had recurrent upper and lower respiratory infections. He was hospitalized twice due to urinary tract infection caused by Klebsiella pneumoniae and human bocavirus associated bronchiolitis, respectively. After diagnosis of IRAK-4 deficiency in the patient, prophylactic treatment with oral amoxicillin was started. But the treatment was later replaced with azithromycin due to allergies. He was vaccinated with meningococcal vaccines to prevent invasive meningococcal disease including meningococcal serogroup A, C, W135 and Y conjugate vaccine, and serogroup B conjugate meningococcal vaccine. Intravenous immunoglobulin (IVIG) replacement therapy was then started at 600 mg/kg/dose in every 4 weeks. During this period, the patient had once SARS-CoV-2 infection with mild symptoms, but he has not had any other infectious diseases to this date.
Whole-Exome Sequencing and Genetic Analysis
The genomic DNA was isolated from the peripheral blood of the patient using the NucleoSpin Blood mini kit for DNA (Macherey–Nagel, Germany) and used for whole-exome sequencing (WES). Collection of raw sequencing data, alignment with the reference human genome, variant calling, and annotations were performed by WES service provider, Genoks, Turkey. Briefly, WES was done using the Human Comprehensive Exome panel (Twist Bioscience, USA), followed by paired-end sequencing performed on DNBSEQ-G400 System (MGI Tech, China), generating 150 base-paired ends. BCL2Fastq2 v2.20 was used for FASTQ generation. BWA-MEM software was used for the alignment with the human genome reference sequence (GRCh37). Genome Analysis Toolkit and SnpEff were used for downstream processing. The gnomAD 2.1.1 (https://gnomad.broadinstitute.org/) and 1000 Genomes Project (http://grch37.ensembl.org/) databases were used to obtain allele frequencies.
In Silico Prediction of Variant Impact
Prediction of the damaging impact of variants was performed via Combined annotation-dependent depletion (CADD) v1.6, Mutation Significance Cutoff (MSC), Sorting intolerant from tolerant (SIFT), and Polymorphism Phenotyping v2 (PolyPhen-2) [14,15,16,17]. Damaging impact of the IRAK-4 mutation was also predicted by molecular dynamics (MD) simulation. Details of MD simulation and analysis of MD trajectories are provided in Supplementary Materials and Methods.
Sanger Sequencing of Genomic DNA
PCR was performed to amplify the region including the target IRAK4 mutation on the genomic DNA (gDNA). Primers are listed in Table S1. The PCR amplicons were sequenced by a service provider (Macrogen, Europe). Unipro UGENE program was used for sequence analysis.
Cell Culture and Cell Isolation
HEK293 cells were cultured in High-Glucose DMEM (Biowest, France) supplemented with 10% FBS (Biological Industries, Israel). Human peripheral blood mononuclear cells (PBMCs) were isolated using Lymphoprep (Stemcell Technologies, USA) from the patient, his family members, and unrelated healthy donors, who were male and between 20 and 25 years of age. Isolated PBMCs were cryopreserved for long-term storage. PBMCs were cultured in RPMI (Biowest) including 10% FBS. Cell cultures were maintained at 37 °C in a humidified incubator with 5% CO2. RNA extraction and details of gene expression analysis are provided in Supplementary Materials and Methods.
Cloning and Site-Directed Mutagenesis
The human canonical IRAK4 cDNA open reading frame (ORF) was PCR amplified using cDNA generated from THP-1 cell line with an in-frame N-terminal FLAG (DYKDDDDK)-tag and cloned into the pcDNA3.1( +) vector through BamHI and XhoI sites. Site-directed mutagenesis (SDM) was performed on wild-type (WT) IRAK-4-encoding plasmid to generate c.G236A:p.C79Y and c.C34T:p.R12C using a modified overlap-extension PCR-based method [18]. PCR-amplified WT and mutant IRAK4 ORF sequences were inserted in frame with 3 × N-terminal FLAG-tag into the pCI-Neo-N-3xFlag vector through EcoRI and XbaI sites. The human canonical IL18RAP cDNA ORF was PCR amplified using cDNA from PBMCs of a healthy donor and cloned into the pCMV6 vector (OriGene, USA) through HindIII and XhoI restriction sites. All constructs generated were confirmed by Sanger sequencing. Primers are provided in Table S1.
Transient Transfection and Immunoblotting
HEK293 cells in 24-well plates were transfected with empty pCI-Neo-N-3xFlag vector, WT, or mutant IRAK4 constructs (350 ng) along with a GFP-encoding plasmid (150 ng) for transfection efficiency, using Lipofectamine 2000 transfection reagent (Invitrogen, USA) according to the manufacturer’s instructions. At 48 h post-transfection, cells were lysed with RIPA buffer including both protease (Roche, USA) and phosphatase (Thermo Fisher Scientific) inhibitors. Whole cell lysates were measured for protein concentration using BCA Protein Assay Kit (TaKaRa Bio, Japan), boiled at 95 °C for 5 min, and subjected to SDS/PAGE (12%). Antibodies are listed in Table S2. Digital images were captured by Amersham Imager 600 (GE Healthcare, Life Sciences). For protein stability assays, HEK293 cells in 12-well plates were transfected with empty pCI-Neo-3xFlag vector, WT, or mutant IRAK4 constructs (700 ng) along with the GFP plasmid (300 ng) using Lipofectamine 2000 reagent. At 24 h post-transfection, cells were treated for 12 h with either cycloheximide (50 μg/mL) (Cayman Chemical, USA) or 0.5% DMSO (PanReac AppliChem, USA) as vehicle control. Treated cells were lysed and subjected to immunoblotting.
Flow Cytometry
PBMCs resuspended at 2 × 106 cells/mL with Dulbecco’s phosphate-buffered saline (DPBS, with no calcium or magnesium) (Biowest) containing %2 FBS were first stained with antibodies against surface proteins, followed by fixation and then intracellular protein staining in permeabilization buffer (Tonbo Biosciences, USA) according to the manufacturer’s instructions. Stained cells were acquired on CytoFLEX Flow Cytometer (Beckman Coulter, USA) or NovoCyte Flow Cytometer (ACEA Biosciences, USA), and analyses were performed with FlowJO (BD Biosciences, USA) software. Antibodies are listed in Table S2.
Generation of IRAK4 knockout HEK293 Cells
Oligonucleotides for gRNAs targeting IRAK4 exon-2 were cloned into the pLentiCRISPR-V2 vector [19,20,21]. Constructs generated were confirmed by Sanger sequencing. HEK293 cells in 6-well plates were transfected with pLentiCRISPR-V2 constructs (1500 ng), pVSV-G (750 ng), and psPAX2 (750 ng) using Lipofectamine 2000 reagent. At 48 h post-transfection, produced lentiviruses in supernatants were concentrated with Lenti-X concentrator (Takara Bio). HEK293 cells in 24-well plates were transduced with concentrated lentiviruses. At 48 h post-transduction, cells were treated with puromycin (InvivoGen) at 1 μg/mL for 2 weeks and checked for gene editing efficiency using T7 Endonuclease I Assay (New England Biolabs, USA). After validation, cells were seeded on 96-well plate at 9 cells/mL for single-cell cloning. Screening of single-cell clones was performed using a competition-based PCR method with Phire Tissue Direct PCR kit (Thermo Fisher Scientific) [22]. Confirmed single-cell clones were further validated by flow cytometry and western blotting for IRAK-4 expression. Two clones (KO no. 1 and KO no. 2) were selected for further studies. Primers are provided in Table S1.
NFκB Reporter Assay
IRAK4 knockout HEK293 cells, KO no. 1 and KO no. 2, in flat-bottom 96-well plates were transfected with pNFκB-Firefly-Luc (120.5 ng), pCMV6-IL18RaP (19 ng), pRLTK (9 ng) and either empty pCI-Neo-N-3xFlag vector, WT, or mutant IRAK4 constructs (1.5 ng) using the Lipofectamine 2000 reagent. At 24 h post-transfection, cells were stimulated with human recombinant IL-18 (R&D Systems, USA) at 100 ng/mL for 12 h. Treated cells were checked for Firefly and Renilla luciferase activities using Dual-Glo® Luciferase Assay System (Promega, USA).
Ex Vivo Stimulation of PBMCs
PBMCs seeded on round-bottom 96-well plate at 3 × 106 cells/mL were stimulated with Pam3CSK4 (100 ng/mL, tlrl-pms), Flagellin (Salmonella typhimurium, 100 ng/mL, tlrl-epstfla), or LPS (Escherichia coli O111:B4, 1 μg/mL, tlrl-3pelps) (InvivoGen) for 6 h in the presence of 1 × Brefeldin A (Biolegend). Treated cells were stained for intracellular TNF in CD14+ monocytes and analyzed via flow cytometry. For bead-based immunoassays, PBMCs seeded on round-bottom 96-well plate at 2 × 106 cells/mL were stimulated with R848 (Resiquimod, 2 μg/mL, tlrl-r848), poly(I:C) (LMW, 10 μg/mL, tlrl-picw), LPS (100 ng/mL), or CpG (ODN 2395, 1 μM, tlrl-2395) and transfected with 2′3′-cGAMP (1 μg/mL, tlrl-nacga23) or poly(dA:dT) (1 μg/mL, tlrl-patn) (InvivoGen) using Lipofectamine 2000 reagent. At 24 h post-treatment, culture supernatants were utilized for multiplex cytokine profiling using LEGENDplex Human Anti-Virus Response Panel (Biolegend) according to the manufacturer’s instructions. Data were analyzed using LEGENDplex Online Data Analysis Software (Biolegend). For enzyme-linked immunosorbent assay (ELISA), PBMCs seeded on round-bottom 96-well plate at 2 × 106 cells/mL were stimulated with R848 (5 μg/mL), poly(I:C) (20 μg/mL), CpG (2.5 μM), or PMA (50 ng/mL, BioGems, USA) and Ionomycin (500 ng/mL, BioGems) and transfected with 2′3′-cGAMP (3 μg/mL) or poly(dA:dT) (3 μg/mL) using X-tremeGene 9 DNA transfection reagent (Roche). At 24 h post-treatment, culture supernatants were utilized for measurement of IFN-α2 and IL-6 by ELISA kits (Biolegend) according to the manufacturer’s instructions. Data were analyzed using 4-parameter logistic curve.
Statistical Analysis
The results of experiments were plotted and analyzed using GraphPad Prism software. For Student’s t test, statistical significance is indicated by asterisks (*P < 0.05, **P < 0.01, and ***P < 0.001), with P values greater than 0.05 considered non-significant (n.s.).
Results
A Private IRAK4 Variation in a Patient with Acute Human Herpesvirus 6 (HHV-6) Encephalitis
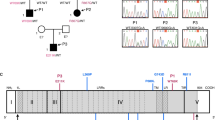
We performed WES on leukocyte gDNA from the patient who presented with encephalitis upon acute HHV-6 infection at the age of 10 months (Fig. 1; Table 1; “Materials and Methods”). He was born to consanguineous parents and had a healthy sister, all of whom had no medical history of severe infection or primary immunodeficiency. We thus hypothesized that a genetic etiology for HHV-6 encephalitis in this patient would display autosomal recessive (AR) inheritance. We searched for rare (Minor allele frequency (MAF) < 0.01) homozygous potential loss-of-function (deletion, essential splicing, insertion, start-loss, stop-gain, and stop-loss) and missense variants present in the patient (Table S3). The variants, for which there were homozygote carriers listed in gnomAD, and the genes, for which there were homozygous potential loss-of function variations listed in gnomAD, were eliminated (Table S4). Next, we prioritized candidate genes based on their expression pattern and function related to viral encephalitis and on the rarity and predicted deleteriousness of variations (Table S4 and S5). IRAK4, encoding the interleukin-1 receptor-associated kinase 4, was the most plausible disease-causing gene, as (i) it encodes a kinase that is involved in TLR/IL-1R signaling, regulating most innate immune responses, and deficiency of which causes recurrent life-threatening bacterial infections in early childhood [11, 12, 23,24,25,26], and (ii) its deficiency was recently reported in anti-NMDAR encephalitis and HHV-6 reactivation in a male infant [10]. Moreover, the IRAK4 variant (NM_016123.4: c.G236A; NP_057207.2: p.C79Y) was not found in any public databases, such as 1000 Genomes, the Single Nucleotide Polymorphism Database (dbSNP), gnomAD, Bravo or the Greater Middle East (GME) Variome Project. Finally, we confirmed, by Sanger sequencing, that the familial segregation of the mutant IRAK4 allele was consistent with an AR mode of inheritance with complete penetrance, as both parents and the healthy sibling were heterozygous for the mutation (Fig. 2a and b).

(Source: NCBI HomoloGene). d Graph showing the predicted CADD scores of the p.C79Y found in the patient (red star) and known disease-causing variations in IRAK4 (blue circles). The CADD-MSC score (95% confidence interval) for IRAK4 is indicated by a dashed line
A novel homozygous missense mutation in IRAK4. a Pedigree of the family affected with acute HHV-6 encephalitis. The patient is shown in black, and the healthy individuals are shown in white. Where available, IRAK4 mutation (p.C79Y) status is indicated. b Familial segregation of the IRAK4 mutation and its homozygous state in the patient were confirmed by Sanger sequencing. c Schematic representation of IRAK-4 and its known domains (Top) and conservation of the p.C79 residue, highlighted in bold, across various species (Bottom).
The p.C79Y is Predicted to be Damaging
The p.C79Y affects an evolutionarily conserved cysteine residue located in the death domain (DD) of IRAK-4 (Fig. 2c) and is in silico predicted to be damaging by CADD, SIFT, and PolyPhen-2 (Table S4). It had a CADD score of 28, which is above the mutation significance cutoff (MSC) score of 15.15 (95% confidence interval) for IRAK4 and comparable to the median CADD score of 29.5 of previously reported disease-causing mutations in IRAK4 (Fig. 2d and Table S6). We also analyzed the impact of p.C79Y on the structure and dynamics of IRAK-4 by molecular dynamics (MD) simulations. The p.C79Y was in silico introduced to the structure of IRAK-4 DD (PDB: 3MOP) [27], and both WT and mutant structures were simulated for 100 ns, followed by analysis of MD trajectories. One-dimension (1D) root-mean-square deviation (RMSD) line of the mutant DD showed a slight increase compared to the WT (Fig. 3a); however, this change, a displacement of 0.2 nm of the Cα traces, was considered insignificant since 1D RMSD plots alone are not sufficient to infer a conformational change due to the stochastic nature of MD simulations [28]. We have also monitored radius of gyration (Rg) of both proteins and found that the p.C79Y did not affect the compactness of the structure, which maintained an Rg of ~ 1.4 nm for both systems (Fig. 3b). Consistently, fluctuations of Cα atoms particularly for those located in the central regions were well-matched in both structures, implying that the p.C79Y did not mobilize specific regions of the IRAK-4 DD (Fig. 3c). Moreover, we observed similar hydrogen bonding and solvent accessibility profiles for WT and mutant IRAK-4 DD (Fig. 3d and e).

Molecular dynamics (MD) simulations and analysis of MD trajectories. The p.C79Y was in silico introduced to the structure of IRAK-4 death domain (DD) (PDB: 3MOP), and both WT and mutant structures were simulated for 100 ns. a The root-mean-square deviation (RMSD) graph shows the backbone flexibility of WT and mutant IRAK-4 DD. b The radius of gyration (RGYR) graph shows the compactness of the proteins. c The root mean square fluctuation (RMSF) graph shows the residual fluctuation. d Graph shows the change in the number of hydrogen atoms in WT and mutant IRAK-4 DD. e The solvent accessibility surface area (SASA) graph shows the change in the hydrophobic core region for WT and mutant IRAK-4 DD. f Gibbs-free energy landscapes (FELs) were calculated based on the Helmholtz-free energy change using the first two principal components, PC-1 and PC-2. FEL shows the different configuration states that can be achieved by protein in simulation. g Figures show illustration of the changes in secondary structures of WT and mutant IRAK-4 DD during the 100-ns MD simulation. h Visualization of WT and mutant IRAK-4 DD structures. Residue 79 is shown by an arrow. i Close-up views of the loop connecting the Helix4 to Helix5 in WT and mutant IRAK-4 DD structures. Some residues are indicated by arrows; some are shown with side chains. Atoms are shown in the following colors: carbon; cyan, oxygen; red, nitrogen; blue, sulfur; yellow. h, i Visuals were produced by using representative structures obtained from the last 1 ns of MD simulations
Free Energy Landscape (FEL) analysis indicated that p.C79Y did not lead to a drastic change in the conformation of the IRAK-4 DD (Fig. 3f). For both WT and mutant DD, only one strong minimal point was observed on the principal component (PC)-1/PC-2 plane, suggesting that both systems converged to a single conformation without any transitions (Fig. 3f). Consistent with the random movements spotted in Fig. 3a and c, the C-terminal of DD was more mobile in mutant model than it was in the WT (Fig. 3g and h). This was due to the alteration of the β-turn flanking the fourth helix in the WT DD structure upon p.C79Y mutation (Fig. 3h). However, the C-terminal of IRAK-4 DD, which contains disordered regions, is expected to undergo conformational changes regardless of any perturbation. Therefore, these findings suggested that the p.C79Y did not affect the global structure and dynamics of IRAK-4 DD (Fig. 3g and h) . Lastly, close-up inspection of the mutated region revealed a novel pi-stacking between Y79 and W74 in the mutant structure (Fig. 3i), which may affect the intermolecular interactions of IRAK-4 DD that recruit W74 and/or nearby residues. In particular, W74 was found at the interface of the complex formed by IRAK-4 DD and MyD88 [27], implying the potential impact of p.C79Y on IRAK-4 function. Moreover, molecular docking of WT and mutant IRAK-4 DD obtained from MD simulation with the Myd88 structure (PDB: 3MOP) was performed. Unlike the WT IRAK-4 DD, most of the docking models for IRAK-4 DD with p.C79Y failed to use the same interface indicated in the crystal structure of IRAK-4-Myd88-IRAK-2 assembly (PDB: 3MOP) (data not shown) [27]. Collectively, these findings suggested that homozygosity for this novel and predicted to be damaging IRAK4:c.G236A:p.C79Y allele can be the underlying cause of HHV-6 encephalitis in this patient.
Severely Diminished IRAK-4 Expression in Patient’s Leukocytes
We assessed expression levels of IRAK-4 in PBMCs from three healthy donors, the patient and his healthy sibling and parents by flow cytometry as previously described [10]. We found that IRAK-4 expression was drastically reduced in CD3+CD4+ T cells, CD3+CD8+ T cells, and CD19+ B cells, but to a lesser extent in CD14+ monocytes of the patient compared to healthy donors and family members (Fig. 4a and b). However, there was no difference found in IRAK4 mRNA levels in between PBMCs from the patient and three healthy controls (Fig. S1a). We also demonstrated that IRAK-4 expression was abolished in patient’s lymphoblastoid B cells by flow cytometry and western blotting (Fig. S1b and c). Next, we generated full-length ORF-constructs encoding WT and mutant IRAK-4 with an in-frame N-terminal 3xFLAG tag or without a tag. We also created an IRAK-4 construct with p.R12C, a morbid allele [29]. There were similar levels of WT and mutant IRAK-4, regardless of being tagged, when transiently over-expressed in HEK293 cells (Fig. 4c); however, we detected more dramatic reduction in the levels of mutant IRAK-4, p.C79Y (72.6%) and p.R12C (83.5%), consistent with previously described damaging impact of p.R12C on IRAK-4 DD stability [30], compared to the WT (32.3%) in transiently transfected HEK293 cells upon treatment with cycloheximide, an inhibitor of protein synthesis, relative to the DMSO treatment used as the vehicle control (Fig. 4d). In addition, we found similar levels of reduction (~ 30%) in the levels of GFP, used as the transfection efficiency control, upon cycloheximide treatment in WT and mutant IRAK-4 transfectants, suggesting that differences in IRAK-4 levels are due to the impact of the mutations. Overall, these findings indicate that p.C79Y reduces the stability of IRAK-4, and therefore leading to diminished IRAK-4 expression in patient’s cells.

Impact of the p.C79Y on IRAK-4 expression. a Representative histograms show intracellular IRAK-4 expression in PBMCs from three healthy controls (C1, C2, and C3), the patient and his family (father, mother, and sibling). PBMCs were identified based on forward and side scatter. Percentage of IRAK-4+ cells was defined based on the CD3+ and CD4+, CD3+ and CD8+, CD14+, or CD19+ gates. Dashed lines indicate isotype control staining. b Graphs show the percentages of IRAK-4 expressing CD3+CD4+ T cells, CD3+CD8+ T cells, CD14+ monocytes, and CD19+ B cells from controls, the patient and his family (father, mother, and sibling). Controls are shown as the average of three healthy controls (C1, C2, and C3). Bars are of the mean ± SEM of two independent experiments using PBMCs from blood sampling at different times. c Representative immunoblot images showing expression levels of WT and mutant IRAK-4 with FLAG-tag (left and middle panels) or without a tag (right panel) in whole cell lysates (30 µg protein/lane) from transiently transfected HEK293 cells. Immunoblotting was performed with the FLAG-tag antibody (Left) or IRAK-4 antibody (Middle and Right panels), followed by GFP and GAPDH antibodies after stripping of the membrane, respectively. Expected molecular weights: IRAK-4; ~ 55 kDa, GFP: ~ 27 kDa, GAPDH: ~ 36 kDa. d Representative immunoblot images showing expression levels of WT and mutant IRAK-4 with FLAG-tag in transiently transfected HEK293 cells upon treatment with cycloheximide (CHX, 50 µg/mL) or DMSO (vehicle control) for 12 h. Immunoblotting was performed with the FLAG-tag antibody, followed by GFP and GAPDH antibodies after stripping of the membrane, respectively. Band intensities were calculated for each lane using the ImageJ software (National Institutes of Health, USA) as detailed (http://rsbweb.nih.gov/ij/). Percent decrease in IRAK-4 levels in the WT and mutant IRAK-4 transfectants were determined by normalizing the IRAK-4/GAPDH ratio of CHX treatment to that of DMSO treatment (vehicle control), which was set as 100. The percent decrease values shown at the bottom are the mean of two independent experiments
The p.C79Y Impairs IRAK-4 Function
IRAK-4 is known to be essential for the early induction of pro-inflammatory cytokines, such as IL-6 and TNF, via TLRs in response to pyogenic bacteria [23, 24, 31]. We stimulated PBMCs from the patient and three healthy donors with Pam3CSK4 (TLR-1/2 agonist), flagellin (TLR-5 agonist), and lipopolysaccharide (LPS; TLR-4 agonist) and assessed intracellular TNF levels in CD14+ monocytes, as previously described [10]. Severely diminished TNF levels were detected in response to Pam3CSK4 and Flagellin, whereas there was around 50% reduction in TNF production upon LPS stimulation in patient’s cells when compared to the healthy controls (Fig. 5a and b). As this reduction in TNF might be partially due to decreased IRAK-4 expression in patient’s CD14+ monocytes, we tested the impact of p.C79Y on IRAK-4 function. We generated 2 different IRAK4 knockout HEK293 cell lines by CRISPR-Cas9 technology to utilize in the IL-18-induced NFκB-dependent luciferase reporter assay as previously described (Fig. S1d, e, f, and g) [10]. Transient over-expression of IRAK-4 with p.C79Y had reduced NFκB-luciferase activity up to 60%, whereas p.R12C had almost abolished NFκB-luciferase activity, as expected [10], compared to the WT in IRAK4 knockout HEK293 cells treated with IL-18 (Fig. 5c). Overall, these findings demonstrate that p.C79Y is hypomorphic, impairing both the stability and function of IRAK-4, and thereby causing AR IRAK-4 deficiency in the patient.

Impact of the p.C79Y on IRAK-4 function. a Representative histograms show intracellular TNF production in CD14+ monocytes from PBMCs of the patient and three healthy controls (C1, C2, and C3) upon stimulation with Pam3CSK4 (TLR-1/TLR-2), Flagellin (TLR-5), and LPS (TLR-4). PBMCs were identified based on forward and side scatter. TNF+ cells were defined based on the CD14+ gate. Dashed lines indicate isotype control staining. NT: no treatment (Medium only). b Bar graph shows the percentages of TNF+ CD14.+ monocytes in PBMCs of the patient and three healthy controls (C1, C2, and C3) upon treatment with indicated stimuli. The values are the mean ± SEM of two independent experiments using PBMCs from blood sampling at different times. c IL18-induced NFκB reporter activity of IRAK4 alleles over-expressed in IRAK4 knockout HEK293 cells. Two IRAK4-knockout HEK293 cell lines, KO no. 1 and KO no. 2, were transfected with plasmids expressing NFκB-dependent Firefly luciferase, Renilla luciferase (as transfection efficiency control), IL-18RaP (IL-18 receptor accessory protein), and either empty vector, WT, or mutant (p.C79Y and p.R12C) IRAK4 constructs. At 24 h post-transfection, cells were stimulated with human recombinant IL-18 for 12 h and harvested to measure luciferase activity. Relative luciferase activity was calculated as normalization of Firefly to Renilla luciferase activity. The values shown are the mean ± SEM of two independent experiments performed in duplicates (*P < 0.05; **P < 0.01; ***P < 0.001; two-tailed unpaired Student’s t test)
Reduced Anti-viral Immune Responses in Patient’s Cells
IRAK4-deficient patients’ leukocytes show remarkably decreased production of type-I interferons (IFNs) in response to induction of TLR-7, TLR-8, or TLR-9 [23, 31]. However, known patients with IRAK-4 deficiency had normal resistance to viruses [11, 13]. As the patient in this study presented with an acute infection with HHV-6, a dsDNA virus, we performed bead-based immunoassay to profile multiple cytokines involved in anti-viral immunity, such as GM-CSF, IFN-α2, IFN-β, IFN-γ, IFN-λ1, IFN-λ2/3, IL-1β, IL-6, IL-8, IL-10, IL-12p70, IP-10, and TNF, from PBMCs of the patient and two healthy donors upon 24-h induction with various agonists: a dsRNA mimic poly(I:C) for TLR-3, LPS for TLR-4, resiquimod (R848) for TLR-7 and TLR-8, CpG Class-C, selected based on potent IFN-α inducing capacity, for TLR-9, a non-canonical cyclic dinucleotide 2′3′-cGAMP for cytosolic DNA sensor STING, and a dsDNA mimic poly(dA:dT) for other cytosolic nucleic acid sensors, such as cGAS, AIM2, DAI, DDX41, IFI16, LRRFIP1, and RIG-I [32, 33] (Fig. 6 and Fig. S2a). Patient’s cells had more than 50% reduction in the levels of IL-1β, IL-6, IL-8, IFN-γ, IFN-λ1, IFN-λ2/3, IP-10, and TNF, but produced very low but similar levels of IFN-α2 and IL-10, when compared to the control cells upon poly(I:C) treatment. Induction of GM-CSF, IFN-γ, IL-1β, IL-6, IL-10, IP-10, and TNF via TLR-4 was severely impaired in patient’s cells, whereas levels of IFN-α2, IFN-β, IFN-λ1, IFN-λ2/3, and IL-12p70 were reduced to a lesser extent (up to 50%) in response to TLR-4 stimulation. Notably, there was markedly diminished production of all cytokines tested, except for IL-8, upon stimulation of TLR-7 and TLR-8 in patient’s cells. TLR-9 induction of patient’s cells had reduced levels of IFN-α2, IFN-β, IL-6, and IP-10, whereas other cytokines were at similar levels compared to the control cells. Moreover, we found decreased production of almost all cytokines tested in response to activation of cytosolic nucleic acid sensors by 2′3′-cGAMP or poly(dA:dT) in patient’s cells (Fig. 6 and Fig. S2a). Finally, we validated by ELISA that patient had decreased IL-6 responses to R848, poly(I:C), CpG, and poly(dA:dT), but similar IL-6 response to PMA/Ionomycin compared to unrelated healthy donors, his healthy parents and 2 years older sister (Fig. S2b). This indicates that diminished IL-6 production from patient’s leukocytes was due to IRAK-4 deficiency rather than defects in IL-6 machinery. 2′3′-cGAMP was less potent compared to other stimuli, leading to variably low or undetectable IL-6 and IFN-α2 responses by ELISA among the patient and healthy controls (Fig. S2b and c). IFN-α2 response to poly(I:C) was also variable, and it was not detected in patient’s and his mother’s cells by ELISA (Fig. S2c). Remarkably, the patient had no IFN-α2 response to R848, CpG, or poly(dA:dT) unlike the healthy donors and his family members (Fig. S2c). Overall, these findings indicate impaired anti-viral immune responses in our IRAK-4-deficient patient.

Diminished anti-viral immune responses in patient cells. Multiplex cytokine profiling was performed using supernatants of PBMCs from the patient (P) and two healthy controls (C1 and C2) upon 24-h treatment with various stimuli indicated. Control average was calculated as the mean of two healthy controls (C1 and C2). Graphs shown on a base-10 logarithmic scale present the mean expression levels (pg/mL) ± SEM of two independent experiments using PBMCs from blood sampling at different times. NT: no treatment (medium only)
Discussion
We report an inherited IRAK-4 deficiency due to a novel homozygous missense mutation in a 10-month-old male child who presented with acute HHV-6 encephalitis. The mutation, p.C79Y, was severely hypomorphic, impairing both the expression and function of IRAK-4, and thereby leading to IRAK-4 deficiency and diminished anti-viral immune responses upon induction of endosomal and cytosolic nucleic acid sensors in the patient. IRAK-4-deficient children are known to be highly susceptible to severe bacterial infections, particularly with Streptococcus pneumoniae, Staphylococcus aureus, and Pseudomonas aeruginosa; however, they are not unduly susceptible to viral infections [11,12,13]. Our patient had only recurrent upper respiratory tract infections before presenting with acute HHV-6 encephalitis. During his follow-up, he had recurrent upper and lower respiratory tract infections, but he was hospitalized twice due to urinary tract infection caused by Klebsiella pneumoniae, which is uncommon in IRAK-4-deficient patients, and acute bronchiolitis associated with human bocavirus, respectively. Following the genetic diagnosis of IRAK-4 deficiency, the patient was given antibiotic prophylaxis, vaccination, and IVIG replacement therapy, owing to all of which he did not have any severe bacterial infection to this date. It is noteworthy that during this period of prophylactic treatment, the patient had once SARS-CoV-2 infection, but recovered with only mild symptoms. Given the clinical features of IRAK-4-deficient patients reported, this is the first study describing inherited IRAK-4 deficiency presenting with a severe viral disease, notably HHV-6 encephalitis upon an acute infection.
Our patient had impaired immune responses upon induction of various TLRs including TLR-1/2, TLR-4, TLR-5, TLR-7, TLR-8, and TLR-9, consistent with the findings of previously reported IRAK-4 deficient patients. Surprisingly, we also found decreased responses in patient’s cells to agonists, such as poly(dA:dT), which activate cytosolic nucleic acid-sensors supposed to signal independently of IRAK-4. However, we did not determine experimentally whether this impairment in patient’s leukocytes was due to defective signaling of cytosolic nucleic-acid sensing pathways per se or merely indirect effects of such synthetic ligands. Similarly, diminished poly(I:C) responses observed in our patient’s cells might be TLR-3-independent. Further studies are needed to corroborate these results and elucidate the involvement of IRAK-4 in intracellular anti-viral immunity, particularly against HHV-6. HHV-6 is known to exert immunomodulatory and immunosuppressive effects on both lymphoid and nonlymphoid cells [34]. In addition, HHV-6 infection was shown to impair TLR-3-, TLR-4-, and TLR-7-mediated inflammatory responses in human PBMC-derived dendritic cells [35]. The first mouse model of HHV-6 infection suggests a role of TLR-9 in HHV-6-associated neuropathology [36]. However, almost no information is available concerning the active viral stage of HHV-6 in the central nervous system (CNS), the brain-intrinsic innate immune responses, and the human genetic basis of acute HHV-6 encephalitis. This study provides that acute HHV-6 encephalitis can be genetically determined in humans. It is also the first report of an inherited immunodeficiency associated with isolated severe HHV-6 infection causing encephalitis. Together with the previous report of AR IRAK-4 deficiency in anti-NMDAR encephalitis and HHV-6 reactivation in an infant [10], our findings expand the genotypic and phenotypic spectrum of human IRAK-4 deficiency and provide strong genetic and immunological evidences for the involvement of IRAK-4 in the pathogenesis of HHV-6 infection/reactivation in the brain. IRAK4 expression is abundant in CNS cells, particularly microglia, of mice and humans [37,38,39], yet further in-depth mechanistic studies including the use of in vitro inflammation/infection models involving primary cells will elucidate the possible role of IRAK-4 in encephalitis and also provide better understanding of allelic and clinical heterogeneity in human IRAK-4 deficiency.
Data availability
Not applicable.
Abbreviations
- AR:
-
Autosomal recessive
- DD:
-
Death domain
- HHV-6:
-
Human herpesvirus-6
- IFN:
-
Interferon
- IRAK-4:
-
Interleukin-1 receptor-associated kinase 4
- MAF:
-
Minor allele frequency
- MD:
-
Molecular dynamics
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor-α
- WES:
-
Whole-exome sequencing
References
Braun DK, Dominguez G, Pellett PE. Human herpesvirus 6. Clin Microbiol Rev. 1997;10(3):521–67.
Brown LS Jr. Seroepidemiology of HIV infection in two cohorts of intravenous drug users in New York City. J Natl Med Assoc. 1988;80(12):1313–7.
Levy JA, Ferro F, Greenspan D, Lennette ET. Frequent isolation of HHV-6 from saliva and high seroprevalence of the virus in the population. Lancet. 1990;335(8697):1047–50.
Okuno T, Takahashi K, Balachandra K, Shiraki K, Yamanishi K, Takahashi M, et al. Seroepidemiology of human herpesvirus 6 infection in normal children and adults. J Clin Microbiol. 1989;27(4):651–3.
Agut H, Bonnafous P, Gautheret-Dejean A. Laboratory and clinical aspects of human herpesvirus 6 infections. Clin Microbiol Rev. 2015;28(2):313–35.
Ongradi J, Ablashi DV, Yoshikawa T, Stercz B, Ogata M. Roseolovirus-associated encephalitis in immunocompetent and immunocompromised individuals. J Neurovirol. 2017;23(1):1–19.
Eliassen E, Lum E, Pritchett J, Ongradi J, Krueger G, Crawford JR, et al. Human herpesvirus 6 and malignancy: a review. Front Oncol. 2018;8:512.
Gewurz BE, Marty FM, Baden LR, Katz JT. Human herpesvirus 6 encephalitis. Curr Infect Dis Rep. 2008;10(4):292–9.
Jouanguy E, Beziat V, Mogensen TH, Casanova JL, Tangye SG, Zhang SY. Human inborn errors of immunity to herpes viruses. Curr Opin Immunol. 2020;62:106–22.
Nishimura S, Kobayashi Y, Ohnishi H, Moriya K, Tsumura M, Sakata S, et al. IRAK4 deficiency presenting with anti-NMDAR encephalitis and HHV6 reactivation. J Clin Immunol. 2021;41(1):125–35.
Picard C, Casanova JL, Puel A. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IkappaBalpha deficiency. Clin Microbiol Rev. 2011;24(3):490–7.
von Bernuth H, Picard C, Puel A, Casanova JL. Experimental and natural infections in MyD88- and IRAK-4-deficient mice and humans. Eur J Immunol. 2012;42(12):3126–35.
Boisson B. The genetic basis of pneumococcal and staphylococcal infections: inborn errors of human TLR and IL-1R immunity. Hum Genet. 2020;139(6–7):981–91.
Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;Chapter 7:Unit7 20.
Itan Y, Shang L, Boisson B, Ciancanelli MJ, Markle JG, Martinez-Barricarte R, et al. The mutation significance cutoff: gene-level thresholds for variant predictions. Nat Methods. 2016;13(2):109–10.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5.
Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc. 2016;11(1):1–9.
Belkaya S, Michailidis E, Korol CB, Kabbani M, Cobat A, Bastard P, et al. Inherited IL-18BP deficiency in human fulminant viral hepatitis. J Exp Med. 2019;216(8):1777–90.
Jordi M, Marty J, Mordasini V, Lunemann A, McComb S, Bernasconi M, et al. IRAK4 is essential for TLR9-induced suppression of Epstein-Barr virus BZLF1 transcription in Akata Burkitt’s lymphoma cells. PLoS ONE. 2017;12(10):e0186614.
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–308.
Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11(8):783–4.
Harayama T, Riezman H. Detection of genome-edited mutant clones by a simple competition-based PCR method. PLoS One. 2017;12(6):e0179165.
Ku CL, von Bernuth H, Picard C, Zhang SY, Chang HH, Yang K, et al. Selective predisposition to bacterial infections in IRAK-4-deficient children: IRAK-4-dependent TLRs are otherwise redundant in protective immunity. J Exp Med. 2007;204(10):2407–22.
Picard C, Puel A, Bonnet M, Ku CL, Bustamante J, Yang K, et al. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science. 2003;299(5615):2076–9.
Picard C, von Bernuth H, Ku CL, Yang K, Puel A, Casanova JL. Inherited human IRAK-4 deficiency: an update. Immunol Res. 2007;38(1–3):347–52.
Medvedev AE, Lentschat A, Kuhns DB, Blanco JC, Salkowski C, Zhang S, et al. Distinct mutations in IRAK-4 confer hyporesponsiveness to lipopolysaccharide and interleukin-1 in a patient with recurrent bacterial infections. J Exp Med. 2003;198(4):521–31.
Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465(7300):885–90.
Grossfield A, Patrone PN, Roe DR, Schultz AJ, Siderius DW, Zuckerman DM. Best practices for quantification of uncertainty and sampling quality in molecular simulations [Article v1.0]. Living J Comput Mol Sci. 2018;1(1).
Hoarau C, Gerard B, Lescanne E, Henry D, Francois S, Lacapere JJ, et al. TLR9 activation induces normal neutrophil responses in a child with IRAK-4 deficiency: involvement of the direct PI3K pathway. J Immunol. 2007;179(7):4754–65.
Yamamoto T, Tsutsumi N, Tochio H, Ohnishi H, Kubota K, Kato Z, et al. Functional assessment of the mutational effects of human IRAK4 and MyD88 genes. Mol Immunol. 2014;58(1):66–76.
Yang K, Puel A, Zhang S, Eidenschenk C, Ku CL, Casrouge A, et al. Human TLR-7-, -8-, and -9-mediated induction of IFN-alpha/beta and -lambda Is IRAK-4 dependent and redundant for protective immunity to viruses. Immunity. 2005;23(5):465–78.
Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O’Rourke K, et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci U S A. 2010;107(21):9771–6.
Unterholzner L. The interferon response to intracellular DNA: why so many receptors? Immunobiology. 2013;218(11):1312–21.
Dagna L, Pritchett JC, Lusso P. Immunomodulation and immunosuppression by human herpesvirus 6A and 6B. Future Virol. 2013;8(3):273–87.
Murakami Y, Tanimoto K, Fujiwara H, An J, Suemori K, Ochi T, et al. Human herpesvirus 6 infection impairs Toll-like receptor signaling. Virol J. 2010;7:91.
Reynaud JM, Jegou JF, Welsch JC, Horvat B. Human herpesvirus 6A infection in CD46 transgenic mice: viral persistence in the brain and increased production of proinflammatory chemokines via Toll-like receptor 9. J Virol. 2014;88(10):5421–36.
McCarthy GM, Bridges CR, Blednov YA, Harris RA. CNS cell-type localization and LPS response of TLR signaling pathways. F1000Res. 2017;6:1144.
Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34(36):11929–47.
Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron. 2016;89(1):37–53.
Acknowledgements
We thank the patient and family for participating in this study. We thank İhsan Gürsel and his laboratory members for their reagent sharing and technical help. We also thank Emel Timuçin for her critical evaluation regarding the molecular dynamics. The numerical calculations reported in this paper were performed using TÜBİTAK ULAKBİM High Performance and Grid Computing Center (TRUBA) resources.
Funding
This study was supported in part by Installation Grant from the European Molecular Biology Organization (EMBO), International Fellowship for Outstanding Researchers from the Scientific and Technological Research Institution of Turkey (TÜBİTAK), and the Science Academy’s Young Scientist Awards Program (BAGEP) (to S. Belkaya). The funders had no role in the design and conduct of this study, in the collection, analysis, and interpretation of the data, or in the preparation, review, or approval of the manuscript.
Author information
Authors and Affiliations
Contributions
S.B. conceived the study. S.B., C.A., and M.O. recruited the patient. S.B., Z.G.T., Y.Y.Y., U.T., and L.I.K. designed and carried out the experiments and performed the analyses. S.B. wrote the manuscript and designed the figures with input from all other authors.
Corresponding author
Ethics declarations
Ethics Approval
This study was conducted in accordance with the institutional, local, and national ethical guidelines, and approved by the İhsan Doğramacı Bilkent University Ethics Committee (#2022_04_28_01).
Consent to Participate
Written informed consent was obtained from all individual participants included in this study and parents of the patient.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tepe, Z.G., Yazıcı, Y.Y., Tank, U. et al. Inherited IRAK-4 Deficiency in Acute Human Herpesvirus-6 Encephalitis. J Clin Immunol 43, 192–205 (2023). https://doi.org/10.1007/s10875-022-01369-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-022-01369-4